Purification and Some Properties of beta-N-Acetyl-D-glucosaminidase from Viscera of Green Crab (Scylla serrata)
Ji-Ping Zhang1,2, Qing-Xi Chen1*, Qin Wang1, and Jin-Jin Xie1
1Department of Biochemistry and Biotechnology, School of Life Sciences, Key Laboratory of Ministry of Education for Cell Biology and Tumor Cell Engineering, Xiamen University, Xiamen 361005, China; fax: 86-592-2185487; E-mail: chenqx@xmu.edu.cn2Department of Animal Science, School of Life Sciences, Foshan Science and Technology College, Foshan 528231, China
* To whom correspondence should be addressed.
Received May 3, 2005; Revision received July 13, 2005
beta-N-Acetyl-D-glucosaminidase was purified from viscera of green crab (Scylla serrata) by extraction with 0.01 M Tris-HCl buffer (pH 7.5) containing 0.2 M NaCl, ammonium sulfate fractionation, and then chromatography on Sephadex G-100 and DEAE-cellulose (DE-32). The purified enzyme showed a single band on polyacrylamide gel electrophoresis, and the specific activity was determined to be 7990 U/mg. The molecular weight of the whole enzyme was determined to be 132.0 kD, and the enzyme is composed of two identical subunits with molecular mass of 65.8 kD. The optimum pH and optimum temperature of the enzyme for the hydrolysis of p-nitrophenyl-N-acetyl-beta-D-glucosaminide (pNP-NAG) were found to be at pH 5.6 and at 50°C, respectively. The study of its stability showed that the enzyme is stable in the pH range from 4.6 to 8.6 and at temperatures below 45°C. The kinetic behavior of the enzyme in the hydrolysis of pNP-NAG followed Michaelis-Menten kinetics with Km of 0.424 ± 0.012 mM and Vmax of 17.65 ± 0.32 µmol/min at pH 5.8 and 37°C, and the activation energy was determined to be 61.32 kJ/mol. The effects of some metal ions on the enzyme were surveyed, and the results show that Na+ and K+ have no effects on the enzyme activity; Mg2+ and Ca2+ slightly activate the enzyme, while Ba2+, Zn2+, Mn2+, Hg2+, Pb2+, Cu2+, and Al3+ inhibit the enzyme to different extents.
KEY WORDS: enzymatic properties, beta-N-acetyl-D-glucosaminidase, purification, Scylla serrataDOI: 10.1134/S0006297906130098
Abbreviations: NAGase) beta-N-acetyl-D-glucosaminidase; NAG) N-acetylglucosamine; pNP-NAG) p-nitrophenyl-N-acetyl-beta-D-glucosaminide; pNP) p-nitrophenol; NaAc-HAc) sodium acetate/acetic acid buffer.
Chitin, a polymer of beta-(1-4)-linked N-acetylglucosamine (NAG),
is mostly distributed in the shell of the crustaceans. Chitin is
digested to NAG by two chitinolytic enzymes. Chitinase cleaves chitin
into dimer and trimer oligomers of NAG, which are further hydrolyzed by
beta-N-acetyl-D-glucosaminidase (NAGase, EC 3.2.1.52) to monomer
NAG [1]. Both chitinase and NAGase are widely
distributed in nature and act in important roles in defense systems
against parasites, molting cycle, digestion of chitinous foods, and so
on [2-5].
Chitinases have been characterized in crustaceans, such as prawn [6, 7], lobsters [8], and shrimp [9, 10]. Recent research on NAGase mostly concerns microorganisms, insects [11, 12], and crustaceans such as the Antarctic krill (Euphausia superba) [5, 13], prawn (Penaeus vannamei) [14], and fiddler crab (Uca pugilator) [15]. Peters et al. [5, 13] found that the NAGase from Antarctic krill is involved in digestion and molting processes. The investigation of enzymatic properties is an essential tool to study physiological adaptations expressed by organism in response to different environmental conditions. Zou et al. [15] found that the NAGase activity in the epidermis and hepatopancreas from fiddler crab (U. pugilator) correlated well with the hemolymph titer of ecdysteroids during the molting cycle, this suggesting that NAGase activity in both tissues is regulated at least in part by the steroid molting hormones.
The green crab (Scylla serrata) is a very important species for mariculture in China. However, the NAGase from green crab (S. serrata) is poorly known. Research on the crab NAGase has very important significance for the breeding and growth of the crab. In this paper, we report the purification and some properties of the enzyme, such as molecular weight, kinetic behavior, pH and thermal stabilities, and the effects of metal ions on the enzyme activity. The enzymatic properties are also compared with the enzyme from other sources.
MATERIALS AND METHODS
Materials. Green crabs (S. serrata) were obtained from the Xiamen Fishery Company, Fujian, China. p-Nitrophenyl-N-acetyl-beta-D-glucosaminide (pNP-NAG) was purchased from the Biochemistry Laboratory of Shanghai Medicine Industry Academy (China). Sephadex G-100 was a Pharmacia (Sweden) product. DEAE-cellulose (DE-32) was from Whatman (UK). Low molecular weight protein markers were purchased from Amersham (UK). p-Nitrophenol (pNP) was purchased from Sigma (USA). All other reagents were local products of analytical grade. The water used was redistilled and ion-free.
Determination of protein concentration and assay of enzymatic activity. The protein concentration was measured by the method of Lowry with bovine serum albumin as the standard. Enzyme activity was determined at 37°C by following the increase in optical density at 405 nm accompanying the hydrolysis of the substrate (pNP-NAG) [6] with pNP molar absorption coefficient of 1.73*104 M-1*cm-1 [16]. A portion of 10 µl of enzyme solution was added to the reaction medium (2 ml) containing 0.2 mM pNP-NAG in 0.05 M NaAc-HAc buffer (pH 5.8). After reaction for 10 min at 37°C, 2 ml of 0.5 M NaOH was added into the reaction mixture to stop the reaction. Absorption was recorded using a Beckman DU-650 spectrophotometer (USA). One unit (U) of enzymatic activity was defined as the amount of enzyme catalyzing the hydrolysis of pNP-NAG to form 1 µmol pNP per min at 37°C.
Purification and determination of homogeneity of the enzyme. NAGase was prepared from viscera of green crab (S. serrata) by extraction with 0.01 M Tris-HCl buffer (pH 7.5) containing 0.2 M NaCl and ammonium sulfate fractionation according to the method of Koga et al. [17]. The crude preparation was further chromatographed on a Sephadex G-100 column (2.5 × 60 cm), which was pre-equilibrated with 0.01 M Tris-HCl buffer (pH 7.5) containing 0.2 M NaCl. The column was eluted with the same buffer with flow rate of 15 ml/h and fraction volume of 3 ml per tube. The active fractions were pooled and dialyzed against 0.01 M Tris-HCl buffer (pH 7.5). The dialyzed solution was loaded onto DEAE-cellulose column (2 × 20 cm) pre-equilibrated with 0.01 M Tris-HCl buffer. The column was eluted with a linear gradient of 0-1 M NaCl at a flow rate of 15 ml/h and fraction volume of 3 ml per tube. Active fractions were pooled. The enzyme homogeneity was tested by polyacrylamide gel electrophoresis (PAGE) with a 4% acrylamide stacking gel and a 12% acrylamide separating gel.
Determination of molecular weight. The molecular weight of the native enzyme was determined using gel filtration on Sephadex G-200, and the subunit molecular mass was determined by polyacrylamide gel electrophoresis (SDS-PAGE). A Sephadex G-200 gel filtration column (2.5 × 70 cm) was pre-equilibrated and eluted with 0.01 M Tris-HCl buffer (pH 7.5) containing 0.2 M NaCl. The standard proteins were chymotrypsin (25 kD), pepsin (35 kD), albumin egg (45 kD), bovine serum albumin (68 kD), alkaline phosphatase from calf intestine (115 kD), and human gamma-globulin (165 kD). SDS-PAGE was done on a discontinuous horizontal thick-layer with a stacking gel of 4% acrylamide in Tris-HCl buffer (pH 6.7) at 10 mA current and a separating gel of 12% acrylamide in Tris-HCl buffer (pH 8.9) at 20 mA current. The gel contained 0.1% SDS. Both the enzyme sample and markers were combined with 1-fold solubilizing mixture containing 4% SDS, 2% 2-mercaptoethanol, 40% sucrose, 0.02% bromophenol blue (tracking dye), and 0.01 M Tris-HCl buffer, pH 6.8. The mixtures were heated in a boiling water bath for 3 min. After electrophoresis, the gel was stained with 0.05% Coomassie Brilliant Blue R-250.
RESULTS AND DISCUSSION
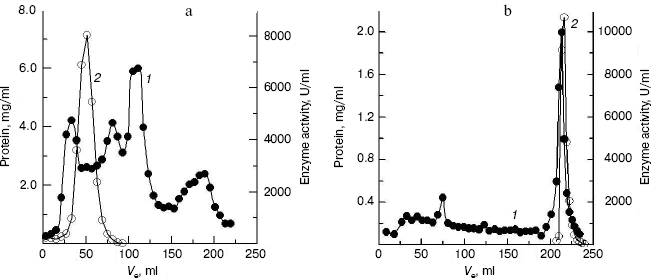
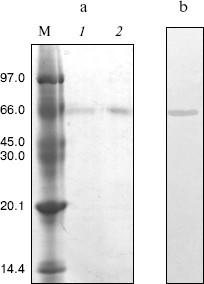
Purification of crab NAGase. The elution diagram of the crab NAGase from a Sephadex G-100 column chromatography is shown in Fig. 1a. Active fractions were pooled and applied to a DEAE-cellulose column (DEAE-32) for further purification. The elution diagram for the DEAE-32 column chromatography is shown in Fig. 1b. The activity peak overlapped with the protein peak, and the active fractions were pooled. The specific activity of the purified enzyme was 7990 U/mg. The final preparation was homogeneous on PAGE (Fig. 2b), indicating the method of purification was effective.
Fig. 1. Elution diagram for column chromatography of NAGase from green crab (S. serrata) on Sephadex G-100 (a) and DEAE-cellulose (b). Curves 1 and 2 represent protein concentration and enzyme activity, respectively.
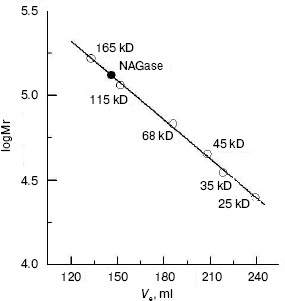
Molecular weight of the native enzyme and its subunit composition. The subunit number of the enzyme was determined by SDS-PAGE and the result showed a single band (Fig. 2a). From a plot of log(Mr) versus the Rf value of the standard proteins, molecular mass of subunits could obtained as 65.8 kD.Fig. 2. Gel electrophoresis of green crab NAGase on SDS-PAGE (a) and on PAGE (b). Lanes: M) molecular markers (kD); 1, 2) NAGase at 20 and 30 µg load, respectively.
The molecular weight of the native enzyme was determined using gel filtration on Sephadex G-200. The result (Fig. 3) shows that the apparent native molecular mass of the NAGase is 132.0 kD. From these results, we conclude that the enzyme is composed of two subunits with the same molecular mass, which was different from that of the enzyme from other sources. For example, NAGase from silkworm (Bombyx mori) is composed of two different subunits of 67.5 and 57.5 kD [17], and the enzyme from Antarctic krill (E. superba) has two distinct forms: one is a single peptide and the other is a heterodimer [5, 13]. In contrast, NAGase from T. cruzi is a single subunit with molecular weight of 48.0 kD [18], and NAGase from T. harzianum is also a single peptide of 72.0 kD [11]. The difference of molecular mass of the enzyme from these different organisms may indicate that the complexities of enzymatic structures are different.
Kinetic parameters and activation energy of the enzyme. The kinetic behavior of the enzymatic hydrolysis of pNP-NAG was studied. Under the conditions employed in the present investigation, the hydrolysis of pNP-NAG by the enzyme follows Michaelis-Menten kinetics. The kinetic parameters were obtained from a Lineweaver-Burk plot with Km of 0.424 ± 0.012 mM and Vmax of 17.65 ± 0.32 µmol/min.Fig. 3. Measurement of molecular weight of native NAGase enzyme of green crab by gel filtration on Sephadex G-200: standard proteins (empty circles); green crab NAGase (filled circle).
At different temperature (25, 30, 35, 37, and 40°C), the maximum velocity (Vmax) was assayed from Lineweaver-Burk plots. The plot of logVmax versus 1/T is a straight line. The activation energy was determined to be 61.32 kJ/mol from the slope of the line.
Effects of pH on the enzyme activity and stability. The effect of pH on the enzyme activity for the hydrolysis of pNP-NAG at 37°C was determined. The result showed that the optimum pH for catalysis is at pH 5.6 (Fig. 4, curve 1), which is close to that of enzyme from fiddler crab (U. pugilator) [15] but higher than that for the prawn enzyme [14]. When the pH value of buffer was higher or lower than the optimum pH, the enzyme activity decreased rapidly. On the other hand, the enzyme was incubated in different pH buffers for 20 h at 4°C, and a portion of 10 µl of the mixture was used for activity assay under the standard system (37°C, pH 5.8). The pH stability result showed (Fig. 4, curve 2) that the enzyme is stable in the pH range of 4.6 to 8.6. When pH was higher than pH 8.6 or lower than pH 4.6, the enzyme activity was lost rapidly.
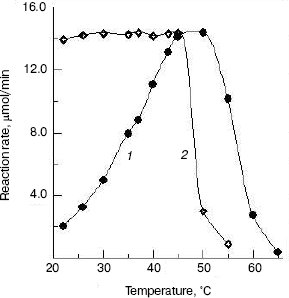
Effects of temperature on the enzyme activity and stability. The effect of temperatures on the enzyme activity was investigated and the result (Fig. 5, curve 1) shows that the optimum temperature is at 50°C. This is different from that of silkworm enzyme [17] and prawn enzyme [14]. The results of study of the thermal stability of the enzyme (Fig. 5, curve 2) show that the enzyme is stable below 45°C, and the enzyme loses activity rapidly at temperatures above 45°C.Fig. 4. Effects of pH on the activity of green crab enzyme (1); the buffer was sodium phosphate/citric acid at different pH values. The pH stability of the enzyme (2). The following buffers were used: Gly-HCl buffer (pH 2.5-4.0), NaAc-HAc buffer (pH 4.0-5.8), Na2HPO4-NaH2PO4 buffer (pH 5.8-8.0), Gly-NaOH buffer (pH 8.0-10.6), and KCl-NaOH buffer (pH 11.2).
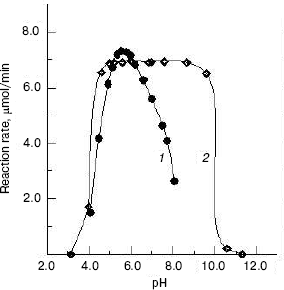
The NAGase from green crab was stable in the pH range from 4.6 to 8.6 and at temperatures below 45°C, indicating that the enzyme has better thermal stability and narrower pH range stability than the prawn enzyme [14]. The NAGase from green crab has wide thermal stability and good pH stability, which might be correlated with the good adaptability of green crab to the environment.Fig. 5. Effects of temperature on the activity of green crab NAGase (1); the enzyme activity was measured at various temperatures in 0.05 M NaAc-HAc buffer (pH 5.8). Thermal stability of the NAGase (2); the enzyme was incubated at different temperature for 30 min in NaAc-HAc buffer (pH 5.8), then portions of the mixture (containing 0.4 µg of the enzyme) were taken for activity assay.
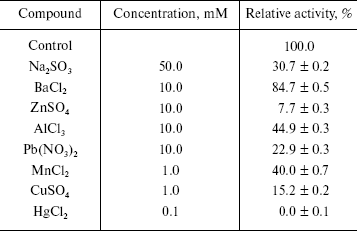
Effects of some metal ions on the enzyme activity. Taking LiSO4, NaCl, NaNO3, Na2SO4, Na2SO3, KCl, KNO3, CaCl2, Ca(NO3)2, MgCl2, BaCl2, ZnSO4, Pb(NO3)2, AlCl3, MnCl2, CuSO4, and HgCl2 as effectors, we probed the effects of these metal ions on the enzyme activity for the hydrolysis of pNP-NAG. Experiments were performed in the standard assay system with various metal ion compounds. Enzyme solution (10 µl containing 0.4 µg of the enzyme) was added for activity assay. The results showed that salts like Li2SO4, NaCl, NaNO3, Na2SO4, KCl, KNO3, CaCl2, Ca(NO3)2, and MgCl2 had less than 10% effect on activity, the others salts causing different extent of inhibition of the enzyme (table). From these results it is concluded that the univalent alkali metal ions Na+ and K+ have no effect on the enzyme activity, this being similar to the case for NAGase from prawn (P. vannamei) [14] and that the counterions SO42-, Cl-, and NO3- do not influence the enzyme activity, while SO32- causes evident inhibition of the enzyme activity. The reason is probably that the green crab lives in sea water for a long time and has better adaptability to normal salinity environment. Bivalent alkaline-earth metal ions Mg2+ and Ca2+ can slightly activate the enzyme, but Ba2+ slightly inhibits the enzyme. Trivalent metal ions Al3+ and transition metal ions Zn2+ and Mn2+ inhibit the enzyme activity. When the concentrations of Zn2+ reached 10 mM and Mn2+ reached 1.0 mM, the enzyme activity decreased by 92.3 and 60.0%, respectively. Adriana et al. reported that Mn2+ does not effect the activity of NAGase from T. foetus [12], while Xie et al. found that Mn2+could slightly activate the prawn NAGase [14]. The differences indicate that NAGases obtained from different organism have different sensitivity to metal ions. The heavy metal ions Hg2+, Pb2+, and Cu2+ clearly inhibited the enzyme. Hg2+ inhibited the enzyme strongly at 0.1 mM, the enzyme activity being almost completely eliminated. But when the concentration of Pb2+ and Cu2+ were 10 and 1 mM, the activity of enzyme remained at 22.9 and 15.2%, respectively. The inhibitions by Pb2+ and Cu2+ of NAGase from green crab are more obvious than those on NAGase from Prawn [14], which may indicate that the harmful effects of heavy metal ions in sea water are different for different crustaceans.
Effects of some metal ions on the beta-N-acetyl-D-glucosaminidase
activity
This investigation was supported by Grant 2004N002 of the Science and Technology Foundation and by Grant 40576066 of the China Natural Science Foundation.
REFERENCES
1.Espie, P. J., and Roff, J. C. (1995) Physiol.
Zool., 68, 727-748.
2.Zen, K. C., Choi, H. K., Krishnamachary, N.,
Muthukrishnan, S., and Kramer, K. J. (1996) Insect. Biochem. Mol.
Biol., 26, 435-444.
3.Ken, A. B., and William, D. S. (1998) J. Mol.
Biol., 280, 913-923.
4.Banat, B. M. A., Kameyama, Y., Yoshioka, T., and
Koga, D. (1999) Insect. Biol., 29, 537-547.
5.Peters, G., Saborowski, R., Buchholz, F., and
Mentlein, R. (1999) Mar. Biol., 134, 697-703.
6.Kono, M., Matsui, T., Shimizu, C., and Koga, D.
(1990) Agric. Biol. Chem., 54, 2145-2147.
7.Watanabe, T., Kono, M., Aida, K., and Nagasawa, H.
(1998) Biochim. Biophys. Acta, 1382, 181-185.
8.Lynn, K. R. (1990) Comp. Biochem. Physiol.,
96B, 761-766.
9.Funke, B., and Spindler, K. D. (1989) Comp.
Biochem. Physiol., 94B, 691-695.
10.Margrethe, E., Bjornar, M., and Ragnar, L. O.
(1996) Comp. Biochem. Physiol., 113B, 717-723.
11.Clemens, K. P., Matteo, L., and Christopher, K.
H. (1996) Curr. Genet., 30, 325-331.
12.Adriana, P. C., and Benedito, P. D. F. (1999)
Parasitol. Res., 85, 256-262.
13.Peters, G., Saborowski, R., Mentlein, R., and
Buchholz, F. (1998) Comp. Biochem. Physiol., 120B,
743-751.
14.Xie, X. L., Chen, Q. X., and Lin, J. C. (2004)
Mar. Biol., 146, 143-148.
15.Zou, E., and Fingerman, M. (1999) Mar.
Biol., 133, 97-101.
16.Chen, Q. X., Zheng, W. Z., Lin, J. Y., Cai, Z.
T., and Zhou, H. M. (2000) Biochemistry (Moscow), 65,
1105-1110.
17.Koga, D., Shimazaki, C., and Yamamoto, K.
(1987) Agric. Biol. Chem., 51, 1679-1681.
18.Brahim, E. L. M., Marie-Helene, R., and
Jean-Louis, J. (1996) Exp. Parasitol., 83, 167-173.