A Comparison of Catalytic Site Intermediates of Cytochrome c Oxidase and Peroxidases
P. R. Rich* and M. Iwaki
Glynn Laboratory of Bioenergetics, Department of Biology, University College London, Gower Street, London WC1E 6BT, UK; E-mail: prr@ucl.ac.uk; m.iwaki@ucl.ac.uk* To whom correspondence should be addressed.
Received June 5, 2007
Compounds I and II of peroxidases such as horseradish peroxidase and cytochrome c peroxidase are relatively well understood catalytic intermediates in terms of their structures and redox states of iron, heme, and associated radical species. The intermediates involved in the oxygen reduction chemistry of the cytochrome c oxidase superfamily are more complicated because of the need for four reducing equivalents and because of the linkage of the oxygen chemistry with vectorial proton translocations. Nevertheless, two of these intermediates, the peroxy and ferryl forms, have characteristics that can in many ways be considered to be counterparts of peroxidase compounds I and II. We explore the primary factors that minimize the generation of unwanted reactive oxygen species products and ensure that the principal enzymological function becomes either that of a peroxidase or an oxidase. These comparisons can provide insights into the nature of biological oxygen reduction chemistry and guidance for the engineering of biomimetic synthetic materials.
KEY WORDS: cytochrome oxidase, horseradish peroxidase, cytochrome c peroxidase, catalytic intermediatesDOI: 10.1134/S0006297907100033
Abbreviations: APX) ascorbic acid peroxidase; CcO) cytochrome c oxidase; CcP) cytochrome c peroxidase; HRP) horseradish peroxidase; ROS) reactive oxygen species.
In early studies of the biochemical nature of cellular respiration,
various controversies arose as to the nature of the oxygen-consuming
pathway. An early view proposed by Chodat and Bach [1] was that the reaction consisted of an oxygenase
that formed an organic peroxide by addition of molecular oxygen,
together with a peroxidase that subsequently utilized this peroxide to
oxidize a variety of reducing species. It was more than twenty years
before the true nature of the reaction was firmly established as a
cytochrome-mediated oxidase reaction [2].
However, as the structural and mechanistic details of the reaction cycles of peroxidases and cytochrome oxidases have emerged, it has become apparent that their catalytic intermediates have significant mechanistic overlap and, indeed, that oxidases have a minor peroxidatic side reaction and that some peroxidases can act as oxidases. It is the purpose of this article to review and compare modern understanding of these enzymes and their intermediates and to assess the design features that confer their biochemical functions whilst minimizing unwanted side reactions and reactive oxygen species (ROS) generation.
STRUCTURES AND REDOX PROPERTIES
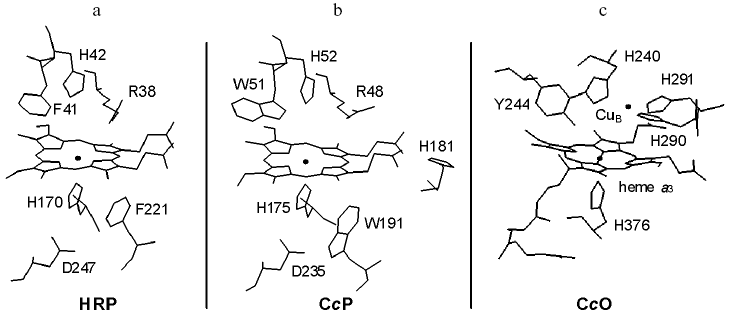
High resolution atomic models of various peroxidases and members of the cytochrome c oxidase (CcO) superfamily have now been resolved in various ligation and redox states. Figure 1 illustrates some key features of the catalytic sites of two of the best characterized peroxidases, horseradish peroxidase (HRP) and cytochrome c peroxidase (CcP), and that of the bovine member of the CcO superfamily of oxidases. The active sites have a high spin heme, either heme B in HRP and CcP or heme A in CcO. In all three, the proximal side of the heme is ligated by the Ntau nitrogen of histidine. In the peroxidases, but not in CcO, the Npi nitrogen of the proximal histidine is hydrogen bonded to an aspartic acid. In CcP, a functionally important tryptophan, W191, is also located close to the proximal heme face but this is replaced by a phenylalanine in HRP. The distal face of the heme in both peroxidases forms the active site cavity with a phenylalanine/tryptophan-histidine-arginine triad that provides essential H-bonding and protonation functions during catalysis [3, 4]. This cavity is separated from the protein surface by a relatively short access distance for hydrogen peroxide.
The active site cavity of CcO also involves the distal heme face and, because of the electronic requirements for O-O bond cleavage (see below), it has an additional redox center, the copper site CuB. CuB is ligated by three histidines, one of which, H240, is linked to tyrosine, Y244, by an unusual covalent bond between the H240 Ntau nitrogen and the ortho ring carbon of Y244 [5]. This creates a contiguously-linked pentameric ring of amino acids H240-P-E-V-Y244 that contains glutamic acid, E242, a residue thought to play a key role in redox-linked proton transfer chemistry [6]. This active site is deeply buried within the protein structure and lies approximately 2/3 of the distance across the membrane from the negative aqueous phase, with long access/exit channels for oxygen, water and protons whose details are not yet fully resolved.Fig. 1. Catalytic sites of HRP, CcP, and CcO. The figures illustrate the heme prosthetic groups and key amino acid residues of: a) recombinant HRP isozyme C; b) yeast CcP; c) bovine CcO. Data are drawn from coordinates in PDB entries 1H58 [64], 2CYP [65] and 2OCC [66].
POTENTIALS, REDOX-LINKED PROTONATIONS, AND
ELECTRONEUTRALITY
An important factor in the chemistry of electron transfer reactions within proteins is the need to minimize the net charge change of stable catalytic intermediates [7]. When occurring in regions of locally low dielectric strength caused by insufficient solvent or protein charge rearrangements, electron transfer can provide a strong driving force for uptake of charge-compensating protons if the protein has appropriately placed protonatable groups whose pKs are linked to the redox changes. From a physical point of view, the requirement for charge compensation originates in free energy considerations and the factors that control redox potentials. These factors have been discussed previously [7].
Empirical studies of protonic changes associated with electronic or ligand state changes in CcO have established that changes within the binuclear center are strictly charge-neutralized by protonation reactions [8, 9]. Such charge-neutralization by protonation changes also occurs when charge changes occur within the iron-heme or distal heme pockets of HRP and CcP [10] although such coupling is less strict than in CcO. In the oxidized ground states of both peroxidases and CcO, the hemes cannot bind oxygen and instead the active sites contain structured waters that can also ligate the metal(s). In the case of CcO, it seems likely that the fourth ligand of CuB is a hydroxide, which provides one protonation site. The 6th, distal, ligand of heme a3 is probably a water at neutral pH. However, it is possible that this water shares a proton “hole” with the phenolic group of the covalently-linked tyrosine and/or with additional structured waters in the cavity. The net result is that the binuclear center cavity can accommodate one or two charge-compensating protons. In peroxidases, the distal histidine can provide a site for charge-compensating protonation when there is a net charge change on the iron-porphyrin system as occurs, for example, on reduction or compound II formation [4, 11] or on binding of anionic ligands [12].
An important difference between peroxidases and CcO metals that governs their primary catalytic roles is redox potentials. Specifically, the ferric/ferrous couples of peroxidases have relatively low midpoint potentials: Em7 is -270 mV for HRP [11] and -194 mV for CcP [13]. Both values are pH-dependent in at least part of the physiological pH range due to redox-linked protonation of the distal histidine [12, 14]. In CcP, a second histidine, H181, located close to a heme propionate, has also been reported to undergo protonation together with the distal H52, linked to the ferric/ferrous heme transition and resulting in two protons bound per electron [13, 15]. However, this unexpected result requires further confirmation since, if purely electrostatic in origin, would require an extreme driving force [16] and instead suggests major structural changes.
The metal sites of the binuclear center of CcO are very much higher than those of peroxidases with Em7 values of roughly +350 mV for both CuB and heme a3 when all other centers are oxidized [17]. Again both are pH-dependent due to redox-linked protonation sites. As noted above, up to two protonation sites may be available within the binuclear center. However, the redox chemistry of the CcO metals is complicated by the fact that both centers interact electrostatically with each other and with heme a [17], and also because they interact with additional shared redox-linked protonation site(s) [18]. These site(s), which are likely to be physically separated from the oxygen intermediates, have been implicated in the coupling of electron transfer and vectorial proton translocation [19-21].
CATALYTIC CYCLES OF PEROXIDASES AND CcO
The overall peroxidase and CcO reaction cycles may be generalized as:
peroxidase: AH2 + H2O2 → A + 2 H2O,
CcO: 4 cyt c2+ + 4H+in + O2 → 4 cyt c3+ + 2 H2O.
Different peroxidases can use various two-equivalent (AH2) or one-equivalent donors. CcO requires four one-equivalent electron donations from ferrocytochrome c, with uptake of the four required protons from the mitochondrial matrix “inside” space. The CcO reaction is further complicated by the linked translocation of four protons across the membrane in which it resides:
CcO: 4 cyt c2+ + 8 H+in + O2 → 4 cyt c3+ + 2 H2O + 4 H+out.
A typical peroxidase reaction cycle is summarized in Fig. 2. The reaction commences with binding and subsequent reaction of hydrogen peroxide with the ferric heme group to produce compound I. The O-O bond has been broken in this intermediate, a reaction that requires two reducing equivalents. The first of these is provided by the ferric iron, which is converted into a ferryl (Fe4+=O) species. In the case of peroxidases such as HRP and myeloperoxidase, the second electron comes from the heme ring itself, forming a pi-cation radical [22-25]. However, the radical species is different in other peroxidases. For example, in CcP, the radical resides on W191 [26, 27]. In APX, the radical resides initially on the porphyrin ring, but migrates slowly to a more distant tryptophan in a pH-dependent manner [28]. It might be noted that there is no net change to the formal +1 charge in the ferric → compound I transition and, therefore, no net protonation change. The next step in the reaction is the one-equivalent reduction of the radical to form compound II. This ferryl state has a net charge of 0 and should result in protonation of the distal histidine to its imidazolium state [4]. H-Bonding of this imidazolium to the ferryl oxygen would account for the H/D-sensitivity of the Fe-O stretch frequency observed by Raman spectroscopy [29]. Finally, a second reducing equivalent re-reduces the ferryl state of compound II back to the ferric form and, with the uptake of a second proton, releases water.
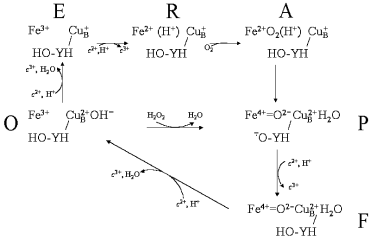
The oxidase reaction cycle is inherently more complicated in that four reductions are required to reduce oxygen to water, the active site is bimetallic and the overall cycle is coupled to four additional proton transfers through the enzyme. Figure 3 summarizes the key intermediates and shows only the binuclear center metal centers, heme a3 and CuB (for simplicity CuA and heme a, through which electrons are delivered singly into the binuclear center, are omitted). The reaction begins from the oxidized form (O) with two separate one-electron transfers, from cytochrome c via the CuA and heme a cofactors, into the binuclear heme-copper binuclear center to form, successively, the singly-electronated (E) and reduced (R) states. These electron transfers are electroneutralized by protonations [8, 9]. The doubly-reduced binuclear center is then able to bind oxygen to form “compound A”, a ferrous-oxygen adduct that can only be observed in stable form at cryogenic temperatures [30]. Conversion of compound A to the next stable intermediate, the misnamed peroxy form (P), is the most complex step chemically. Contrary to the original assumption that it contains a peroxide species, it is now clear that the O-O bond is already broken to form ferryl heme a3 in P [31-36]. However, this O-O bond cleavage requires four electrons: two are provided by the ferrous/ferryl transition of heme a3, one is provided by the cuprous/cupric transition of CuB and the fourth donor must be a radical-forming species. This donor is still not known definitively, but most likely is the covalently-linked histidine-tyrosine that is close to the oxygen binding site [5, 37-41]. The subsequent catalytic steps are then equivalent to those of the peroxidase cycle: the next electron donated from cytochrome c re-reduces the radical site to form the ferryl (F) intermediate in which ferryl heme a3 remains. Finally, a fourth electron transfer from cytochrome c reduces the ferryl species back to the ferric state with formation of water. The P → F → O steps, as with peroxidase compound I → compound II → ferric, are each associated with net proton consumption [8].Fig. 2. The peroxidase reaction cycle of HRP. Hydrogen peroxide reacts with the ferric state to produce compound I. In the case of HRP, the radical resides on the porphyrin ring but, as discussed in the text, may be otherwise located in different peroxidases. Compound I is reduced to compound II, and compound II back to the ferric state by two one-electron, one-proton transfers (see text).
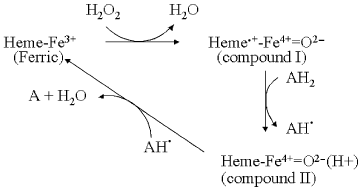
Fig. 3. The oxidase and peroxidase reaction cycles of CcO. The oxidase reaction cycle consists of the outer ring of reactions O → E → R → A → P → F → O. The lower triangle of reactions, O → P → F → O, represent a cytochrome c peroxidase reaction cycle. Some details are oversimplified for clarity, such as showing the electron wholly on CuB in E and wholly on the binuclear center in R, and the precise steps where water is released are not fully known. Further details are given in the text.
PEROXIDASE AND OXIDASE SIDE-REACTIONS
Besides the principal reaction cycles described above, both peroxidases and CcO undergo a range of additional reactions that can produce different types of net reaction cycles, albeit at much lower maximal velocities. For example, compounds I and II of HRP and CcP can react with a further molecule of hydrogen peroxide, extracting a single electron (and, presumably, a proton) and releasing superoxide. HRP compound I can also be reduced back to its ferric state with hydrogen peroxide, resulting in a slow catalatic type of reaction cycle. If the heme of HRP or CcP is reduced to the ferrous state, oxygen is able to bind to form “compound III”, a species equivalent to oxyferrous hemoglobin or compound A of CcO. However, because of the low redox potential of the heme, the species has significant Fe3+-O2.- character and dissociation into ferric heme and free superoxide, O2.-, is relatively rapid. The net result is an oxidase type of reaction that can consume oxygen with the production of the reactive oxygen species (ROS) superoxide and, after dismutation, hydrogen peroxide.
The reaction of fully oxidized CcO with hydrogen peroxide to form the peroxy (P) intermediate is well known [42, 43]. The hydrogen peroxide provides the two electrons, two protons, and diatomic oxygen that would normally be provided individually to generate P. This reaction is shown in Fig. 3. It is equivalent to formation of compound I by reaction of ferric peroxidase with hydrogen peroxide. The P state can react with a second hydrogen peroxide to form the ferryl (F) intermediate, again by a reaction that resembles one possible mechanism for conversion of peroxidase compound I into compound II. Since both P and F can accept electrons from cytochrome c or other electron donors, CcO can, in the absence of oxygen, act as a cytochrome c peroxidase. This peroxidatic cycle of CcO is represented by the lower reaction triangle shown in Fig. 3. It has been directly demonstrated as a ferrocyanide peroxidase activity [44] and the peroxidase “short-circuit” has been exploited to study the details of its coupling mechanism [45].
SPECTROSCOPIC FEATURES OF INTERMEDIATES OF PEROXIDASES AND
CcO
Both peroxidases and CcO have been studied intensively by a range of spectroscopic methods including UV/visible/near-IR, CD and MCD, EPR and ENDOR, vibrational mid-IR and Raman, and other spectroscopies. Review of this large body of spectroscopic data is beyond the present scope and we focus here only on some pertinent UV/visible features that are summarized in the table. Also included where available are equivalent bands of cytochrome bo intermediates. This bacterial oxidase, which oxidizes ubiquinol rather than cytochrome c, is otherwise highly homologous to CcO in sequence around, and structure of, its oxygen reduction site, but the heme in this case is heme O [46], a derivative that is spectrally very similar to heme B of the peroxidases and, therefore, is easier to compare. The absorption bands of compound I/P in relation to those of their equivalent ferric and compound II/F states are particularly relevant to the present discussion since they can be used to deduce whether the radical in compound I/P is located on the heme or on an amino acid.
Comparison of principal UV/visible characteristics of peroxidase and CcO
intermediates*
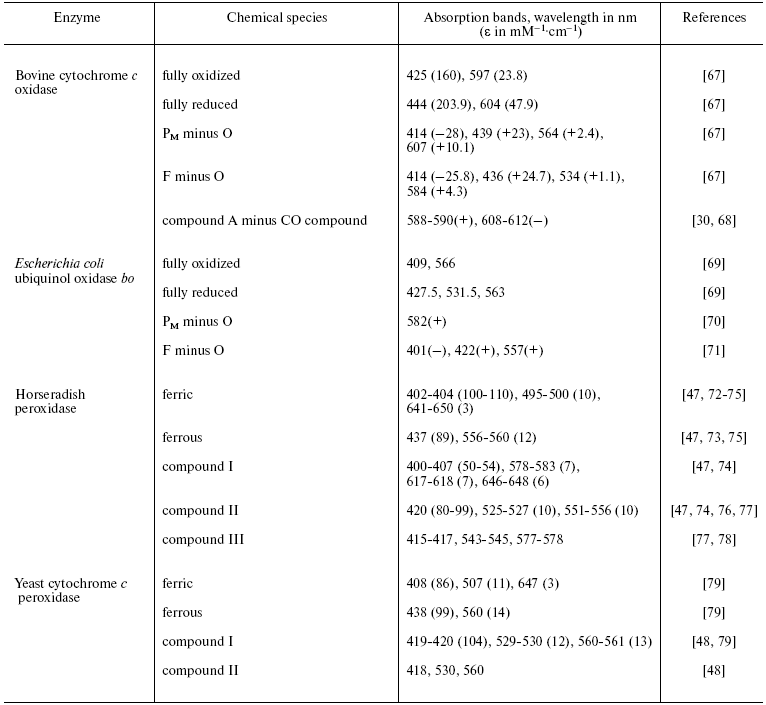
*Where available, absolute absorption spectra and their extinction
coefficients are quoted. However, in some instances, it has been
necessary instead to quote difference spectra between two states and in
these cases both maxima (+) and minima (-) are given.
PEROXIDASE COMPOUND I AND THE CcO PEROXY INTERMEDIATE
There are clear similarities between compound I of the peroxidases and the peroxy (P) intermediate of CcO. Both can be formed from the fully oxidized ferric state by reaction with hydrogen peroxide and both result in O-O bond cleavage by taking one electron from the surroundings to form a radical and a second electron from the ferric heme to produce a ferryl, Fe4+=O2-, state. As noted above, the location of the radical is different in different peroxidases. When the radical is on the porphyrin ring as in HRP, the effects on the UV/visible bands are very characteristic (table). Specifically, the Soret band remains at roughly the same wavelength as that of the ferric state, but its extinction coefficient decreases dramatically and the visible region lacks prominent features [47]. This contrasts with compound I of peroxidases such as CcP (table), or in the slowly forming secondary product of APX, where the radical is on an amino acid residue. In this case, relative to the ferric state the Soret band is red-shifted and of at least equal intensity, and two prominent visible bands are evident [48].
The location of the radical in the P state of CcO has been harder to specify because it cannot be observed by EPR/ENDOR spectroscopy. However, the red-shift and intensity of the Soret band, and the appearance of prominent visible bands (table), argue strongly against its location on the porphyrin ring. Instead, the vast body of data now point to its location on the covalently-linked histidine-tyrosine structure [5, 37-41]. The H240 is itself a ligand to CuB and spin coupling with the paramagnetic CuB2+ could account for its EPR silence [49, 50]. It has also been shown, however, that lowering of pH results in a dramatic alteration of the visible spectrum of P, with the 607 nm band shifting to 575 nm [51], although the Soret band remains roughly constant. This form has been termed F* (“F dot”) [52] as it is spectrally similar to the F intermediate but, because it is isoelectronic with P, should also contain a radical species. Again, the UV/visible features are inconsistent with a porphyrin pi-cation radical. F* formation is accompanied by appearance of a low level (5%) of an EPR-detectable radical that has been proposed to reside on a tryptophan [50] or tyrosine [53, 54]. Hence, at least to some extent, it appears likely that the radical itself can redistribute in response to local charge change caused by protonation changes, as is seen in APX [28] and, more dramatically, in myoglobin where the radical is rapidly lost altogether [55]. In the case of CcO, radical migration is not known to have any mechanistic significance and a role in peroxidases also remains uncertain [3].
PEROXIDASE COMPOUND II AND THE CcO FERRYL INTERMEDIATE
The equivalence of peroxidase compound II and the ferryl (F) intermediate of CcO is even more striking. In both cases, reduction of the radical present in compound I or in P leaves only the ferryl, Fe4+=O2-, heme. Compound II and F can be converted back into their ferric ground states by donation of a single electron (and uptake of an associated proton). Relative to their ferric forms, peroxidase compound II and F have the red-shifted Soret bands of undiminished intensity and strong visible features that are characteristic of ferryl states without an associated porphyrin radical.
PEROXIDASE COMPOUND III AND THE CcO COMPOUND A
INTERMEDIATE
Comparisons can also be made between the ferrous-oxygen adducts of peroxidases (compound III) and CcO (compound A). From a spectroscopic point of view, these species are characterized by a strong Soret band that lies between the ferric and ferrous Soret bands and prominent visible bands that are shifted relative to the ferrous alpha-band. However, compounds III and A differ dramatically in important respects. In the case of compound III of peroxidases, the heme potential is sufficiently low that the species will have significant Fe3+-O2- character and, therefore, will easily dissociate into ferric heme and free superoxide, providing the ROS-producing oxidase pathway of peroxidases described above. In practice, however, this undesirable pathway is minimized in peroxidases by the very low potential of the ferric/ferrous couple so that the ferrous state should not readily form.
In contrast, oxygen can only bind to CcO to form compound A when both heme a3 and CuB are reduced, whereas oxygen binds readily to the ferrous state of peroxidases. The precise reason for this behavior of CcO is not established. It is becoming apparent that CuB may have a somewhat higher midpoint potential than heme a3 [56]. Hence, in the one-electron state the electron should be mostly on CuB (as indicated in Fig. 3). However, carbon monoxide is also unable to bind unless both heme a3 and CuB are reduced [57] and, since, carbon monoxide should cause even a single electron to reside predominantly on heme a3, it seems unlikely that the distribution of a single electron can be the primary factor that prevents oxygen/CO binding. The reason instead most likely resides in the hydroxide/water ligands of the oxidized metals that preclude binding of other ligands and only become exchangeable with oxygen when both metals are reduced. Even if oxygen did bind transiently to the E state, the much higher ferric/ferrous potential would minimize superoxide release, as is the case in myoglobins and hemoglobins [58]. When oxygen does bind to the two-electron reduced CcO, ROS-producing reactions are prevented by the extremely rapid four-electron reaction that breaks the O-O bond and forms P.
STABLE AND TRANSIENT INTERMEDIATES IN CcO
Many of the properties of the intermediates that are discussed above have been measured in conditions in which the intermediates are stable for seconds or minutes. When working in vivo, both peroxidases and CcO can turn over many times per second and the catalytic intermediates might exist only for tens of milliseconds or less. In general for peroxidases, apart from the issue of radical migration in compound I [3], the static and transient forms of the intermediates are likely to be equivalent.
However, this is not necessarily the case for CcO intermediates, where the reaction cavity is deeply buried and with different access/exit channels for substrates and protons, and with coupling between the oxygen reduction chemistry and addition proton translocating reactions. One case in point is the R intermediate, a species that can be formed in a stable state by reaction of the ferric enzyme with carbon monoxide and oxygen. The carbon monoxide acts as a donor of two electrons and two protons to the binuclear center to form R and oxygen then binds to form compound A that spontaneously decays to form P:
heme a33+/CuB2+ + CO + H2O → heme a32+/CuB+(2H+) + CO2 (R state),
heme a32+/CuB+(2H+) + O2 → heme a32+/CuB+(2H+)-O2 (compound A),
heme a32+/CuB+(2H+)-O2 → compound P.
The P state formed in this manner (or by reaction with hydrogen peroxide [42, 43]) is usually termed PM as it is formed from the R or “mixed-valence state” in which the binuclear center metals are reduced but heme a and CuA are oxidized. There is quite reasonable evidence that a species with the same spectral characteristics can be formed during the natural catalytic cycle. However, in the electrostatic model of coupling of proton translocation with electron transfer, forward electron transfer from heme a to the binuclear center should protonate a proton trap site which subsequently loses its proton to the positive aqueous phase when the oxygen intermediate proceeds to the next stage and gains a proton. Hence, the R state in the natural cycle might be expected to have a loaded trap site that deprotonates to the positive phase when R reacts with oxygen to produce PM. However, as noted recently [20], this is clearly not the case when the R state formed with CO reacts with oxygen to form PM since the R → PM transition has no electrogenic components [59] and, therefore, the two protons required for PM production are already in the oxygen binding site. Hence, it is quite possible that the static R state formed with carbon monoxide has both protons within the binuclear center (perhaps because of the nature of the CO redox chemistry) whereas the R state formed during the natural catalytic cycle has only one proton within the binuclear center with the other in the trap site ready for expulsion into the positive phase.
A second type of P intermediate is formed transiently when the fully-reduced (FR) form of CcO reacts with oxygen, as has been studied intensively by flow-flash methods [59, 60]. In this case, oxygen binds and reacts rapidly to produce a transient species that is optically the same as PM, but has had an additional electron donated into the oxygen site from heme a that reduces the radical. This species has been termed PR [61] and differs from PM in lacking the radical. Subsequent protonation of PR converts it into the F intermediate. The fact that the radical site has little or no effect on the optical signature of PM versus PR is again consistent with the radical being located at a site other than the heme ring. In fact, it is only protonation of PM (to form F* at low pH) or of PR (to form F), rather than presence of the radical, that results in the marked change of the visible band from 607 to 575 (F*) or 580 nm (F). In studying these intermediates, however, consideration must again be given to the problem of whether their overall protonation pattern is the same as that of an equivalent intermediate formed in the natural forward reaction cycle; this is particularly pertinent when beginning with fully reduced CcO, a species that would never occur naturally.
It is clear that there are fundamental similarities between the key compounds I and II intermediates of the peroxidases and the P and F intermediates of CcO. Indeed, these and other intermediates can result in various peroxidatic, oxidatic, and catalatic side reactions. What makes the peroxidatic cycle dominate in peroxidases is the low redox potential of the ferric/ferrous couple and low rate constant for oxygen binding (k = 4.5·104 M-1·sec-1 for CcP) that minimize compound III formation and a high rate constant (k = 1.7·107 M-1·sec-1 [62]) for reaction of hydrogen peroxide with the ferric state to form compound I. In the case of CcO, the key factors that make the oxidase/water cycle dominate are the higher redox potentials of the metal centers so that they can be reduced and bind oxygen, and the low reaction rate constant for reaction of the oxidized state with hydrogen peroxide (k = (3-8)·102 M-1·sec-1 [43]). The restriction of the ability of oxygen to bind only when the binuclear center has two electrons means that binding to the R state (which is very fast (~108 M-1·sec-1 [63]) is followed, without the need to wait for further electron transfers from cytochrome c, by very rapid conversion to the P state (tau ~ 50 µsec). This rapid sequence from oxygen binding to O-O cleavage precludes ROS formation.
These differences govern the very different physiological roles that these proteins play, even though their key intermediates show many similarities. Comparison of these intermediates in relation to the natural catalytic cycles provides insights into the nature of biological oxygen reduction chemistry and suggests principles for the engineering of synthetic biomimetic devices.
This overview arises from the many projects and coworkers since being encouraged to enter the oxidase area by Peter Mitchell in 1986.
Currently, the work is supported by BBSRC project grants BB/C50747X/1 and BB/C51715X/1.
REFERENCES
1.Chodat, R., and Bach, A. (1903) Ber. Dtsch.
Chem. Ges., 36, 606-608.
2.Keilin, D. (1966) The History of Cell
Respiration and Cytochrome, Cambridge University Press,
Cambridge.
3.Hiner, A. N. P., Raven, E. L., Thorneley, R. N. F.,
Garcia-Cánovas, F., Neptuno, J., and Rodríguez-López,
J. N. (2002) J. Inorg. Biochem., 91, 27-34.
4.Jones, P., and Dunford, H. B. (2005) J. Inorg.
Biochem., 99, 2292-2298.
5.Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki,
T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., and
Yoshikawa, S. (1996) Science, 272, 1136-1144.
6.Rich, P. R. (2003) Biochem. Soc. Trans.,
31, 1095-1105.
7.Rich, P. R. (1996) in Protein Electron Transfer
(Bendall, D. S., ed.) BIOS Scientific Publishers Ltd., Oxford, pp.
217-248.
8.Mitchell, R., Mitchell, P., and Rich, P. R. (1992)
Biochim. Biophys. Acta, 1101, 188-191.
9.Mitchell, R., and Rich, P. R. (1994) Biochim.
Biophys. Acta, 1186, 19-26.
10.Meunier, B., Rodriguez-Lopez, J. N., Smith, A.
T., Thorneley, R. N. F., and Rich, P. R. (1995) Biochemistry,
34, 14687-14692.
11.Harbury, H. A. (1957) J. Biol. Chem.,
225, 1009-1024.
12.Meunier, B., Rodriguez-Lopez, J. N., Smith, A.
T., Thorneley, R. N. F., and Rich, P. R. (1998) Biochem. J.,
330, 303-309.
13.Conroy, C. W., Tyma, P., Daum, P. H., and Erman,
J. E. (1978) Biochim. Biophys. Acta, 537, 62-69.
14.Yamazaki, I., Araiso, T., Hayashi, Y., Yamada,
H., and Makino, R. (1978) Adv. Biophys., 11, 249-281.
15.Iizuka, T., Makino, R., Ishimura, Y., and
Yonetani, T. (1985) J. Biol. Chem., 260, 1407-1412.
16.Junge, W., Haumann, M., Mulkidjanian, A., and
Clausen, J. (2002) Phil. Trans. R. Soc. Lond. B, 357,
1407-1418.
17.Wikström, M., Krab, K., and Saraste, M.
(1981) Cytochrome Oxidase A Synthesis, Academic Press,
London.
18.Moody, A. J., and Rich, P. R. (1990) Biochim.
Biophys. Acta, 1015, 205-215.
19.Rich, P. R. (1995) Aust. J. Plant.
Physiol., 22, 479-486.
20.Iwaki, M., and Rich, P. R. (2007) J. Am. Chem.
Soc., 129, 2923-2929.
21.Wikström, M., and Verkhovsky, M. I. (2007)
Biochim. Biophys. Acta. Bioenergetics, in press.
22.Schulz, C. E., Devaney, P. W., Winkler, H.,
Debrunner, P. G., Doan, N., Chiang, R., Rutter, R., and Hager, L. P.
(1979) FEBS Lett., 103, 102-105.
23.Roberts, J. E., Hoffman, B. M., Rutter, R., and
Hager, L. P. (1981) J. Biol. Chem., 256, 2118-2121.
24.Rutter, R., Valentine, M., Hendrich, M. P.,
Hager, L. P., and Debrunner, P. G. (1983) Biochemistry,
22, 4769-4774.
25.Furtmüller, P. G., Zederbauer, M.,
Jantschko, W., Helm, J., Bogner, M., Jakopitsch, C., and Obinger, C.
(2006) Arch. Biochem. Biophys., 445, 199-213.
26.Hoffman, B. M., Roberts, J. E., Kang, C. H., and
Margoliash, E. (1981) J. Biol. Chem., 256, 6556-6564.
27.Huyett, J. E., Doan, P. E., Gurbiel, R.,
Houseman, A. L. P., Sivaraja, M., Goodin, D. B., and Hoffman, B. M.
(1995) J. Am. Chem. Soc., 117, 9033-9041.
28.Hiner, A. N. P., Martínez, J. I., Arnao, M.
B., Acosta, M., Turner, D. D., Raven, E. L., and
Rodríguez-López, J. N. (2001) Eur. J. Biochem.,
268, 3091-3098.
29.Behan, R. K., and Green, M. T. (2006) J.
Inorg. Biochem., 100, 448-459.
30.Chance, B., Saronio, C., and Leigh, J. S. (1975)
J. Biol. Chem., 250, 9226-9237.
31.Proshlyakov, D. A., Pressler, M. A., and Babcock,
G. T. (1998) Proc. Natl. Acad. Sci. USA, 95,
8020-8025.
32.Fabian, M., and Palmer, G. (1999)
Biochemistry, 38, 6270-6275.
33.Proshlyakov, D. A., Ogura, T., Shinzawa-Itoh, K.,
Yoshikawa, S., Appelman, E. H., and Kitagawa, T. (1994) J. Biol.
Chem., 269, 29385-29388.
34.Proshlyakov, D. A., Ogura, T., Shinzawa-Itoh, K.,
Yoshikawa, S., and Kitagawa, T. (1996) Biochemistry, 35,
76-82.
35.Proshlyakov, D. A., Ogura, T., Shinzawa-Itoh, K.,
Yoshikawa, S., and Kitagawa, T. (1996) Biochemistry, 35,
8580-8586.
36.Fabian, M., Wong, W. W., Gennis, R. B., and
Palmer, G. (1999) Proc. Natl. Acad. Sci. USA, 96,
13114-13117.
37.Ostermeier, C., Harrenga, A., Ermler, U., and
Michel, H. (1997) Proc. Natl. Acad. Sci. USA, 94,
10547-10553.
38.Mitchell, D. M., Ädelroth, P., Hosler, J.
P., Fetter, J. R., Brzezinski, P., Pressler, M. A., Aasa, R.,
Malmström, B. G., Alben, J. O., Babcock, G. T., Gennis, R. B., and
Ferguson-Miller, S. (1996) Biochemistry, 35, 824-828.
39.Michel, H. (1999) Nature, 402,
602-603.
40.Proshlyakov, D. A., Pressler, M. A., DeMaso, C.,
Leykam, J. F., DeWitt, D. L., and Babcock, G. T. (2000) Science,
290, 1588-1591.
41.Uchida, T., Mogi, T., and Kitagawa, T. (2000)
Biochemistry, 39, 6669-6678.
42.Wrigglesworth, J. M. (1984) Biochem. J.,
217, 715-719.
43.Jünemann, S., Heathcote, P., and Rich, P. R.
(2000) Biochim. Biophys. Acta, 1456, 56-66.
44.Konstantinov, A. A., Vygodina, T., Capitanio, N.,
and Papa, S. (1998) Biochim. Biophys. Acta, 1363,
11-23.
45.Pecoraro, C., Gennis, R. B., Vygodina, T. V., and
Konstantinov, A. A. (2001) Biochemistry, 40,
9695-9708.
46.Wu, W., Chang, C. K., Varotsis, C., Babcock, G.
T., Puustinen, A., and Wikström, M. (1992) J. Am. Chem.
Soc., 114, 1182-1187.
47.Blumberg, W. E., Peisach, J., Wittenberg, B. A.,
and Wittenberg, J. B. (1968) J. Biol. Chem., 243,
1854-1862.
48.Pond, A. E., Bruce, G. S., English, A. M., Sono,
M., and Dawson, J. H. (1998) Inorg. Chim. Acta, 275/276,
250-255.
49.Fabian, M., and Palmer, G. (1995)
Biochemistry, 34, 13802-13810.
50.Rich, P. R., Rigby, S. E. J., and Heathcote, P.
(2002) Biochim. Biophys. Acta, 1554, 137-146.
51.Vygodina, T., and Konstantinov, A. (1989)
Biochim. Biophys. Acta, 973, 390-398.
52.Moody, A. J., and Rich, P. R. (1994) Eur. J.
Biochem., 226, 731-737.
53.Budiman, K., Kannt, A., Lyubenova, S., Richter,
O.-M. H., Ludwig, B., Michel, H., and MacMillan, F. (2004)
Biochemistry, 43, 11709-11716.
54.Svistunenko, D. A. (2005) Biochim. Biophys.
Acta. Bioenergetics, 1707, 127-155.
55.Cooper, C. E., Jurd, M., Nicholls, P., Wankasi,
M. M., Svistunenko, D. A., Reeder, B. J., and Wilson, M. T. (2005)
Dalton Transactions, 3483-3488.
56.Gorbikova, E. A., Vuorilehto, K., Wikström,
M., and Verkhovsky, M. I. (2006) Biochemistry, 45,
5641-5649.
57.Wilson, D. F., and Miyata, Y. (1977) Biochim.
Biophys. Acta, 461, 218-230.
58.Brunori, M., Saggese, U., Rotilio, G. C.,
Antonini, E., and Wyman, J. (1971) Biochemistry, 10,
1604-1609.
59.Jasaitis, A., Verkhovsky, M. I., Morgan, J. E.,
Verkhovskaya, M. L., and Wikström, M. (1999) Biochemistry,
38, 2697-2706.
60.Namslauer, A., Aagaard, A., Katsonouri, A., and
Brzezinski, P. (2003) Biochemistry, 42, 1488-1498.
61.Morgan, J. E., Verkhovsky, M. I., Palmer, G., and
Wikström, M. (2001) Biochemistry, 40, 6882-6892.
62.Dunford, H. B., and Hewson, W. D. (1977)
Biochemistry, 16, 2949-2957.
63.Babcock, G. T., and Wikström, M. (1992)
Nature, 356, 301-309.
64.Berglund, G. I., Carlsson, G. H., Smith, A. T.,
Szöke, H., Henriksen, A., and Hajdu, J. (2002) Nature,
417, 463-468.
65.Finzel, B. C., Poulos, T. L., and Kraut, J.
(1984) J. Biol. Chem., 259, 13027-13036.
66.Yoshikawa, S., Shinzawa-Itoh, K., Nakashima, R.,
Yaono, R., Yamashita, E., Inoue, N., Yao, M., Fei, M. J., Libeu, C. P.,
Mizushima, T., Yamaguchi, H., Tomizaki, T., and Tsukihara, T. (1998)
Science, 280, 1723-1729.
67.Rich, P. R., and Moody, A. J. (1997) in
Bioelectrochemistry: Principles and Practice (Gräber, P.,
and Milazzo, G., eds.) Birkhäuser Verlag AG, Basel, pp.
419-456.
68.Ogura, T., Yoshikawa, S., and Kitagawa, T. (1989)
Biochemistry, 28, 8022-8027.
69.Tsubaki, M., Mogi, T., Hori, H., Hirota, S.,
Ogura, T., Kitagawa, T., and Anraku, Y. (1994) J. Biol. Chem.,
269, 30861-30868.
70.Morgan, J. E., Verkhovsky, M. I., Puustinen, A.,
and Wikström, M. (1995) Biochemistry, 34,
15633-15637.
71.Puustinen, A., Verkhovsky, M. I., Morgan, J. E.,
Belevich, N. P., and Wikström, M. (1996) Proc. Natl. Acad. Sci.
USA, 93, 1545-1548.
72.Adak, S., Mazumdar, S., and Banerjee, R. (1997)
J. Biol. Chem., 272, 11049-11056.
73.Howes, B. D., Feis, A., Raimondi, L., Indiveri,
C., and Smulevich, G. (2001) J. Biol. Chem., 276,
40704-40711.
74.Hewson, W. D., and Hager, L. P. (1979) J.
Biol. Chem., 254, 3182-3186.
75.Yamazaki, I., Yokota, K., and Tamura, M. (1966)
in Hemes and Hemoproteins (Chance, B., Estabrook, R. W., and
Yonetani, T., eds.) Academic Press, London, pp. 319-326.
76.Adediran, S. A., and Lambeir, A.-M. (1989)
Eur. J. Biochem., 186, 571-576.
77.Kawano, T., Muto, H., Adachi, M., Hosoya, H., and
Lapeyrie, F. (2002) Biosci. Biotechnol. Biochem., 66,
646-650.
78.Wittenberg, J. B., Noble, R. W., Wittenberg, B.
A., Antonini, E., Brunori, M., and Wyman, J. (1967) J. Biol.
Chem., 242, 626-634.
79.Yonetani, T., and Ray, G. S. (1965) J. Biol.
Chem., 240, 4503-4508.