Molecular Model and ATPase Activity of Carboxyl-Terminal Nucleotide Binding Domain from Human P-Glycoprotein
Feng Qian, Dongzhi Wei*, Jianglan Liu, and Shengli Yang
State Key Laboratory of Bioreactor Engineering, New World Institute of Biotechnology, East China University of Science and Technology, Shanghai 200237, P. R. China; fax: 8621-64250068; E-mail: dzhwei@ecust.edu.cn* To whom correspondence should be addressed.
Received October 8, 2004; Revision received December 28, 2004
ATP binding and hydrolysis are required for P-glycoprotein mediated multidrug resistance. To investigate the molecular mechanism involved in ATP binding and hydrolysis, a three-dimensional model of the carboxyl-terminal nucleotide binding domain (NBD2) was built by homology modeling. Modeling revealed the human P-glycoprotein ATP-binding site and the possible role of conserved Gln1118 residue. Recombinant NBD2 was overexpressed in Escherichia coli and the conserved Gln1118 residue was mutated to an alanine residue. The Vmax for ATP hydrolysis by the mutant NBD2 was ~56% of the Vmax of wild-type NBD2. But both proteins displayed similar affinity for ATP, with Km of 479 and 466 µM for mutant and wild-type NBD2, respectively. These results suggest that the possible role of Gln1118 is as an activating residue for ATP hydrolysis. The molecular model also provided structural information about the interactions between NBD2 and the chemosensitizer quercetin. The complex indicated that quercetin was tightly bound to the ATP-binding site and competed for binding. The three-dimensional model of NBD2 can be used to both guide enzymological studies and provide a theoretical basis for the design of potential multidrug resistance reversers.
Key words: molecular modeling, multidrug resistance, nucleotide binding domain, P-glycoprotein, site-directed mutagenesisDOI: 10.1134/S0006297906130037
Abbreviations: MDR) multidrug resistance; Pgp) P-glycoprotein; NBD1) N-terminal nucleotide binding domain; NBD2) carboxyl-terminal nucleotide binding domain.
The ATP-binding cassette (ABC) transporter superfamily is the largest
family of transmembrane proteins. These proteins bind ATP and use its
energy to drive the transport of various molecules across cell
membranes [1]. One of the distinctive
characteristics of ABC transporters is the presence of a nucleotide
binding domain (NBD). The NBDs of ABC transporters contain several
conserved regions such as the Walker-A (GXXGXGKS/T [X, any amino acid])
and Walker-B (XXXXDE/D [X, hydrophobic residue]) motifs [2] and, in between, the Signature motif (LSGGQ) [3].
P-Glycoprotein (Pgp, the product of MDR1 gene) that is a member of the ABC transporter superfamily can confer multidrug resistance (MDR) to tumor cells by pumping chemotherapeutic drugs from the cytoplasm [4]. The key role of ATP binding and hydrolysis in Pgp-mediated drug transport has been demonstrated in transport studies with membrane vesicles from Pgp positive cells and with purified Pgp reconstituted in liposome [5]. Chemosensitizer, which blocks Pgp ATPase activity, can revert cell MDR phenotype. NBDs are responsible for ATP binding and hydrolysis. Several recombinant proteins containing N-terminal and C-terminal NBDs (NBD1 and NBD2, respectively) derived from various ABC transporters have been expressed to investigate the ATP binding and hydrolysis [6, 7]. Single point mutations at the key lysine reside in the Walker-A motif and at the key aspartic acid in the Walker-B motif of NBD have been shown to reduce or abrogate the ATP hydrolysis activity and in some cases impair nucleotide binding [8, 9]. No high resolution structural data of Pgp is available, and reports of expression and biochemical study of NBD2 from human Pgp are scarce, these facts limiting our understanding of molecular mechanisms involved in ATP binding and hydrolysis.
In this study, we performed homology modeling of NBD2 from human P-glycoprotein. The functional role of Gln1118 was investigated by the studying the biochemical properties of recombinant NBD2 and its mutant Q1118A. The chemosensitizer quercetin was docked in the modeled structure of NBD2, and the interactions with human P-glycoprotein were investigated.
MATERIALS AND METHODS
Alignment and molecular modeling. Multiple alignment was built by ClustalX [10] software using the Gonnet series protein weight matrix. The template Haemolysin B, 1MT0 [11] was used to build the model of nbd1 with the identity of 48%. The template human cTAP1, 1JJ7 [12] was used to build the model of nbd2 with the identity of 46%. The program MODELLER [13] was used to search for the optimal side chain packing in the NBD structural model and to refine the main chain positions as well using the simulated annealing molecular dynamics protocol. The ideal bond lengths and angles were used as a restraint during the simulated annealing optimization process. To avoid being trapped in local minimum of the global energy landscape, 50 independent runs of optimization starting from a random seed were carried out to generate 50 models. The model with the lowest value of the MODELLER objective function was thus chosen as the final model for the NBD structure. The final model was analyzed by structure validation program, PROCHECK [14], which evaluated stereochemical quality of a protein structure. The dimer model of NBDs was modeled based on the Rad50 dimer PDB structure (1F2U) [15]. The models of NBDs were superimposed onto the Rad50 dimer using the Magic Fit option of Swiss-Pdb Viewer 3.7 [16]. The resulting dimer model of NBDs was submitted to Swiss Model optimize mode where energy minimization is performed using Gromos96. Structures were visualized with Swiss-Pdb Viewer 3.7.
Molecular docking. The molecular docking program AutoDock [17], which uses the Lamarckian genetic algorithm method for conformational search and docking, was applied to perform the automated molecular docking simulations. The number of generations, energy evaluations, and docking runs were set to 370,000, 1,500,000, and 20, respectively. The kinds of atomic charges were taken as Kollman-all-atom for receptor and Gasteiger-Hückel for the ligands.
Construction of the expression plasmid encoding NBD2 and its mutant Q1118A. The NBD2 DNA fragment of MDR1 (encoding 254 amino acids; Met1027-Gln1280 of human P-glycoprotein) was amplified from cDNA of MDR cell line KB-A1 by polymerase chain reaction (PCR) with the following primers: primer 1, 5´-CGGGATCCATGCCGAACACATTG-3´; primer 2, 5´-CGGAATTCTCACTGGCGCTTTG-3´. These primers contained BamHI and EcoRI sites (underlined) for cloning of the DNA fragment, in frame, into expression vector pGEX-4T-1. The PCR fragment digested with BamHI and EcoRI was cloned into pGEX-4T-1. Escherichia coli strain BL21 was transformed with the expression plasmid and grown on agar plates containing 100 µg/ml ampicillin. Sequences of plasmid clones were confirmed by DNA sequencing. For site-directed mutagenesis to introduce a Q1118A mutation, nucleotide mutations at positions 3352 (C→G) and 3353 (A→C) were achieved by sequence overlap PCR methodology. Two internal complementary primers (primer 3, 5´-ATCGTGTCCGCGGAGCCCAT-3´; primer 4, 5´-ATGGGCTCCGCGGACACGAT-3´) were used in conjunction with two outer primers (primer 1, primer 2). The resulting fragment was recloned into the pGEX4T-1 vector. The sequence of plasmid containing NBD2 Q1118A mutant was confirmed by sequencing.
Expression, renaturation, and purification of NBD2 and its mutant Q1118A. Escherichia coli BL21 cells containing NBD2 and its mutant Q1118A were cultured in 200 ml of 2YTA medium (16 g/liter tryptone, 10 g/liter yeast extract, 5 g/liter NaCl, and 100 µg/ml ampicillin) at 37°C with vigorous shaking until the absorbance at 600 nm reached 1.2 units. The culture was then induced with 10 g/liter lactose and continued for an additional 12 h at 30°C. Cells were harvested by centrifugation at 5000g for 10 min at 4°C and resuspended in lysis buffer (20 mM Tris-HCl, pH 8.0, 1 mM EDTA, and 1 mg/ml lysozyme). The cells were lysed at room temperature for 30 min, and the suspension was sonicated. Most of the recombinant protein was insoluble and recovered as inclusion bodies. The pellet from centrifugation at 20,000g for 20 min at 4°C was resuspended in wash buffer (20 mM Tris-HCl, pH 8.0, 1 mM EDTA, and 1% (v/v) Triton X-100). Inclusion bodies were homogenized in solubilization buffer (20 mM Tris-HCl, pH 8.0, 8 M urea, 2 mM DTT) and incubated at room temperature for at least 2 h. Renaturation was initiated by a rapid 10-fold dilution of the solubilized inclusion bodies into refolding buffer (20 mM Tris-HCl, pH 8.0, 10 mM NaCl, 2 M urea, 5% (v/v) glycerol, 1 mM reduced glutathione, 0.1 mM oxidized glutathione) and maintained for 12 h. Renatured protein was dialyzed against 20 mM Tris-HCl, pH 8.0, 10 mM NaCl overnight, and then applied to a DEAE column. The column was washed with wash buffer (20 mM Tris-HCl, pH 8.0, 10 mM NaCl) and the retained proteins were then eluted with 20 ml of a linear NaCl gradient (10-200 mM) at a flow rate of 30 ml/h. Absorbance of the column eluent was monitored continuously at 280 nm. Protein fractions were analyzed by SDS-PAGE.
Circular dichroism (CD) measurement. NBD2 was expressed in pET expression system and purified with an AKTA explorer 100. CD spectra were measured for the purified NBD2 with a Jasco J715 spectropolarimeter at room temperature with a path length of 1 mm. The molar ellipticity of the NBD2 was measured in the range 190-250 nm. Sample spectrum without protein was subtracted. From the CD spectrum, the secondary structure was calculated by J-700 Secondary Structure Estimation.
Measurement of ATPase activity using a colorimetric assay. ATPase activity of recombinant proteins was determined by the release of inorganic phosphate using a colorimetric assay [18]. Protein solution (100 µg/ml) was incubated at 37°C for 5 min in reaction mixture containing 50 mM Tris-HCl, pH 7.5, 5 mM sodium azide, 2 mM EGTA, pH 7.0, 2 mM ouabain, 50 mM NaCl, and 6 mM MgCl2. The reactions were initiated by adding 5 µl of ATP in a 50-µl final volume and incubated at 37°C for 20 min. The reactions were terminated by adding 50 µl of 6% (w/v) SDS, 3% (w/v) ascorbate, and 0.5 M sodium molybdate in 0.5 M HCl. Products were stabilized by adding 50 µl of 2% (w/v) sodium citrate, 2% (w/v) sodium meta-arsenite, and 2% (v/v) acetic acid. After incubation at 37°C for 10 min, the absorbance was read at 850 nm.
RESULTS
Sequence alignment and quality of the structural model. The highest similarity among all ABC transporters is found within the nucleotide-binding domain [19]. A sequence alignment of NBDs between human P-glycoprotein and other ABC transporters shows five conserved regions (Fig. 1). Walker-A motif (GXXGXGKS/T [X, any amino acid]) and Walker-B motif (XXXXDE/D [X, hydrophobic residue]) are common to a wide variety of nucleotide binding proteins. The Signature motif (LSGGQ), just upstream of the Walker-B motif, is distinctive to the ABC family. The Glutamine Loop (Gln-Loop), containing a well-conserved glutamine residue, is located between the Walker-A and Signature motifs. The final conserved motif is the Histidine Loop (His-Loop), situated 30-40 amino acids C-terminal to the Walker-B motif and containing an almost invariant histidine.
The modeled three-dimensional structure of NBD2 was analyzed by the structure validation program PROCHECK. Analysis of the Ramachandran plot of the model shows that 92.4% of the residues lie in the most favored regions, 6.7% in the additional allowed regions, 0.4% in the generously allowed regions (Lys1164), and 0.4% in the disallowed regions (Asp1171). The Ramachandran plot for the template 1JJ7 was also generated. The overall rating for the model is similar to that obtained for the template.Fig. 1. Sequence alignment of NBDs of human P-glycoprotein and other ABC transporters. The sequences of 1JJ7, 1MT0, and 1F2U were obtained from the Protein Data Bank (PDB). The five conserved regions are indicated by boxes.

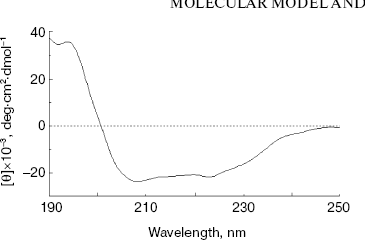
Secondary structure of NBD2. The secondary structure of NBD2 was determined by CD spectral analysis. The CD spectra were measured between 190 and 250 nm. The spectrum exhibits two minima at 208 and 222 nm (Fig. 2), which are typical of alpha-helical conformation. From the CD spectrum, NBD2 was calculated to consist of 36% alpha-helix, 21% beta-strand, 11% turn, and 32% others. The results are similar to the secondary structure prediction of the NBD2 three-dimensional model (33% alpha-helix, 25% beta-strand, 8% turn, and 34% others).
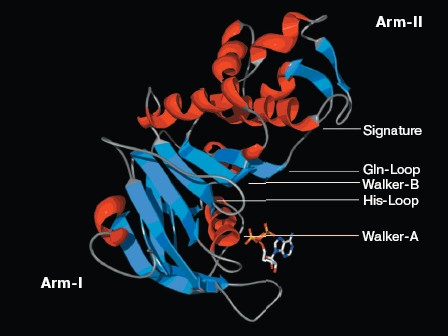
Structure analysis of the ATP-binding site. Molecular modeling was performed using the program MODELLER. NBD2 of human P-glycoprotein forms an L-shaped molecule with two arms (Fig. 3, see color insert, p. 1). Arm-I contains the larger subdomain, which includes the Walker-A motif, Walker-B motif, Gln-Loop, and His-Loop, and forms the ATP-binding site. Arm-II contains the smaller subdomain within which is situated the Signature motif.Fig. 2. Circular dichroism spectra of NBD2. The plot of molar ellipticity (Theta) versus wavelength (nm) is depicted.
There is good conservation for the fold of the ATP-binding site of the ABC transporters [19]. In the model of NBD2, the positions of the residues interacting with ATP are inherited from the templates. The environment of the ATP-binding site of NBD2 is illustrated in Fig. 4 (see color insert, p. 1). Residues in contact with ATP in the model of NBD2 were analyzed with the Ligand-Protein Contacts software [20].Fig. 3. Structural model of the NBD2 of human P-glycoprotein. The two arms of the NBD2 and five conserved regions are indicated. alpha-Helices are shown in red and beta-sheets in blue. Bound ATP is displayed in stick format.
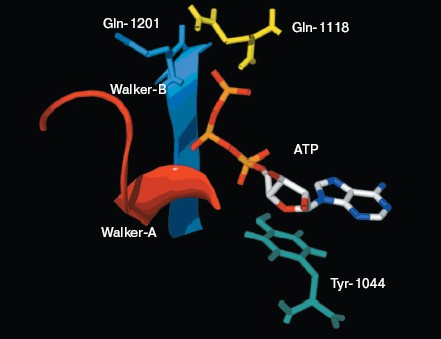
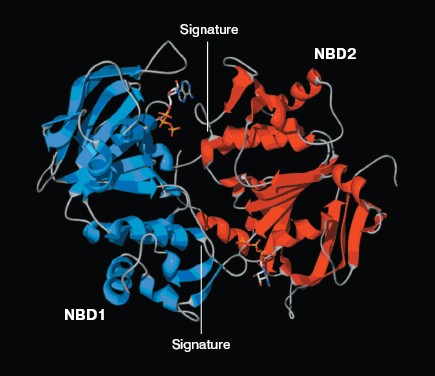
The ATP molecule forms contacts with the Walker-A and Walker-B motifs that are typical of those observed in other ATP-binding proteins such as Ras and adenylate kinase [21]. The Walker-A motif including residues Gly1069 to Lys1077 forms the phosphate binding loop, or P loop, which wraps around and provides tight binding for the beta-phosphate of ATP. The Walker-B motif forms a beta-strand and its Asp residue is involved in the coordination of Mg2+, a mandatory cofactor in the hydrolysis of ATP [22]. Tyr1044 provides an additional stabilizing force towards ATP by way of hydrophobic interaction. Its plane of the aromatic ring is parallel with the adenine ring of the nucleotide molecule. The Signature motif makes no contact with nucleotide in the NBD2 model. But in the dimer model of NBDs that was modeled based on the geometry of Rad50 (Fig. 5, see color insert, p. 2), the Signature motif of one NBD forms a composite ATP-binding site with the Walker A and B motifs of the other NBD. The dimer model is consistent with the data of Loo et al. [23] on the involvement of signature sequence into ATP binding. Two conserved residues are in close proximity to the ATP nucleotide, positioned so that they could interact with gamma-phosphate of ATP. These include Gln-Loop residue Gln1118 and Walker-B residue Glu1201. In HisP and Rad50, these two residues have been postulated to modulate the polarization of the attacking water molecule and could be the activating residue. In NBD2, they might be an initiator for hydrolysis.Fig. 4. ATP-binding site of NBD2. ATP and the conserved residues forming interactions with the nucleotide are displayed. The residues and regions are shown in red ribbon (Walker-A), blue ribbon (Walker-B), green stick (Tyr1044), yellow stick (Gln1118), and blue stick (Glu1201).
Expression and purification of NBD2 and its mutant Q1118A. Mutagenesis was used to investigate the function of the conserved Gln1118 residue that was predicted from the three-dimensional model to participate in interaction with ATP. The cDNA encoding NBD2 was amplified by reverse transcriptase PCR and cloned into the pGEX4T-1 expression vector. The Q1118A mutation was introduced into the NBD2 sequence by site-directed mutagenesis. Recombinant proteins encoded by pGEX4T-1-NBD2 and its mutant Q1118A-based plasmids were expressed in E. coli cells. Expression and purification of the NBD2 and its mutant Q1118A are illustrated in Fig. 6. Lanes 1 and 3 represent the total bacterial protein for each recombinant protein prior to induction. After induction with IPTG, each protein was overexpressed and constituted the main component among total bacterial proteins. The vast majority of each protein was localized in inclusion bodies. Solubilization of inclusion body protein was achieved under denaturing conditions. Each recombinant protein was renatured by rapid dilution and then loaded onto a DEAE column for final purification. NBD2 protein and mutant Q1118A protein yielded equal purities and in similar amounts.Fig. 5. Putative dimer model of NBDs. The dimer was modeled based on the geometry of the Rad50 dimer structure. NBD1 is shown in blue, NBD2 is shown in red, and the Signature motif is indicated.
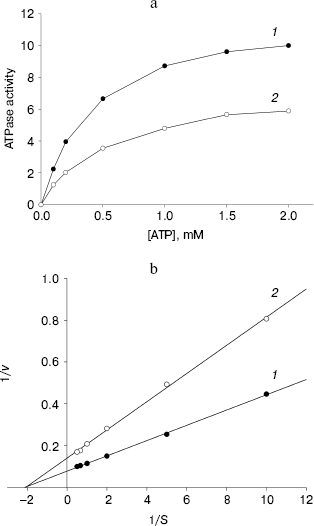
ATPase activity of NBD2 and its mutant Q1118A. ATPase activity of the recombinant proteins was determined by the release of inorganic phosphate using a colorimetric assay. The ATPase activity of NBD2 was dependent on the presence of Mg2+ and linearly related to the amount of protein. Typical Michaelis-Menten behavior was exhibited, yielding estimations of Km of 466 µM, as determined by a Lineweaver-Burk plot (Fig. 7b). The ATPase activities of the Q1118A mutant and that of the wild-type NBD2 were compared. The Vmax for ATP hydrolysis by the mutant NBD2 was ~56% of the Vmax of the wild-type NBD2 (Fig. 7a). But both proteins displayed similar affinity for ATP, with Km of 479 and 466 µM for mutant NBD2 and wild-type NBD2, respectively (Fig. 7b).Fig. 6. Expression and purification of the NBD2 and its mutant Q1118A. Expression of NBD2 protein before (lane 1) and after (lane 2) IPTG induction. Expression of mutant Q1118A protein before (lane 3) and after (lane 4) IPTG induction. Purified NBD2 protein (lane 5) and its mutant Q1118A protein (lane 6).
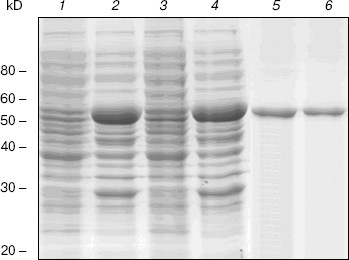
Fig. 7. Kinetics of ATPase activity. a) ATP dependence of the ATPase activities of the isolated NBD2 (1) and its mutant Q1118A (2). ATPase activities are given in mol/min per mol protein. b) Lineweaver-Burk plot of dependence of ATPase activity of NBD2 (1) and its mutant Q1118A (2). Vmax and Km were calculated from the plot of 1/v versus 1/S.
DISCUSSION
Multidrug resistance (MDR) of cancer cells is often associated with overexpression of Pgp, which functions as an ATP-driven efflux pump for chemotherapeutic drugs [24]. ATP binding and hydrolysis are required for the Pgp mediated drug transport. An inhibitory effect on Pgp ATPase activity could result in interference with the drug transport capability of Pgp [25]. A structural model for Pgp has been used to elucidate the molecular mechanism for the open conformation of Pgp [26] and the interface between the two transmembrane domains [27]. But these models did not provide information on the molecular mechanism involved in ATP binding and hydrolysis. In this study, we performed homology modeling of the carboxyl-terminal nucleotide binding domain (NBD2) from human Pgp. This molecular model provided the structural basis for the key catalytic residues involved in ATP hydrolysis and the interactions between chemosensitizer quercetin and the ATP binding site of NBD2. It provided useful information for enzymological studies and a new approach for MDR reverser (chemosensitizer) design.
The role of Gln1118. Residue Gln1118 in Pgp (corresponding to Gln100 in HisP and Gln140 in Rad50) is highly conserved. In the NBD2 molecular model, Gln1118 is in close proximity to the ATP nucleotide, positioned so that it could interact with gamma-phosphate of ATP. This residue makes a water-bridged hydrogen bond to the gamma-phosphate of ATP in HisP and a direct hydrogen bond to the gamma-phosphate of AMP-PNP in Rad50 [15]. It was predicted to be an activating residue for ATP hydrolysis. Site-directed mutagenesis was used to validate the predicted function of Gln1118. The Q1118A mutant was affected in its ATPase activity but this residue might not participate the binding of ATP because the mutation had no influence on the affinity for ATP. A similar result was also shown in the Q1114A mutant of mouse MDR3 P-glycoprotein by Urbatsch et al. [28]. The specific ATPase activity decreased from 0.32 to 0.039 µmol/min per mg protein. But they displayed a similar affinity for ATP, with Km of 0.69 and 0.70 mM for mutant and wild-type mouse MDR3 P-glycoprotein, respectively. These results indicate that the glutamine residue participates in ATP hydrolysis but it might not participate the binding of ATP. However, this mutagenesis was not found to completely abolish ATPase activity. It indicates that other residues can also be responsible for activation of hydrolysis. Structural analysis of the ATP-binding site of NBD2 in the molecular model shows that Glu1201 also makes contact with the gamma-phosphate of ATP and might play the role of an initiator for hydrolysis. Our model is consistent with experimental data and provides a structural basis of the effects of Gln1118 on ATPase activity.
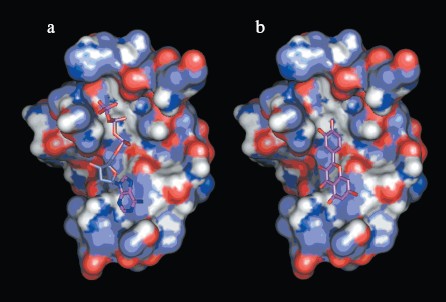
Interactions between chemosensitizer quercetin and NBD2. The NBD2 molecular model also provides evidence that the chemosensitizer quercetin affects Pgp mediated transport of anticancer drugs by direct inhibition of the ATPase activity of Pgp. It was reported that quercetin inhibited efflux of Hoechst 33342 and enhanced its accumulation in multidrug resistant CHRC5 cells by inhibiting the ATPase activity of Pgp [29]. In this study, quercetin was docked to the modeled structure of NBD2 using the program Autodock. The structural model of the complex indicated that quercetin was tightly bound to NBD2. Fourteen putative hydrogen bonds were observed between NBD2 and quercetin involving the residues Tyr1044, Thr1046, Arg1047, Gly1073, Gly1075, Lys1076, Ser1077, Tyr1087, and Glu1201. There were close shape complementarities between the quercetin and ATP binding sites of Pgp. The adenine region, phosphate binding region, and sugar pockets of the ATP binding site were occupied by quercetin. Figure 8 (see color insert, p. 2) shows that quercetin and ATP would occupy overlapping spatial coordinates and compete for binding.
Application of NBD2 molecular model. Since ATPase activity is critical for the transporter function of Pgp, the specific inhibition of ATP binding and hydrolysis within the NBDs should constitute a good tool to reverse MDR. So NBD2 may be an attractive target for design of new MDR reversers (chemosensitizers or modulators). Previously, a structure-based approach has identified inhibitors to a variety of protein systems, including inhibitors of HIV-1 protease [30], influenza hemagglutinin [31], and SARS_CoV 3CL proteinase [32]. Virtual screening is an efficient method for structure-based drug design, which can be performed employing the molecular docking method to identify possible inhibitors from small molecule electronic databases. Specificity and affinity can then be improved using structural information from the receptor and binding mode of the inhibitor in a cycle of structure-based design [33]. The three-dimensional model of NBD2 can be used for the structure-based virtual screening of potential MDR reversers. Work is in progress concerning virtual screening on the NCI Plated Compounds database (approximately 140,000 compounds).Fig. 8. a) Binding site of NBD2 complexed with ATP. b) Binding site of NBD2 complexed with quercetin. The binding site of NBD2 is represented by a molecular surface diagram. Bound ATP and quercetin are displayed in stick format.
In conclusion, we constructed a molecular model of the carboxyl-terminal nucleotide binding domain (NBD2) from human P-glycoprotein. Structural analysis and site-directed mutagenesis suggest that the conserved Gln1118 residue might be an initiator for ATP hydrolysis. The interactions of chemosensitizer with NBD2 provided structural basis of inhibition of P-glycoprotein ATPase activity by quercetin. The three-dimensional model of NBD2 can be used both to guide biochemical study of properties and to screen potential MDR reversers.
This work was supported by the National Natural Science Foundation of China (No. 30171088), Shanghai Key Disciplinary Foundation, and “863” Project of China (No. 2001 AA215261).
REFERENCES
1.Dean, M., Rzhetsky, A., and Allikmets, R. (2001)
Genome Res., 11, 1156-1166.
2.Walker, J. E., Saraste, M., Runswick, M. J., and
Gay, N. J. (1982) EMBO J., 1, 945-951.
3.Bianchet, M. A., Ko, Y. H., Amzel, L. M., and
Pedersen, P. L. (1997) J. Bioenerg. Biomembr., 29,
503-524.
4.Sharom, F. J. (1997) J. Membr. Biol.,
160, 161-175.
5.Ambudkar, S. V., Dey, S., Hrycyna, C. A.,
Ramachandra, M., Pastan, I., and Gottesman, M. M. (1999) Annu. Rev.
Pharmacol. Toxicol., 39, 361-398.
6.Hartman, J., Huang, Z., Rado, T. A., Peng, S.,
Jilling, T., Muccio, D. D., and Sorscher, E. J. (1992) J. Biol.
Chem., 267, 6455-6458.
7.Dayan, G., Baubichon-Cortay, H., Jault, J. M.,
Cortay, J. C., Deleage, G., and Di Pietro, A. (1996) J. Biol.
Chem., 271, 11652-11658.
8.Ramjeesingh, M., Li, C., Garami, E., Huan, L. J.,
Galley, K., Wang, Y., and Bear, C. E. (1999) Biochemistry,
38, 1463-1468.
9.Hrycyna, C. A., Ramachandra, M., Germann, U. A.,
Cheng, P. W., Pastan, I., and Gottesman, M. M. (1999)
Biochemistry, 38, 13887-13899.
10.Thompson, J. D., Gibson, T. J., Plewniak, F.,
Jeanmougin, F., and Higgins, D. G. (1997) Nucleic Acids Res.,
25, 4876-4882.
11.Schmitt, L., Benabdelhak, H., Blight, M. A.,
Holland, I. B., and Stubbs, M. T. (2003) J. Mol. Biol.,
330, 333-342.
12.Gaudet, R., and Wiley, D. C. (2001) EMBO
J., 20, 4964-4972.
13.Sali, A., and Blundell, T. L. (1993) J. Mol.
Biol., 234, 779-815.
14.Morris, A. L., MacArthur, M. W., Hutchinson, E.
G., and Thornton, J. M. (1992) Proteins, 12, 345-364.
15.Hopfner, K. P., Karcher, A., Shin, D. S., Craig,
L., Arthur, L. M., Carney, J. P., and Tainer, J. A. (2000) Cell,
101, 789-800.
16.Guex, N., and Peitsch, M. C. (1997)
Electrophoresis, 18, 2714-2723.
17.Morris, G. M., Goodsell, D. S., Halliday, R. S.,
Huey, R., Hart, W. E., Belew, R. K., and Olson, A. J. (1998) J.
Comput. Chem., 19, 1639-1662.
18.Chifflet, S., Torriglia, A., Chiesa, R., and
Tolosa, S. (1988) Analyt. Biochem., 168, 1-4.
19.Geourjon, C., Orelle, C., Steinfels, E.,
Blanchet, C., Deleage, G., Di Pietro, A., and Jault, J. M. (2001)
Trends Biochem. Sci., 26, 539-544.
20.Sobolev, V., Sorokine, A., Prilusky, J., Abola,
E. E., and Edelman, M. (1999) Bioinformatics, 15,
327-332.
21.Saraste, M., Sibbald, P. R., and Wittinghofer, A.
(1990) Trends Biochem. Sci., 15, 430-434.
22.Abrahams, J. P., Leslie, A. G., Lutter, R., and
Walker, J. E. (1994) Nature, 370, 621-628.
23.Loo, T. W., Bartlett, M. C., and Clarke, D. M.
(2002) J. Biol. Chem., 277, 41303-41306.
24.Gottesman, M. M., and Pastan, I. (1993) Annu.
Rev. Biochem., 62, 385-427.
25.Horio, M., Gottesman, M. M., and Pastan, I.
(1988) Proc. Natl. Acad. Sci. USA, 85, 3580-3584.
26.Seigneuret, M., and Garnier-Suillerot, A. (2003)
J. Biol. Chem., 278, 30115-30124.
27.Stenham, D. R., Campbell, J. D., Sansom, M. S.,
Higgins, C. F., Kerr, I. D., and Linton, K. J. (2003) FASEB J.,
17, 2287-2289.
28.Urbatsch, I. L., Gimi, K., Mounts, S. W., and
Senior, A. E. (2000) Biochemistry, 39, 11921-11927.
29.Shapiro, A. B., and Ling, V. (1997) Biochem.
Pharmacol., 53, 587-596.
30.Rutenber, E., Fauman, E. B., Keenan, R. J., Fong,
S., Furth, P. S., Ortiz de Montellano, P. R., Meng, E., Kuntz, I. D.,
DeCamp, D. L., and Salto, R. (1993) J. Biol. Chem., 268,
15343-15346.
31.Bodian, D. L., Yamasaki, R. B., Buswell, R. L.,
Stearns, J. F., White, J. M., and Kuntz, I. D. (1993)
Biochemistry, 32, 2967-2978.
32.Xiong, B., Gui, C. S., Xu, X. Y., Luo, C., Chen,
J., Luo, H. B., Chen, L. L., Li, G. W., Sun, T., Yu, C. Y., Yue, L. D.,
Duan, W. H., Shen, J. K., Qin, L., Shi, T. L., Li, Y. X., Chen, K. X.,
Luo, X. M., Shen, X., Shen, J. H., and Jiang, H. L. (2003) Acta
Pharmacol. Sin., 24, 497-504.
33.Kuntz, I. D. (1992) Science, 257,
1078-1082.