The Role of beta1 Integrin Subfamily in Anchorage-Dependent Apoptosis of Breast Carcinoma Cells Differing in Multidrug Resistance
G. E. Morozevich1, N. I. Kozlova1, M. E. Preobrazhenskaya1, N. A. Ushakova1, I. A. Eltsov1, A. A. Shtil2, and A. E. Berman1*
1Orekhovich Institute of Biomedical Chemistry, Russian Academy of Medical Sciences, ul. Pogodinskaya 10, 119121 Moscow, Russia; fax: (7-495) 245-0857; E-mail: berman@ibmc.msk.ru2Blokhin Russian Cancer Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia
* To whom correspondence should be addressed.
Received November 8, 2005; Revision received January 6, 2006
Integrin expression was investigated in MCF-7 human breast adenocarcinoma line and in the MCF-7Dox line, which was selected from MCF-7 by a resistance to multiple antitumor drugs (MDR). We have shown that acquisition of MDR was accompanied by a drastically reduced expression of some integrins of the beta1-subfamily (alpha2beta1, alpha3beta1, alpha6beta1) and of alphavbeta5 intergin in the adenocarcinoma cells. In contrast, expression of alpha5beta1 integrin was markedly increased in the MDR cells. Along with multiple antitumor drug resistance, MCF-7Dox cells demonstrate elevated resistance to anchorage-dependent apoptosis (anoikis) and enhanced in vitro invasive activity. To elucidate the implication of beta1-integrins in the above phenotypic modifications, the effect of beta1-integrin signaling was assayed. Stimulation of beta1-mediated signaling was accomplished by treating of the cells with antibodies to the beta1-subunit common for members of the beta1-subfamily. These data show that activation of beta1-integrin signaling markedly upregulated anoikis of the adenocarcinoma cells.
KEY WORDS: integrins, extracellular matrix, anoikis, metastasis, invasionDOI: 10.1134/S000629790605004X
Abbreviations: MDR) resistance to multiple antitumor drugs; polyHEM) poly(2-hydroxyethyl methacrylate); PCR) polymerase chain reaction; RT) reverse transcription.
Drug resistance of tumor cells is one of the main factors hindering
effective chemotherapy of cancer. The phenomenon of multidrug
resistance (MDR) has been described in which cells surviving one
pharmaceutical acquire resistance to others. Among numerous mechanisms
of MDR, the role of a plasma membrane glycoprotein with molecular mass
of 140-170 kD (P-glycoprotein, Pgp170) is the most studied. This
protein transports from the cell a number of natural metabolites and
xenobiotics including pharmaceuticals used in chemotherapy of tumors
[1, 2].
Other mechanisms are known that are not mediated by Pgp-dependent transport [3]. Recently, resistance has been reported against antitumor agents upon cell adhesion to extracellular matrix proteins [4-6]. These data are indicative of possible involvement of integrins in MDR; these cell surface matrix-specific receptors mediate both the cell-matrix interaction and signal transduction controlling various intracellular processes including those associated with cell survival [7, 8].
Integrins comprise a large family of heterodimers composed of alpha- and beta-subunits linked together with non-covalent bonds. Many beta-subunits can form dimers with different alpha-subunits, which is the basis for classification of integrin subfamilies (such as beta1, beta2, beta3, and so on) [8, 9]. Most integrins possess crossed ligand specificity, that is, each receptor can bind two or more matrix proteins, and each matrix protein can form complexes with at least two receptors [8, 9].
Integrins may be involved in drug resistance via their role in anchorage-dependent apoptosis, anoikis. The mechanism of anoikis is that integrins bound to the extracellular matrix provide signals hindering cell death, whereas disruption of these bonds leads to apoptosis [10, 11]. This mechanism is supposed to underlie higher resistance of cells attached to a substrate in comparison with the cells suspended in a medium [5, 12, 13].
Down-regulation of integrin signal activity that controls apoptotic cell death might be a mechanism accounting for drug resistance [14, 15]. It was shown, for instance, that sensitivity of some tumor cell types to cytosar, cisplatin, and camptothecin is due to integrin-mediated activation of the tumor suppressor p53, which stimulates apoptosis, and the oncogene c-Abl [14]. On the other hand, integrin alpha4beta1 induces resistance of B-cellular lympholeukosis cells to fludarabine by activation of the anti-apoptotic protein Bcl-xL and inhibition of p53 activity [16].
However, pathways of transduction of integrin-mediated signals controlling apoptosis require further investigation. In particular, it is unclear whether integrins can only transmit protective (anti-apoptotic) signal, and cell death in anoikis is due to the blocking of this signal. Can these receptors generate pro-apoptotic signals inducing cell death upon detaching of cell-matrix contacts? In a gut carcinoma cell line, we first found that the vitronectin-specific integrin alphavbeta3 generates a signal enhancing apoptosis, upon disruption of cell-substrate contacts [17]. Ability to stimulate apoptotic cell death was also found in some other integrins [18, 19]. Hence, the cells that do not express these integrins might have higher vitality. One can expect that one of the mechanisms forming drug-resistant cell population is selection of cells that do not express one or more such integrins. This suggestion is confirmed by our data demonstrating decreased expression of alphavbeta3 integrin in tumor cells upon acquisition of MDR [20].
In the present study, we have shown that human breast adenocarcinoma cells acquiring MDR during selection in the presence of the antitumor drug doxorubicin compared with the initial cells are characterized by drastically decreased expression of most beta1-subfamily integrins. Multidrug-resistant cells compared with parental cells exhibit significantly increased resistance to anoikis and are significantly more active in invasion in vitro. We have first found that up-regulation of beta1-integrin signaling in drug-sensitive cells leads to a marked enhancement of anchorage-dependent apoptosis.
MATERIALS AND METHODS
Chemicals. Monoclonal antibodies BHA2.1 against integrin alpha2beta1 and P3G8 against the alphav-subunit and polyclonal antibodies against the cytodomains of subunits alpha1, alpha2, alpha4, alpha5, beta1, beta3, and beta5 were purchased from Chemicon Int. (USA). Monoclonal antibodies P1B5 against the receptor alpha3beta1 were purchased from Dako Corp. (USA). Monoclonal antibodies ICO-53 against the histocompatibility complex HLA-ABC were kindly provided by Dr. Yu. A. Baryshnikov (Blokhin Oncology Research Center, Russian Academy of Medical Sciences, Moscow). Biotinamidocaproate N-hydroxysuccinimide ester (NHS-biotin), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrasolium bromide (MTT), and poly(2-hydroxyethyl methacrylate) (polyHEM) were purchased from Sigma (USA). Oligodeoxyribonucleotides were synthesized by Syntol (Moscow, Russia). Reverse transcription (RT) and polymerase chain reaction (PCR) kits were purchased from Gibco (USA).
Cell lines. Human breast adenocarcinoma cell line MCF-7 was purchased from ATCC (American Type Culture Collection, USA). Its variant MCF-7Dox selected in the presence of doxorubicin was kindly provided by Prof. T. N. Ignatova (University of Illinois at Chicago, USA). The cells were cultured in the DMEM medium containing 10% fetal calf serum (HyClone, USA), 2 mM L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin at 37°C in an atmosphere of 5% CO2. The MCF-7Dox cells were cultured in presence of 1.7 µM doxorubicin. Cells from logarithmic growth-phase were used in all experiments.
Biotinylation of cell surface proteins, immunoprecipitation of integrins, and electrophoresis of proteins were carried out as described previously [21]. Immunoblots were treated with streptavidin-peroxidase and developed in a ECL (Enhanced Chemiluminescence) system (Amersham, UK).
RT-PCR was performed as described previously [17]. Primer sequences and PCR conditions for integrin subunits are given in Table 1.
Table 1. PCR conditions for amplification of
integrin cDNAs
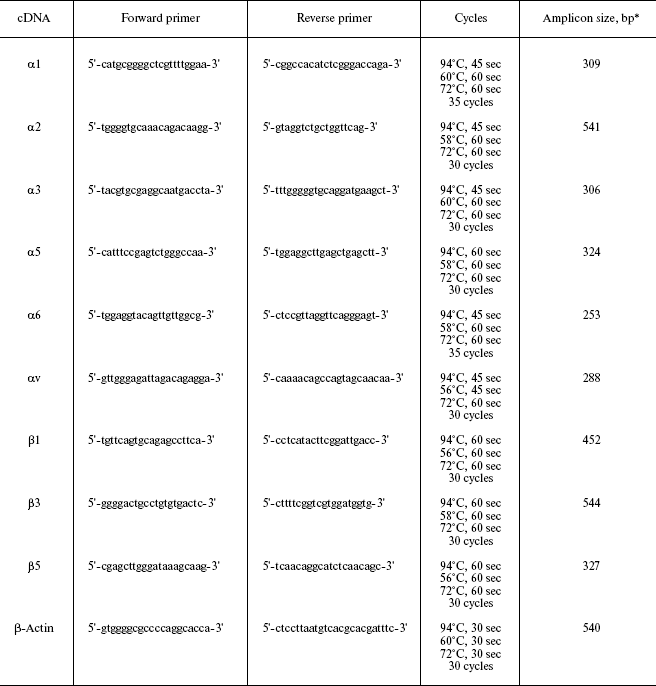
*bp, base pairs.
Invasion was determined in Transwell (Costar) as described previously [22].
Anoikis determination was based on apoptotic signs (intranucleosomal DNA degradation) occurring when cell were grown on the non-adhesive substrate polyHEM prepared as described elsewhere [23]. DNA degradation was determined using a Cell Death Detection ELISA Kit (Roche Diagnostic GmbH, Germany) according to the manufacturer's protocol. This assay is based on quantitative enzyme-linked determination of histones in mono- and oligonucleosomes accumulating during DNA fragmentation.
In experiments on the effect of antibody-induced activation of integrins on anoikis, apoptosis was determined from incorporation of radioactive thymidine into intact and degraded DNA [24] in our modification. Cells were incubated in a medium containing [14C]thymidine (2 µCi/2*105 cells) for 17 h at 37°C, washed with phosphate buffer saline (PBS) for removal of the unincorporated label, and incubated for 1 h at 37°C. Then the cells were detached with EDTA-trypsin, suspended in medium containing 2.5% fetal serum, and incubated on substrates composed of immobilized antibodies (see below) at 37°C for 3.5 h. Then the cells were collected with EDTA-trypsin, suspended in medium containing 10% fetal serum, and incubated on polyHEM for 24 h at 37°C. Following the incubation, the cells were lysed in 20 mM Tris-HCl buffer, pH 7.4, containing 4 mM EDTA and 0.4% Triton X-100. The lysate was centrifuged at 15,000 rpm, the pellet was dissolved in buffer containing 1% SDS, and radioactivity was determined both in the pellet (intact DNA) and supernatant (fragmented DNA). Apoptosis (%) = radioactivity of supernatant/(radioactivity of pellet + radioactivity of supernatant).
To prepare substrates from immobilized antibodies, 24-well plates were treated with rabbit antibodies against mouse IgG (25 µg/ml) for 2 h at 37°C, washed with PBS, blocked with albumin (10 mg/ml in PBS, 1 h, 37°C) followed by washing with the culture medium, and the plates were incubated for 18 h at 4°C with antibodies against beta1, alphav, and histocompatibility complex HLA-ABC, dilution 1 : 100. Then the plates were washed three times with the medium and used for cell plating as described above.
Statistical analysis of data was performed by Student's t-test using Sigma-Plot software. Difference between cell lines was considered to be statistically significant at p . 0.05.
RESULTS AND DISCUSSION
Substrate-anchorage apoptosis of drug-resistant and drug-sensitive cells. Examination of drug resistance of MCF-7Dox cells has shown that they are resistant not only to doxorubicin, but also to the antitumor drugs vincristine, taxol, and mitoxantrone (data not shown). Thus, these cells possess multidrug resistance (MDR). Moreover, unlike MCF-7, MCF-7Dox cells express the active form of Pgp170 protein (data not shown).
Since the role of integrins in MDR may be determined by their role in anoikis (see introduction), we have studied this type of cell death in these cells.
Quantitative analysis of anoikis was performed using enzyme-linked assay of histones associated with mono- and oligonucleosomal DNA (Fig. 1). It is seen from the figure that, before plating on non-adhesive substrate, the cells of both lines do not differ in contents of degraded DNA. However, after 24 h in suspension, the degraded DNA contents in MCF-7Dox cells were about 3.5-fold less than in MCF-7 cells. So, acquisition of MDR by cells is accompanied by significant elevation of resistance to anchorage-dependent apoptosis.
Expression and role of integrins in anchorage-dependent apoptosis. Since integrins play a key role in transduction of signals controlling anoikis [10, 11], one can suppose that modifications in expression of these receptors underlie the found difference between the analyzed cell lines.Fig. 1. Sensitivity of MCF-7 and MCF-7Dox cells to anoikis. Lysate prepared from about 3000 cells of each type was used in ELISA. Light columns, cells before incubation on polyHEM; hatched columns, cells after 24 h of incubation on polyHEM. * p < 0.001 (MCF-7 after incubation on polyHEM in relation to MCF-7 before incubation on poly HEM).

Immunoblotting (Fig. 2) and RT-PCR (Fig. 3) show that drug-resistant and sensitive cells strongly differ in the spectrum of expressed integrins. The resistant line demonstrates drastic down-regulation of most integrins of beta1 and alphav subfamilies--both at the level of cell surface expression and transcription of genes encoding integrin subunits. It is seen that unlike MCF-7, MCF-7Dox cells do not express the collagen-specific receptor alpha2beta1, collagen- and laminin-specific receptor alpha3beta1, laminin-specific integrin alpha6beta1, and vitronectin-specific integrin alphavbeta5. At the same time, fibronectin-specific receptor alpha5beta1 is a major integrin in drug resistant cells whereas in the wild type MCF-7 cells its expression is negligible. In addition, some enhancement of collagen-specific integrin alpha1beta1 expression was found in the MCF-7Dox cells.
It is seen from Fig. 3 that alterations in gene expression of integrin subunits accompanying MDR completely correspond to alterations in surface expression of these receptors. In MCF-7Dox cells, the genes encoding alpha5 and beta1 subunits that form alpha5beta1 dimer are the most active. In parental line, the genes encoding alpha2, alpha3, alphav, as well as beta1 and beta5 subunits, i.e., the subunits comprising dimers alpha2beta1, alpha3beta1, and alphavbeta5, possess the maximum activity.Fig. 2. Surface expression of integrins in MCF-7 (1) and MCF-7Dox (2) cells. Cell membrane proteins were biotinylated (see “Materials and Methods”), and the integrin dimers were precipitated by antibodies against individual subunits or (in the case of alphavbeta5) against the complete dimer.
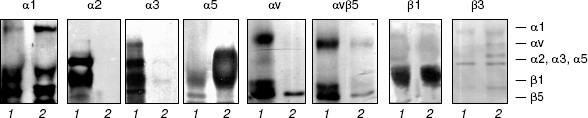
The observed changes in integrin expression might be considered as an accidental feature not related to anoikis mechanisms. One can suggest, however, that the parental cells, under disruption of their contacts with matrix, realize a signal to apoptosis, which is mediated by some integrins of beta1 and/or alphav family. In this case, the absence of these receptors in MCF-7Dox cells could explain their elevated resistance to anchorage-dependent apoptosis. Moreover, resistance of these cells to anoikis can be an additional mechanism to the well-characterized Pgp170-mediated efflux of a variety of drugs out of the cell.Fig. 3. RT-PCR analysis of genes encoding integrin subunits in MCF-7 (1) and MCF-7Dox (2) cells. Primer sequences and reaction conditions are given in “Materials and Methods”.

To test this suggestion, the MCF-7 cells were plated on antibodies to beta1 or alphav subunits immobilized on plastic. Adhesion to such substrate results in clustering of corresponding cell surface receptors and activation of their signaling function [17].
The data of Table 2 show that the activation of MCF-7 cells by immobilized beta1-subunit antibodies enhances anoikis by 70% in comparison with the control, in which anoikis was analyzed after adhesion on “neutral” antibodies (against the HLA histocompatibility complex) or on poly-L-lysine. Activation by antibodies against the alphav-subunit had no visible effect on anoikis of MCF-7 cells.
Table 2. Activation of signaling function of
beta1-integrins enhances anoikis of MCF-7 cells
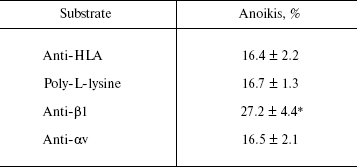
Note: The cells were incubated on the indicated substrates followed
by plating on the non-adhesive substrate (see “Materials and
Methods”). Anoikis was determined as a ratio (%) of degraded DNA
radioactivity to the sum of native and degraded DNA radioactivities of
cells plated on non-adhesive substrate. The results are mean ±
s.e.m. of three independent experiments. * p < 0.05 relative
to controls (anti-HLA and poly-L-lysine).
Invasion in vitro of drug-resistant and drug-sensitive cells. The ability of tumor cells to survive in the absence of matrix contacts is a prerequisite of their invasion into surrounding tissues and metastasis formation, a hallmark of malignant progression [10, 25]. To this end, one may suppose that MCF-7Dox cells differ from the parental ones by a more severe malignant phenotype. This suggestion was confirmed by an in vitro invasion assay demonstrating that invasive activity of drug-resistant cells is 10-fold higher than of their sensitive counterparts (Table 3).
Table 3. Invasion in vitro of MCF-7
and MCF-7Dox cells
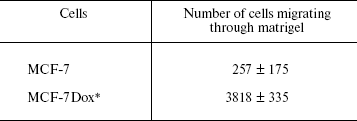
Note: Matrigel (150 µg) was polymerized on Transwell
membranes and 5*104 cells in 300 µl DMEM medium
containing 0.5% fetal serum were applied into the upper chamber. The
cells that migrated into the lower chamber were counted after
120 h of incubation at 37°C. The data are mean ±
s.e.m. of three independent experiments.
There are conflicting data in the literature concerning expression and the role of integrins in apoptosis, drug resistance, and progression of tumor cells. For example, drug-resistant cells of ovary carcinoma are different from their sensitive precursors in their elevated expression level of integrins alpha2beta1 and alpha6beta1, as well as enhancement of invasion in vitro [26]. Higher expression levels of subunits alpha2, alpha5, alpha6, beta1, and beta4, collagenases MMP-2 and MMP-9, as well as enhanced invasive activity were observed in a melphalan-resistant subline of nasopharyngeal cancer, as compared with the initial sensitive line, whereas taxol-resistant subline was characterized by decreased expression of alpha2beta1 and was indistinguishable from the initial one in invasion in vitro [27]. Our results confirm the data of other authors demonstrating the up-regulation of alpha5beta1 receptor [28] and down-regulation of integrin alpha2beta1 [29] in MCF-7 cells acquiring MDR, while a decrease in integrin alpha5beta1 expression was detected in melanoma cells with various levels of Pgp-mediated MDR [30].
The ambiguity of these results can be explained by the fact that, as indicated above, many integrins possess overlapping ligand affinity and, hence, various receptors can transmit physiologically equal signals when interacting with the same matrix protein [8, 22]. One can anticipate that evolution of a particular cell population is not associated with changes in a single integrin but it is rather a spectrum of these receptors that ensures the optimal adaptation of cells to microenvironment and development of the resulting phenotype: MDR, invasiveness, and metastasis formation.
In the present work, using a model of breast carcinoma cells we have shown for the first time the ability of beta1 family integrins to stimulate anoikis, while alphavbeta5 receptor has no such capability. Furthermore, we have previously demonstrated that integrin alphavbeta3 similar in its ligand properties to integrin alphavbeta5 induces the anchorage-dependent apoptosis in gut carcinoma cells [17]. The ability to induce opposite effects on apoptosis in various cells types has been reported for integrins alpha6beta4 [19, 31] and alpha4beta1 [18, 32].
Our finding that stimulation of beta1-integrin signaling resulted in an enhanced anoikis of drug-sensitive cells correlates with the drastic down-regulation of the most beta1-receptors and markedly increased resistance to anoikis in MDR cells. Of interest, the decreased expression of collagen- and laminin-specific integrins alpha2beta1 and alpha3beta1 known to be involved in cell differentiation, and enhanced expression of vitronectin- and fibronectin-specific alphavbeta3 and alpha5beta1 receptors implicated in invasion and proliferation are often observed in primary breast carcinomas [12]. A correlation between integrin-controlled resistance to anoikis and drug-resistance of tumor cells was documented in a number of studies [16, 32, 33].
The mechanisms of integrin-mediated apoptosis are poorly known. In endothelial cells, alphavbeta3 and beta1-integrins were shown to generate pro-apoptotic signal provided that none of them interacts with its own ligand, although cell attachment to a substrate is not disrupted [11]. The authors coined the term “integrin-mediated death” for such type of apoptosis. Non-ligated integrin proved to interact with and activate caspase 8. Mechanisms of integrin-mediated death and anoikis are apparently different [11].
Of substantial interest are the data that integrin activation induced by cell-matrix interaction up-regulates p53 and enhances sensitivity to antitumor agents damaging DNA (doxorubicin, etopozide, etc.) [14]. Detachment of cells from substrate results in decrease in p53 level and increase in drug resistance. This effect appeared to be cell type specific. If the cell lines studied in the present work are of such type, one can anticipate that decrease in expression of beta1-integrins in MCF-7Dox cells is associated with their diminished sensitivity to anoikis and higher drug resistance in comparison with MCF-7.
In conclusion, some suggestion may be made concerning the role of integrin alpha5beta1 in MCF-7Dox cells. Enhancement of expression of this receptor in cells with MDR apparently has a dual effect: 1) many investigators have documented a protective effect of alpha5beta1 from anoikis [34, 35], and this finding allows a supposition that this receptor contributes to higher resistance of MCF-7Dox cells both to anchorage-dependent apoptosis and MDR; 2) the signals mediated by the receptor alpha5beta1 stimulate production of matrix-specific metalloproteinases [36] and, hence, favor invasion and metastasis formation in tumor cells. These data are in agreement with the results obtained in the present study.
This study was supported by the Russian Foundation for Basic Research (project Nos. 03-04-48968, 05-04-49695, and 06-04-49066).
REFERENCES
1.Roninson, I. B., Abelson, H., Housman, D. E.,
Howell, N., and Varshavsky, A. (1984) Nature, 309,
626-628.
2.Roninson, I. B. (1997) Encyclopedia of
Cancer, 2, 1095-1107.
3.Barzilay, G., and Hickson, I. D. (1997)
Encyclopedia of Cancer, 1, 532-548.
4.Damiano, J. S., Cress, A. E., Hazlehurst, L. A.,
Shtil, A. A., and Dalton, W. S. (1999) Blood, 93,
1658-1667.
5.Elliott, T., and Sethi, T. (2002) Expert. Rev.
Anticancer Ther., 2, 449-459.
6.Hazlehurst, L. A., Landowski, T. H., and Dalton, W.
S. (2003) Oncogene, 22, 7396-7402.
7.Rintoul, R. C., and Sethi, T. (2002) Clin.
Sci., 102, 417-424.
8.Berman, A. E., Kozlova, N. I., and Morozevich, G.
E. (2003) Biochemistry (Moscow), 68, 1284-1299.
9.Eble, J. A. (1997) in Molecular Biology
Intelligence Unit. Integrin-Ligand Interaction (Eble, J. A., and
Kunn, K., eds.) Chapman and Hall, New York, pp. 1-40.
10.Frisch, S. M., and Screaton, R. A. (2001)
Curr. Opin. Cell Biol., 13, 555-562.
11.Cheresh, D. A., and Stupack, D. G. (2002)
Nature Med., 8, 193-194.
12.Chrenek, M. A., Wong, P., and Weaver, V. M.
(2001) Breast Cancer Res., 3, 224-229.
13.Damiano, J. S. (2002) Curr. Cancer Drug
Targets, 2, 37-43.
14.Truong, T., Sun, G., Doorly, M., Wang, J. Y. J.,
and Schwartz, M. A. (2003) Proc. Natl. Acad. Sci. USA,
100, 10281-10286.
15.Yeh, Y. Y., Chuang, S. E., Yeh, K. H., Song, Y.
C., Chang, L. L. Y., and Cheng, A. L. (2004) Oncogene,
23, 3580-3588.
16.Fuente, M. T., Casanova, B., Cantero, E., del
Cerro, M. H., Garcia-Marco, J., Silva, A., and Garcia-Pardo, A. (2003)
Biochem. Biophys. Res. Commun., 311, 708-712.
17.Kozlova, N. I., Morozevich, G. E., Chubukina, A.
N., and Berman, A. E. (2001) Oncogene, 20, 4710-4717.
18.Marco, R. A. W., Diaz-Montero, C. M., Wygant, J.
N., Kleinerman, E. S., and McIntyre, B. W. (2003) J. Cell.
Biochem., 88, 1038-1047.
19.Bachelder, R. E., Marchetti, A., Falcioni, R.,
Soddu, S., and Mercurio, A. M. (1999) J. Biol. Chem.,
274, 20733-20737.
20.Kozlova, N. I., Morozevich, G. E., Shtil, A. A.,
and Berman, A. E. (2004) Biochem. Biophys. Res. Commun.,
316, 1173-1177.
21.Kozlova, N. I., Morozevich, G. E., Solovyeva, N.
I.,Vinokurova, S. V., and Berman, A. E. (1997) Biochem. Mol. Biol.
Int., 43, 529-539.
22.Morozevich, G. E., Kozlova, N. I., Chubukina, A.
N., Eltsov, I. A., and Berman, A. E. (2004) Biochemistry
(Moscow), 69, 665-673.
23.Frisch, S. M., and Francis, H. (1994) J. Cell
Biol., 124, 619-626.
24.Rozzo, C., Chiesa, V., Caridi, G., Pagnan, G.,
and Ponzoni, M. (1997) Int. J. Cancer, 70, 688-698.
25.Yawata, A., Adachi, M., Okuda, H., Naishiro, Y.,
Takamura, T., Hareyama, M., Takayama, S., Reed, J. C., and Imai, K.
(1998) Oncogene, 16, 2681-2686.
26.Sedlak, J., Sedlakova, O., Hlavcak, P., Hunakova,
L., Bizik, J., Grofova, M., and Chorvath, B. (1996) Neoplasma,
43, 389-395.
27.Liang, Y., Meleady, P., Cleary, I., McDonnel, S.,
Connoly, L., and Clynes, M. (2001) Eur. J. Cancer, 37,
1041-1052.
28.Nista, A., Leonetti, C., Bernardini, G.,
Mattioni, M., and Santoni, A. (1997) Int. J. Cancer, 72,
133-141.
29.Narita, T., Kimura, N., Sato, M., Matsuura, N.,
and Kannagi, R. (1998) Anticancer Res., 18, 257-262.
30.Meyer, N., Duensing, S., Anastassiou, G., Brevis
Nunez, F., Grosse, J., Ganser, A., and Atzpodien, J. (1999)
Anticancer Res., 19, 1509-1512.
31.Dowling, J., Yu, Q., and Fuchs, E. (1996) J.
Cell Biol., 134, 559-572.
32.Matsunaga, T., Takemoto, N., Sato, T., Takimoto,
R., Tanaka, I., Fujimi, A., Akiyama, T., Kuroda, H., Kawano, Y.,
Kobune, M., Kato, J., Hirayama, Y., Sakamaki, S., Kohda, K., Miyake,
K., and Niitsu, Y. (2003) Nature Med., 9, 1158-1165.
33.Schmidt, M., Hovelmann, S., and Becckers, T. L.
(2002) Brit. J. Cancer, 87, 924-932.
34.Lee, J. W., and Juliano, R. L. (2000) Mol.
Biol. Cell, 11, 1973-1987.
35.Matter, M. L., and Ruoslahti, E. (2001) J.
Biol. Chem., 276, 27757-27763.
36.Werb, Z., Tremble, P. M., Behrendtsen, O.,
Crowley, E., and Damsky, C. H. (1989) J. Cell Biol., 109,
877-889.