Peroxidase Activity of Mitochondrial Cytochrome c Oxidase
T. V. Vygodina and A. A. Konstantinov*
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-3181; E-mail: konst@genebee.msu.ru* To whom correspondence should be addressed.
Received May 23, 2007
Mitochondrial cytochrome c oxidase is able to oxidize various aromatic compounds like o-dianisidine, benzidine and its derivatives (diaminobenzidine, etc.), p-phenylenediamine, as well as amidopyrine, melatonin, and some other pharmacologically and physiologically active substances via the peroxidase, but not the oxidase mechanism. Although specific peroxidase activity of cytochrome c oxidase is low compared with classical peroxidases, its activity may be of physiological or pathophysiological significance due to the presence of rather high concentrations of this enzyme in all tissues, as well as specific localization of the enzyme in the mitochondrial membrane favoring accumulation of hydrophobic aromatic substances.
KEY WORDS: the oxygenase theory of A. N. Bach, respiratory chain, peroxidase activity, aromatic substratesDOI: 10.1134/S0006297907100045
High oxidant activity of molecular oxygen and products of its partial reduction (especially hydrogen peroxide) is widely used by living organisms both for energy conservation and in the “plastic metabolism” for oxidative synthesis or degradation of various substances.
The main forms of oxygen involved in enzymatic oxidation include molecular oxygen (O2), superoxide radical (O2-), and hydrogen peroxide (H2O2).
Enzymes utilizing these oxygen forms as electron acceptors are referred to as oxidases (and also as monooxygenases), superoxide dismutases, and peroxidases.
Oxidases (donor:O2-oxidoreductases) are numerous and diverse enzymes that utilize molecular oxygen as terminal electron acceptor. Some of them (usually flavoproteins) catalyze the two-electron reduction of oxygen followed by formation of hydrogen peroxide. Others can catalyze the four-electron reduction of molecular oxygen to two molecules of water without release of toxic intermediates (products of partial oxygen reduction such as superoxide anion and hydrogen peroxide). These include all known terminal oxidases of respiratory chains of aerobic bacteria, plants, and animals, for example, cytochrome c oxidase.
Cytochrome c oxidase is the key enzyme of aerobic metabolism of eukaryotes and many bacteria. The enzyme catalyzes the four-electron reduction of oxygen to water coupled to generation of DeltaµH+ onto the inner mitochondrial membrane or cytoplasmic membrane of bacteria; it corresponds to a so-called third site of energy coupling of the mitochondrial respiratory chain [1-5]. Significant progress has been achieved in studies of cytochrome oxidase; this includes elucidation of three-dimensional structure of the enzyme from bovine heart mitochondria [6] and three bacterial species [7-9]. Using various spectral methods and methods of fast kinetics, the main intermediates formed in the oxygen reductase center of cytochrome oxidase during the catalytic cycle have been identified [10]. Figure 1 shows the catalytic cycle of cytochrome oxidase.
Several schemes of “separation” of the catalytic cycle of cytochrome oxidase into partial reactions have been described in the literature. Most frequently authors subdivide total reaction into reductive and oxidative parts of the cycle [3, 5, 11, 12]. However, this approach adequately describes only an artificial experimental situation, when initially oxidized enzyme is completely reduced under aerobic conditions (i.e. by four electrons) by means of strong artificial reductant such as dithionite (reduction phase) and then this completely reduced enzyme is mixed with oxygen (oxidation phase). Using the natural electron donor, cytochrome c, complete reduction of the enzyme never occurs and transfer of each of four electrons through the enzyme includes both the reduction phase of redox centers of cytochrome oxidase and subsequent oxidation phase by oxygen bound to the enzyme.
Konstantinov et al. [13-15] have proposed a different approach. According to this approach the catalytic cycle of cytochrome oxidase consists of two different reactions: oxidase (eu-oxidase) reaction, in which molecular oxygen undergoes the two electron reduction to peroxide bound at the active site and peroxidase reaction, in which endogenous peroxide undergoes further two electron reduction to two H2O molecules (Fig. 1). During the work on this paper, we have found that such consideration of the catalytic cycle exactly corresponds to the initial hypothesis on respiration mechanism proposed by A. N. Bach [16]. Bach and Chodat suggested the two-step nature of the respiration mechanism, which includes the oxygenation phase (interaction of catalyst with molecular oxygen resulting in formation of bound peroxide) followed by subsequent peroxidase phase, in which endoperoxide acts as electron acceptor for an oxidized substrate (see [16] and detailed discussion in the paper by P. Nichols in this issue). In the scheme of Fig. 1 the eu-oxidase phase (which would be defined as the oxygenase within Bach's terminology) begins with the transfer of two electrons to the oxygen reductase center of oxidized cytochrome oxidase, containing high spin heme and a copper ion, and these centers are reduced to a32+ and CuB+. In such state high spin heme a32+ can bind an oxygen molecule (similar to myoglobin), and the reduced state CuB is required for oxygen transport to heme a32+ via a special channel; this stage involves intermediate binding with reduced copper ion playing the role of a “gate”.
In the presence of two electrons in the oxygen reductase center (on heme and copper), the bound oxygen molecule is immediately reduced to peroxide (one electron comes from the a32+ heme and the other from CuB+). This results in formation of the ferri-peroxy complex P (P for peroxy), in which peroxide bound to the ferri-heme a3 is doubly protonated. If there is an access to two protons, the ferri-peroxy complex is very unstable and the doubly protonated peroxide P0, an analog of the short-term enzyme-substrate peroxidase complex (Compounds 0) [17] is formed as the short-term unstable intermediate, which immediately undergoes heterolytic cleavage (by analogy with heme peroxidases) [15, 18]. The distal oxygen atom of the peroxide retains both electrons accepted by the O2 molecule (thus explaining why such cleavage of the O-O bond is defined as heterolytic) and together with two protons this distal oxygen atom forms a water molecule. The proximal oxygen atom remains bound to the heme iron and accepts two electrons from the enzyme. One electron comes directly from the a3 heme iron, which becomes tetravalent. As in the case of yeast cytochrome c peroxidase, the second electron comes from an aromatic amino acid residue of cytochrome oxidase located near the a3 heme; this results in formation of an amino acid radical (R*). Certain evidence exists that the electron donor amino acid residue is the conservative tyrosine located near the heme and forming a covalent bond with imidazole of one of three histidines forming a coordination sphere of an “invisible copper” (CuB) [6, 7, 19, 20]. The cleavage of the O-O bond results in formation of FI similar to so-called Compound I of peroxidases. In this intermediate heme a3 iron exists as the oxoferryl complex (Fe4+=O2-), which may be considered as the complex of ferryl iron of a3 heme with a doubly deprotonated water molecule, and the second electron vacancy is at the tyrosine radical [20]. Thus, the intermediate FI has two electrons less than the initial oxidized state of the enzyme (O). Intermediate FI with characteristic an intensive absorption maximum at 607 nm was designated for a long time (and is sometimes designated now) as intermediate P (P for peroxy), suggesting that it contains a bound molecule of doubly deprotonated hydrogen peroxide forming a bridge between a33+ and CuB2+. Since it is clear that intermediate 607 lacks the O-O bond, this old-fashioned terminology misleads. It is better to define the ferryl-oxo complex as FI-607, where the index I indicates the existence of homology with peroxidase Compound I. This index also indicates that in the catalytic cycle of cytochrome oxidase, index I suggests that this intermediate precedes formation of the other oxoferryl complex FII with maximum at 580 nm; the latter is homologous to peroxidase Compound II (FII-580) usually defined in the literature just as F.Fig. 1. Catalytic cycle of cytochrome oxidase. The scheme shows characteristics of the oxygen reductase center of cytochrome oxidase (heme a3 and “invisible” CuB) in various intermediates: O, oxidized (Fe3+/Cu2+); R, reduced (Fe2+/Cu+); Oxy, oxycomplex (Fe2+-O2/Cu+). In other intermediates, CuB exists as Cu2+. Species that are in rectangular frames can be observed experimentally; those in parentheses denote the states that have not been resolved but can be presumed as analogous to the well investigated enzymes. The chemical mechanism of the catalytic cycle of cytochrome oxidase is clearly subdivided into two phases--eu-oxidase and peroxidase; this perfectly coincides with the hypothesis on the mechanism of oxygen reduction proposed by Bach and Chodat [16]; in Bach's terminology the “eu-oxidase” phase would be defined as the “oxygenase” phase.
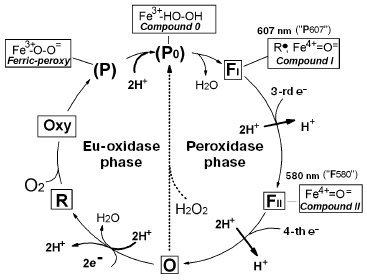
The peroxidase phase of the catalytic cycle of cytochrome oxidase (transfer of the third and fourth electrons) is basically analogical to the sequence of events in the peroxidase reaction. The third electron reduces an amino acid residue in the oxygen reductase center, whereas the fourth electron reduces the oxoferryl complex of heme a3, and thus returns cytochrome oxidase to the initial state. Thus, the full catalytic cycle of cytochrome oxidase falls into two well defined chemical phases--oxidase (eu-oxidase) and peroxidase.
On addition of hydrogen peroxide to oxidized cytochrome oxidase, the enzyme undergoes transition from the state O (via the hydroperoxide complex P0) into FI-607 (indicated by the dotted arrow in Fig. 1), bypassing the stages of the catalytic cycle related to addition of the first two electrons, oxygen, and two protons (all these ingredients are pooled together in the hydrogen peroxide molecule), i.e. the eu-oxidase phase of the cytochrome oxidase reaction. Using these circumstances, we have separately investigated the partial peroxidase activity of mitochondrial and bacterial cytochrome oxidases under aerobic conditions by means of the artificial electron donor ferrocyanide [14, 21], as well as the natural donor cytochrome c [22]. The possibility in principle for a cytochrome c peroxidase reaction catalyzed by the mitochondrial enzyme has been demonstrated under strictly anaerobic conditions by Orii's group [23-26]. However, particular experimental conditions used by Orii and coworkers suggest demonstration of so-called “pseudo-peroxidase activity” (see [14]), the direct two-electron oxidation of R state to O by hydrogen peroxide (Fig. 1) [27], excluding the peroxidase phase of the catalytic cycle and formation of the oxoferryl intermediates of cytochrome oxidase.
It was found that the peroxidase phase of the cytochrome oxidase reaction is coupled to proton translocation [21, 22] and the delivery of protons (both pump and substrate ones) occurs via D-proton channel, whereas substitutions of critical amino acids in proton channel K do not affect peroxidase activity [22].
The characteristic feature of one electron redox transitions FI → FII and FII → O is rather high values of half-reduction potential E0´ = 0.8-1.0 V; which is typical for both the peroxidase phase of cytochrome oxidase catalytic cycle and peroxidases [28, 29]. This feature allows peroxidase to oxidize a large number of compounds exhibiting electron-acceptor properties. We have suggested that cytochrome oxidase may nonspecifically oxidize various compounds that are not substrates of its oxidase activity due to peroxidase activity of the enzyme.
In this study, we have demonstrated that numerous organic aromatic compounds, which are not oxidized by cytochrome oxidase via the oxidase mechanism (i.e. using molecular oxygen as the terminal acceptor), can undergo a low rate oxidation by cytochrome oxidase via the peroxidase mechanism.
MATERIALS AND METHODS
Reagents, buffers, and aromatic substrates of high purity used in this study were purchased from Sigma (USA). The commercial preparation of hydrogen peroxide (about 30%) of high purity was kept in a refrigerator. Concentration of hydrogen peroxide was checked before experiments using the molar extinction coefficient Deltaepsilon240 = 40 M-1·cm-1 [30], and then solutions of required concentration were prepared. All buffers and solutions were prepared using MilliQ purified water. Poorly water soluble aromatic substrates (paracetamol and melatonin) were dissolved in doubly distilled ethanol.
Preparations of cytochrome oxidase were isolated from bovine cardiac mitochondria using the modified method of Fowler et al. [31] followed by its subsequent purification [32]. Cytochrome oxidase was solubilized by dodecyl maltoside (SOL-GRADE; Anatrace, USA). Concentration of cytochrome oxidase was determined from the differential absorption spectrum (sample reduced by dithionite minus the oxidized one) using the coefficient of molar extinction of this enzyme Deltaepsilon605-630_= 27 mM-1·cm-1.
Spectrophotometric studies were carried out in a standard cuvette with light path of 10 mm. The basic incubation medium contained 50 mM MES/Tris buffer, pH 6.5 (if a particular pH value is not specified), and 0.05% dodecyl maltoside. These conditions are subsequently indicated as the standard ones. Absorption spectra were registered using a Cary Bio 300 spectrophotometer (Varian, USA) at the scanning rate of 2 nm/sec. The absolute spectrum of a substrate was recorded versus the reference cuvette containing the same buffer, then hydrogen peroxide and cytochrome oxidase were added to both cuvettes and spectra were registered after certain time intervals. Kinetics of substrate peroxidation were registered using the dual-wavelength mode of an SLM-Aminco DW-2000 spectrophotometer (SLM Instruments, USA).
RESULTS
We initially investigated the interaction of bovine mitochondrial cytochrome oxidase with a set of classical substrates used during studies of peroxidases. Table 1 shows structural formulas of these compounds and Table 2 summarizes the results of our experiments.
Table 1. Aromatic compounds tested as
substrates for assay of peroxidase activity of mitochondrial cytochrome
oxidase
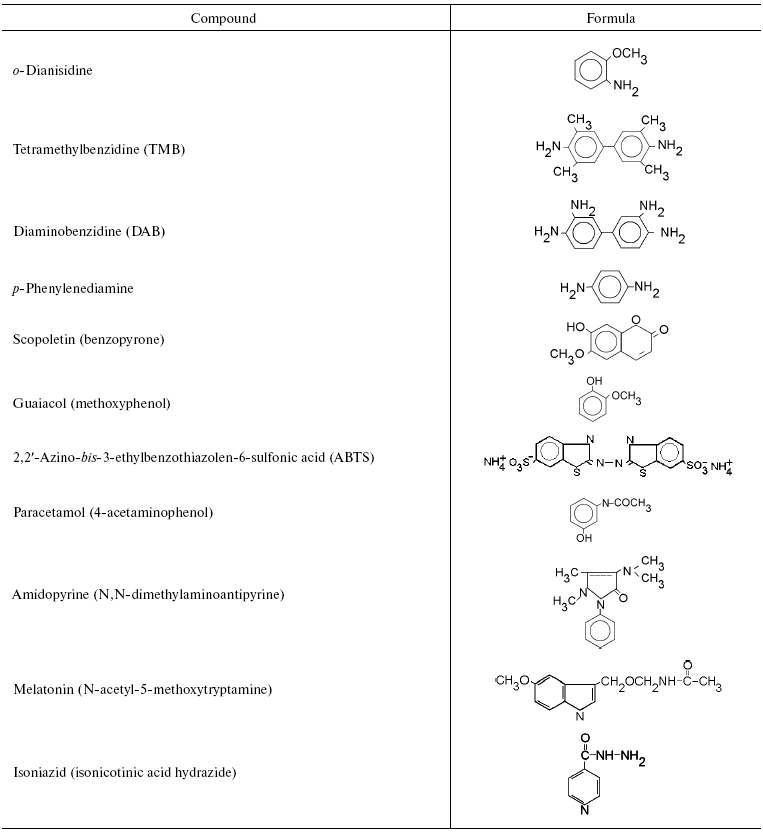
Table 2. Peroxidation of aromatic compounds
catalyzed by bovine heart cytochrome oxidase

Note: The reaction was carried out under the standard conditions
(see “Materials and Methods”) in the presence of
1 µM cytochrome oxidase, 1 mM
H2O2, and 0.2 mM oxidation substrate (except
amidopyrine and paracetamol used in 1 mM concentration).
Experiments have shown that cytochrome oxidase can catalyze peroxidation of o-dianisidine, benzidine and its derivatives (tetramethylbenzidine and diaminobenzidine), and also p-phenylenediamine. However, the latter was partially oxidized by hydrogen peroxide in the absence of cytochrome oxidase. Obviously, interaction of substrates with the “entrance” center of cytochrome oxidase (CuA) plays an essential role in the peroxidase reaction. The environment of CuA carries negative charge and therefore the enzyme exhibits better interaction with hydrophilic positively charged one-electron donors (cytochrome c, hexamine ruthenium), and also with compounds which can form cations during protonation or one electron oxidation (aromatic amines, quinonimines, etc.). Anions (ferrocyanide, ascorbate) and also quinols generated during one electron oxidation, negatively charged semiquinone-anion are usually poor electron donors for the enzyme [33]. The reaction of cytochrome oxidase with ABTS (2,2´-azino-bis-3-ethylbenzothiazolen-6-sulfonic acid) is probably complicated by total negative charge of this compound, while scopoletin and guaiacol cannot effectively interact with the hydrophilic environment of the “entrance” CuA center of cytochrome oxidase due to their relative hydrophobic nature and lack of a positive charge. It should be noted that bd-type quinol oxidase effectively catalyzed guaiacol peroxidation but cannot react with other more hydrophilic substrates (data not shown).
For detailed characterization of peroxidase activity of cytochrome oxidase, we have investigated the interaction of this enzyme with o-dianisidine. The overall equation of this reaction is:
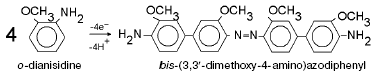
Fig. 2. Peroxidation of o-dianisidine catalyzed by bovine heart cytochrome c oxidase. a) Sample and control cuvettes contain standard incubation medium, pH 6.5. Initial spectrum 1 was recorded after addition of 0.2 mM o-dianisidine to the sample cuvette. Then H2O2 and cytochrome oxidase (final concentrations of 1 mM and 1 µM, respectively) were added to both cuvettes and after the indicated time intervals (2-40 min) spectra 2-7 were registered. b) Kinetics of o-dianisidine peroxidation: 1) the cuvette contained standard incubation medium, pH 6.5, and also 0.2 mM o-dianisidine and 1 µM cytochrome oxidase. Additions: 1 mM H2O2 and 2 mM KCN; 2) all components are the same as in (1) but without cytochrome oxidase.
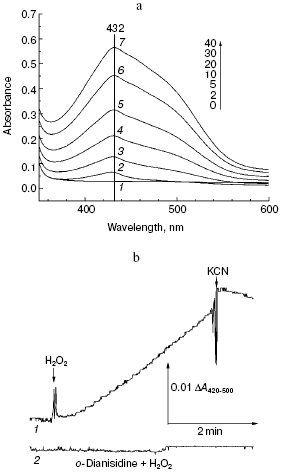
Aerobic incubation of o-dianisidine with cytochrome oxidase for at least 30 min did not result in any oxidation of this compound (data not shown). However, addition of hydrogen peroxide caused progressive oxidation of o-dianisidine, accompanied by the increase of absorption at 432 nm. Figure 2a shows a typical series of spectra reflecting time-dependent o-dianisidine oxidation. Addition of a cytochrome oxidase inhibitor, cyanide, completely blocked the oxidation of o-dianisidine (Fig. 2b, curve 1) and it was not observed during addition of H2O2 in the absence of cytochrome oxidase (Fig. 2b, curve 2). Thus, there is evidence that enzymatic oxidation of o-dianisidine occurred via the peroxidase mechanism. The reaction rate initially increased with the increase of enzyme concentration but then it reached saturation (Fig. 3c); this may be explained by interaction of cytochrome oxidase with primary reaction products.Fig. 3. Dependence of the rate of peroxidase reaction on concentration of o-dianisidine (a), hydrogen peroxide (b), and cytochrome c oxidase (c). The main conditions are the same as indicated in Fig. 2.
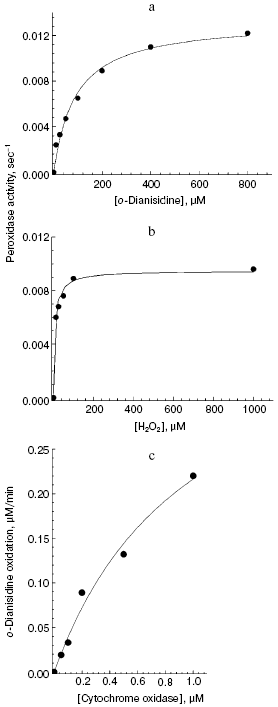
The dependence of peroxidase activity of cytochrome oxidase on concentration of o-dianisidine added (Fig. 3a) is characterized by a typical saturation curve. At 1 mM H2O2, the values of Km for o-dianisidine and Vmax (expressed as turnover number) were 90 ± 13 µM and 0.013 ± 0.0006 sec-1, respectively. At constant o-dianisidine concentration of 0.1 mM the reaction rate constantly increased with the increase of hydrogen peroxide (Fig. 3b), reaching half-maximal value at 10 µM; this is consistent with the value of 10-5 M calculated from the kinetic parameters:
Vmax = 0.01 sec-1/kv,
where kv is the rate of H2O2 binding in the active site of cytochrome oxidase, equal to 1000 M-1·sec-1. Decreasing pH from 8.0 to 6.4 was accompanied by a 5-fold increase of cytochrome oxidase activity (from 0.002 to 0.01 sec-1, respectively). Oxidation of o-dianisidine via the peroxidase mechanism was also demonstrated for aa3 type bacterial cytochrome oxidase from Rhodobacter sphaeroides (data not shown). The bacterial enzyme was two times more active than bovine heart cytochrome oxidase. This is consistent with the observation that the enzyme from R. sphaeroides has two times higher specific cytochrome c oxidase activity.
In subsequent experiments, we explored the possibility of peroxidation by cytochrome oxidase of some arbitrarily selected compounds possessing either physiological activity or acting as pharmacological drugs. (In this case, their oxidation catalyzed by cytochrome oxidase may have biomedical importance.)
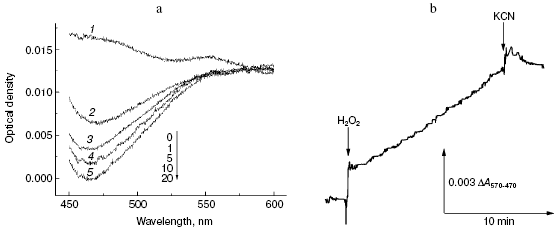
First, we compared the resistance of two well-known antipyretics possessing rather simple structure: paracetamol (Tylenol, 4-acetaminophenol) and amidopyrine (N,N-dimethyl-4-aminoantipyrine). One electron oxidation of amidopyrine via the peroxidation mechanism resulted in radical formation, which can be monitored spectrophotometrically, e.g. by DeltaA570-470 [35]. Incubation of 1 mM amidopyrine with cytochrome oxidase in the presence of 2 mM H2O2 was accompanied by decrease in absorption at 470 nm (Fig. 4a) implying its oxidation via the peroxidase mechanism. Kinetic traces of amidopyrine peroxidation using the dual-wavelength mode DeltaA570-470 are shown in Fig. 4b. Addition of cytochrome oxidase to 1 mM aqueous solution of amidopyrine did not cause any changes under aerobic conditions; this shows that substance cannot be oxidized via the oxidase pathway. Subsequent addition of 2 mM hydrogen peroxide caused amidopyrine oxidation, which was completely blocked by cyanide. Maximal values of specific activity of cytochrome oxidase for amidopyrine peroxidation at 2 mM H2O2 and 20°C did not exceed 0.001 sec-1, this values being one order of magnitude less than that observed for o-dianisidine. In contrast to amidopyrine, paracetamol was completely resistant to peroxidation by cytochrome oxidase. Isonicotinic acid hydrazide (isoniazid, one of the most important basic drugs for treatment of tuberculosis) was resistant to peroxidation catalyzed by cytochrome oxidase; however, it was effectively oxidized by a special catalase-peroxidase isolated from bacillus tuberculosis [36].
Second, we investigated the interaction of cytochrome oxidase with melatonin, which attracts much attention in various biomedical studies [37]. It is formed in the pineal gland of vertebrates and is a hormone of very high hierarchy, which is involved in regulation of biorhythms. Melatonin may be also formed in cells of other tissues, where it acts as a signaling molecule in many processes; it also acts as a potent anti-apoptotic factor. There is growing interest in anticancer activity of melatonin [37]. Melatonin may easily undergo oxidation-reduction conversion; for example, it is readily oxidized by myeloperoxidase [38] and is one of the most active antioxidants involved in quenching of reactive oxygen species and therefore in the antioxidant protection of the cells [37].Fig. 4. Peroxidation of amidopyrine catalyzed by cytochrome oxidase. Sample and control cuvettes contain standard incubation medium, pH 6.5. a) Initial spectrum 1 was recorded after addition of 1 mM amidopyrine to the sample cuvette. Then H2O2 and cytochrome oxidase (final concentrations of 2 mM and 1 µM, respectively) were added to both cuvettes, and after the indicated time intervals spectra 2-5 were registered. b) Kinetics of amidopyrine peroxidation. The cuvette contained 1 mM amidopyrine and 1 µM cytochrome oxidase in the basic incubation medium. Additions: 2 mM H2O2 and KCN.
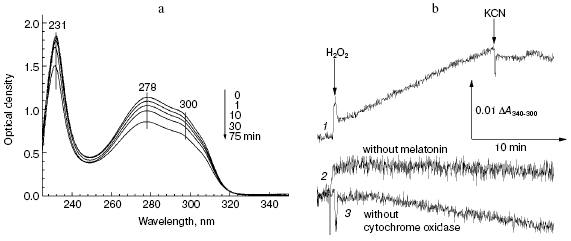
The data of Fig. 5a show that addition of hydrogen peroxide to bovine heart cytochrome oxidase caused the decrease in intensity of melatonin absorption bands in the near and far UV regions at 300, 280, and 231 nm. Melatonin oxidation required the presence of cytochrome oxidase and hydrogen peroxide and it was totally blocked by cyanide (Fig. 5b, curve 1). If the molar extinction of melatonin is about 5 mM-1·cm-1 (at 300 nm), the specific activity of its peroxidation in the presence of 1 mM H2O2 was about 0.0032 sec-1.
Fig. 5. Melatonin peroxidation catalyzed by cytochrome oxidase. a) Sample and control cuvettes contained the standard buffer, pH 6.5. Spectrum 1 was recorded after addition of 0.2 mM melatonin to the sample cuvette. Then 1 mM H2O2 and 0.5 µM cytochrome oxidase were added and after the indicated time intervals spectra 2-5 were registered. b) Kinetics of melatonin peroxidation. Curves: 1) cuvette contained 0.2 mM melatonin and 0.5 µM cytochrome oxidase in the basic incubation medium. Additions 1 mM H2O2, 2 mM KCN; 2, 3) same as (1) but without melatonin or cytochrome oxidase, respectively.
DISCUSSION
The major conclusion of this study is the following: cytochrome oxidase (like peroxidases and cytochrome P450) can catalyze the peroxidation reaction (i.e. reaction involving formation of intermediates FI and FII, homologous to peroxidase Compounds I and II) of various aromatic substrates including physiologically important substances.
This is quite clear because one electron oxidation of substituted aromatic compounds to corresponding free radicals requires a rather potent electron acceptor in the enzyme with the half-reduction potential E0´ of about +1 V. Such values of the half-reduction potential are typical for heme a3 at the highest oxidation state of heme iron present in intermediates FI and FII of the catalytic cycle of this enzyme; these intermediates are homologous to peroxidase Compounds I and II [39]. (In the usual oxidized enzyme form, the values of E0´ of the redox centers do not exceed 0.35-0.4 V.)
Compounds FI and FII of cytochrome oxidase may also appear during enzyme interaction with hydrogen peroxide constantly formed (in small quantities) within cells. According to [40] formation of the compound FI-607 of cytochrome oxidase reaches saturation even at micromolar concentrations of hydrogen peroxide. Besides marked steady-state concentrations of the FI-607 and FII-580 intermediates are accumulated during cytochrome oxidase turnover in an energized membrane [28, 41].
It is clear that turnover number of cytochrome oxidase during peroxidation of aromatic compounds usually does not exceed one turnover per tens of thousands of seconds; it is incomparable with its main cytochrome c oxidase activity as well as specific activity of peroxidases or cytochrome P450, for which oxidation of aromatic substrates is the main catalytic function. This means that peroxidase activity is not a massive catabolic process involving cytochrome oxidase.
Reasons underlying low specific activity of cytochrome oxidase in the peroxidase reaction are quite clear. First, oxidized cytochrome oxidase rather slowly interacts with hydrogen peroxide (kv = 500-1000 M-1·sec-1), which is similar to metmyoglobin but far below the values (kv ~ 107 M-1·sec-1) characteristic for rapidly functioning peroxidases. The main reason for such difference is vacancy of the sixth coordination bond of heme iron in rapidly functioning peroxidases, whereas in cytochrome oxidase or in methemoglobin the sixth coordination position (distal axial coordination bond) is occupied by a water molecule or even more tightly bound hydroxyl (depended on pH). Slow binding of H2O2 to cytochrome oxidase limits enzyme peroxidase activity even in the case of such readily oxidizable substrates as the natural donor, cytochrome c.
Considering oxidation of less effective (than cytochrome c) donors, derivatives of benzidine or other aromatic compounds, an additional reason for cytochrome oxidase low rate in peroxidase reaction is determined by the fact that peroxidase molecules contain special substrate binding sites closely positioned (<10 Å) to the heme iron whose higher oxidation states generated due to interaction with H2O2 are directly involved in one electron oxidation of aromatic or any other donor to the corresponding radical. In contrast, binuclear oxygen reducing center of cytochrome c oxidase including heme a3 is located deeply inside the enzyme and oxidation of donors involves the “entrance” redox center CuA located on the enzyme surface and positioned at the distance of about 23 Å from heme a3. The half-reduction potential of CuA (about +0.24 V) is rather low so that the energy profile of multistage reaction of oxidation-reduction (which is thermodynamically favorable in general),
donor → CuA → heme a → heme a34+=O2-,
is characterized by rather sharp increase at the first stage, and this inhibits reaction.
However, from our viewpoint the small “parasitic” activity of cytochrome oxidase should not be ignored as it may cause formation of side (potentially toxic) products of pharmacological drugs and also contribute to degradation of useful compounds. Cytochrome oxidase is a ubiquitous enzyme that is present in all tissues of the body. Peroxidases or cytochromes P450 have not been found in all tissues; they are almost absent in myocardium, where the concentration of mitochondria and therefore cytochrome oxidase is quite high. Besides, cytochrome oxidase has particular localization in the mitochondrial membrane, which readily accumulates hydrophobic aromatic compounds inside its phospholipid bilayer and also absorbs amphiphilic compounds on its surface. Thus, one should take into consideration possibility of nonspecific peroxidation of various substances catalyzed by cytochrome oxidase via the peroxidase mechanism; this may contribute to intracellular metabolism of biologically active drugs (under conditions of cardiotoxicity) and other compounds.
The authors are grateful to Professor P. Nicholls for the opportunity to read his manuscript prepared for this issue before publication and to discuss the historical aspect of this problem.
This work was supported by a grant from the Russian Foundation for Basic Research (No. 06-04-48185) (T.V.V.) and also by grants from the Howard Hughes Medical Institute (No. 55005615) and CRDF (No. RUB-1-2836) (A.A.K.).
REFERENCES
1.Babcock, G. T., and Wikstrom, M. (1992)
Nature, 356, 301-309.
2.Ferguson-Miller, S., and Babcock, G. T. (1996)
Chem. Rev., 7, 2889-2907.
3.Richter, O.-M. H., and Ludwig, B. (2003) Rev.
Physiol. Biochem. Pharmacol., 147, 47-74.
4.Gennis, R. B. (2005) in Biophysical and
Structural Aspects of Bioenergetics (Wikstrom, M., ed.) RSC
Publishing, Norfolk, pp. 1-24.
5.Wikstrom, M. (2004) Biochim. Biophys. Acta,
1655, 241-247.
6.Tsukihara, T., Aoyama, H., Yamashita, E., Takashi,
T., Yamaguichi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., and
Yoshikawa, S. (1996) Science, 272, 1136-1144.
7.Iwata, S., Ostermeier, C., Ludwig, B., and Michel,
H. (1995) Nature, 376, 660-669.
8.Svensson-Ek, M., Abramson, J., Larsson, G.,
Tornroth, S., Brzezinski, P., and Iwata, S. (2002) J. Mol.
Biol., 321, 329-339.
9.Soulimane, T., Buse, G., Bourenkov, G. B.,
Bartunik, H. D., Huber, R., and Than, M. E. (2000) EMBO J.,
19, 1766-1776.
10.Einarsdottir, O., and Szundi, I. (2004)
Biochim. Biophys. Acta, 1655, 263-273.
11.Bloch, D., Belevich, I., Jasaitis, A., Ribacka,
C., Puustinen, A., Verkhovsky, M. I., and Wikstrom, M. (2004) Proc.
Natl. Acad. Sci. USA, 101, 529-533.
12.Jancura, D., Berka, V., Antalik, M., Bagelova,
J., Gennis, R. B., Palmer, G., and Fabian, M. (2006) J. Biol.
Chem., 281, 30319-30325.
13.Konstantinov, A. A., Siletskiy, S. A., Mitchell,
D., Kaulen, A. D., and Gennis, R. B. (1997) Proc. Natl. Acad. Sci.
USA, 94, 9085-9090.
14.Konstantinov, A. A., Vygodina, T. V., Capitanio,
N., and Papa, S. (1998) Biochim. Biophys. Acta, 1363,
11-23.
15.Konstantinov, A. (1998) J. Bioenerg.
Biomembr., 30, 121-130.
16.Bach, A. N., and Chodat, R. (1902) Ber.
Deutsch. Chem. Gesell., 35, 2466-2470.
17.Baek, H. K., and van Wart, H. E. (1989)
Biochemistry, 28, 5714-5719.
18.Blomberg, M., Siegbahn, P. E. M., Babcock, G. T.,
and Wikstrom, M. (2000) J. Inorg. Chem., 80,
1238-1243.
19.Qin, L., Hiser, C., Mulichak, A., Garavito, R.
M., and Ferguson-Miller, S. (2006) Proc. Natl. Acad. Sci. USA,
103, 16117-16122.
20.Proshlyakov, D. A., Pressler, M. A., DeMaso, C.,
Leykam, J. F., DeWitt, D. L., and Babcock, G. L. (2000) Science,
2000, 1588-1591.
21.Vygodina, T. V., Capitanio, N., Papa, S., and
Konstantinov, A. A. (1997) FEBS Lett., 412, 405-409.
22.Vygodina, T. V., Pecoraro, C., Mitchell, D.,
Gennis, R., and Konstantinov, A. A. (1998) Biochemistry,
37, 3053-3061.
23.Orii, Y. (1982) J. Biol. Chem.,
257, 9246-9248.
24.Orii, Y. (1982) in Oxygenases and Oxygen
Metabolism (Nozaki, M., et al., eds.) Academic Press, N. Y., pp.
137-149.
25.Miki, T., and Orii, Y. (1986) J. Biochem.,
100, 735-745.
26.Miki, T., and Orii, Y. (1986) J. Biol.
Chem., 261, 3915-3918.
27.Zaslavsky, D., Smirnova, I. A., Adelroth, P.,
Brzezinsky, P., and Gennis, R. B. (1999) Biochemistry,
38, 2307-2311.
28.Wikstrom, M. (1989) Nature, 338,
776-778.
29.Wikstrom, M., and Morgan, J. E. (1992) J.
Biol. Chem., 267, 10266-10273.
30.Bergmayer, H. U., Gawehn, K., and Grassl, M.
(1970) in Methoden der Enzymatischen Analyze (Bergmayer, H. U.,
ed.) Verlag Chemie, Weinheim, pp. 440.
31.Fowler, L. R., Richardson, S. H., and Hatefi, Y.
(1962) Biochim. Biophys. Acta, 64, 170-173.
32.MacLennan, D. H., and Tzagoloff, A. (1965)
Biochim. Biophys. Acta, 96, 166-168.
33.Musatov, A. P., Berka, V., Ksenzenko, M. Y.,
Vygodina, T. V., and Konstantinov, A. A. (1991) Biol. Membr.
(Moscow), 8, 229-234.
34.Metelitza, D., Shibaev, V., Eryomin, A., Melnik,
V., and Zhilina, Z. (1995) Biochemistry (Moscow), 60,
257-268.
35.Kachurin, A. M., Kropachev, E. V., Iogannsen, M.
G., and Petrov, A. S. (1991) Biokhimiya, 56,
1768-1778.
36.Chouchane, S., Lippai, I., and Magliozzo, R. S.
(2000) Biochemistry, 39, 9975-9983.
37.Pandi-Perumal, S. R., Srinivasan, V., Maestroni,
G. J. M., Cardinali, D. P., Poeggeler, B., and Hardeland, R. (2006)
FEBS J., 273, 2813-2838.
38.Allegra, M., Furtmuller, P. G., Regelsberger, G.,
Turco-Liveri, M. L., Tesoriere, L., Peerretti, M., Livrea, M. A., and
Obinger, C. (2001) Biochem. Biophys. Res. Commun., 282,
380-386.
39.Arnhold, J., Furtmuller, P. G., Regelsberger, G.,
and Obinger, C. (2001) Eur. J. Biochem., 268,
5142-5148.
40.Vygodina, T. V., and Konstantinov, A. A. (1987)
FEBS Lett., 219, 387-392.
41.Wikstrom, M. (1981) Proc. Natl. Acad. Sci.
USA, 78, 4051-4054.