REVIEW: Primary Mechanisms of Photoactivation of Molecular Oxygen. History of Development and the Modern Status of Research
A. A. Krasnovsky, Jr.1,2
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninskii pr. 33, 119071 Moscow, Russia2Biological Faculty, Lomonosov Moscow State University, 119899 Moscow, Russia; E-mail: phoal@mail.ru
Received June 1, 2007
This review starts from a brief historical account devoted to the principles of the Bach-Engler peroxidation theory and experiments and ideas which led A. N. Bach to its creation. Then, the discovery of photodynamic action is described, which was shown to result from pigment photosensitized activation of molecular oxygen. The dramatic history of mechanistic studies of oxygen photoactivation is reviewed starting from the Bach-Engler peroxidation theory to the hypothesis of moloxide, discovery of singlet oxygen and free radicals and, then, to modern views on the primary photoactivation processes. The origin of widely used division of photodynamic processes into type I and type II and the relation of these processes to the nature of the primary photochemical reactions of photosensitizers are discussed. New definitions of these reactions are proposed on the basis of the mechanisms of oxygen photoactivation. Photographs of the scientists who greatly contributed to the development of this field of research are presented.
KEY WORDS: peroxidation of organic compounds, photodynamic action, singlet oxygen, free radicals, oxygen dimols, triplet state, oxygen activationDOI: 10.1134/S0006297907100057
Bach-Engler peroxidation theory. A conception stating that molecular oxygen is chemically rather passive and special activation mechanisms are needed to involve it into chemical and biological processes appeared in the scientific literature in the middle of XIX century. The author of this conception was probably Shönbein, the discoverer of ozone [1]. At least, the term “active (excited) oxygen” was widely used in his papers and in works of his followers. The peroxidation theory formulated independently by A. N. Bach and C. Engler in 1897 was an important step in understanding mechanisms of oxygen activation [2-4]. Bach came to this theory from investigation of photosynthesis. Engler's studies dealt with oil and oil products. As this journal is devoted to the 150 birthday of A. N. Bach, it is of interest to describe the development of Bach's ideas and the sense of his conceptions. Initially, Bach was interested in mechanisms of CO2 fixation and oxygen evolution in photosynthesis. He proposed that light caused the reaction between CO2 and H2O whose primary products were a peroxide-type compound “percarbonic acid” and formaldehyde. Formaldehyde was then condensed into carbohydrates, and percarbonic acid was decomposed with formation of hydrogen peroxide, the cleavage of which led to O2 evolution [5]. To prove this idea, Bach started to study hydrogen peroxide formation in plants. At first, he used methods of peroxide detection which were previously described in the scientific literature. As a result, he concluded that these methods did not work in plant materials because the chemicals, which were required for detection of hydrogen peroxide, interacted with metabolites of plant cells. Therefore, he developed a new method of hydrogen peroxide detection based on the use of potassium dichromate, aniline, and oxalic acid. In the presence of hydrogen peroxide, these compounds formed a relatively stable brightly colored violet product. Using this method, Bach concluded that plants actively accumulated hydrogen peroxide upon illumination [6]. This effect was finally proved by Mehler, who came to a similar conclusion almost half a century later [7]. This observation stimulated Bach to think about roles of peroxides in plant and animal cells. As a result, he claimed the following: “Organic food products--carbohydrates, lipids, and proteins--which are consumed by a living organism are fully oxidized in it by oxygen during a relatively short time. However, these organic compounds are almost indifferent to free or passive oxygen... It is clear that ... an organism ... must have a mechanism that causes activation of oxygen, which comes from the atmosphere... Activation of oxygen might occur due to intermediate formation of peroxides, which always appear during slow oxidation processes, no matter what is the nature of the oxidizing compound... A term “peroxide” I apply to those oxygen-containing compounds whose function is similar to that of hydrogen peroxide and whose molecules contain at least one -O-O- group. The transformation from passive to active oxygen can only occur owing to decomposition of the oxygen molecule (O=O). It is obvious that destruction of one of these bonds and transformation of O=O into -O-O- requires less energy than the destruction of two bonds” [2]. The development of this idea allowed Bach to propose that primary reactions of oxygen with readily oxidized organic compounds lead to accumulation of peroxides, which play a role of active oxygen and oxidize molecules, which are more chemically stable:

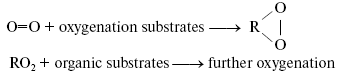
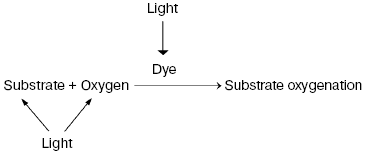
Discovery of photodynamic action. Three years after the first papers by Bach and Engler devoted to the principles of peroxidation of organic compounds, Oscar Raab and Hermann von Tappeiner in the Munich Pharmacological Institute discovered an important phenomenon, which Tappeiner later named photodynamic action [8, 9]. Using a microscope illuminated by sunlight, they noticed that strong light killed cells stained with fluorescing dyes. The action spectrum of cell killing corresponded to the absorption spectra of the dyes. It was shown soon that photodynamic action occurred due to dye-photosensitized photooxygenation of cell components, which was accompanied by peroxide accumulation [10, 11]. Hence, it was demonstrated that photoexcited dye molecules can initiate oxygenation of biological substrates. It was shown later in numerous papers of many research groups that in natural conditions, photodynamic action is a reason of many destructive, signaling, and protective processes in living cells, tissues, and whole ecosystems. Moreover, photodynamic action is used for photo- and laser medicine [12-17]. At present, the term “photodynamic action” is mainly applied to the processes where dyes are photosensitizers, i.e. they trigger the reaction cascade, which leads to oxygenation of organic substrates, but they are not destroyed themselves. This term is often applied also to the photooxygenation reactions, which occur due to photoexcitation of substrates or oxygen molecules (Fig. 1).
Moloxide hypothesis. Bach's and Engler's ideas were used for the first explanation of the primary mechanisms of photodynamic action. In 1904, Straub proposed that the oxygenation of biological substrates is determined by unstable peroxide, later named moloxide, which is formed upon illumination of dyes [11]. In 1867, formation of such peroxide was observed by Frizsche upon photochemical oxygenation of tetracene by air oxygen [18].Fig. 1. Definition of the photodynamic action. The classic definition claims that the term “photodynamic action” is equivalent to the term “dye-photosensitized oxygenation of organic matter”. However, the processes where the role of photosensitizer belongs to oxygen or substrate molecules have similar mechanisms.
![]()
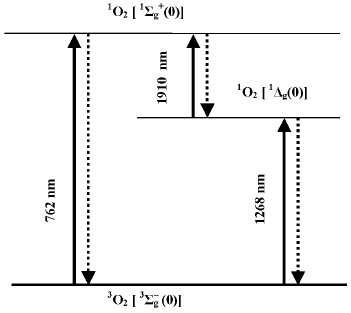
Discovery of singlet oxygen and free radicals. The trend of discussion about the mechanisms of photodynamic action was strongly changed after 1928 when Mulliken applied to oxygen the molecular orbit theory [21, 22]. He concluded that oxygen molecules are triplet in the ground state. This explained paramagnetism of gaseous oxygen, which was discovered by Faraday in 1848. Mulliken also claimed that oxygen molecules have two relatively low-lying singlet levels. Electronic transition from the ground to one of these singlet levels corresponded to the dark red Fraunhofer line (762 nm) in the spectrum of solar radiation, which was found by Wollaston and Fraunhofer in the beginning of the XIX century [23, 24]. As shown by Kirchhoff in 1862, this line belongs to the absorption spectrum of oxygen in the Earth's atmosphere [25]. Mulliken proposed that the second singlet level should have lower energy and predicted the existence of one more oxygen absorption band at about 1500 nm, which was not known at that time. In 1933-1934, Mulliken's suggestion was experimentally confirmed. A new band was observed at ~1270 nm in the absorption spectra of the Earth's atmosphere and liquid oxygen [26, 27]. Analysis of the absorption spectra of liquid oxygen revealed also the absorption bands of oxygen dimols, (O2)2 [26]. These discoveries were very important for investigation of molecular oxygen. At the same time, it was a triumph of the molecular orbit theory.

According to the modern terminology, the ground state of molecular oxygen is denoted by spectroscopic symbols 3Sigmag-, and the lowest singlet states by symbols 1Sigmag+ and 1Deltag (Fig. 2). It is noteworthy that in the gas phase, the intensities of the oxygen absorption bands corresponding to the triplet-singlet transitions are very weak because these transitions are highly forbidden by spin, symmetry, and angular orbital moment [28, 29]. Kasha noted that the transition of the oxygen molecule from the ground to the singlet 1Deltag state is probably most forbidden in nature [29]. This causes extraordinary metastability of this state, whose lifetime is rather long in chemical systems.
In 1931, only three years after the first papers by Mulliken, Kautsky proposed that singlet molecules of oxygen (1O2), which appeared owing to energy transfer from excited photosensitizer molecules (Sens*) to O2, could initiate photodynamic reactions:Fig. 2. Scheme of electronic transitions between the ground and the lowest singlet states of molecular oxygen. Numbers in brackets denote the vibrational transitions. The wavelengths indicate the main maxima of the absorption and luminescence spectra in the gas phase.
Sens* + O2 → Sens + O2e.
To prove this idea, Kautsky performed a well-known experiment, during which he observed photosensitized oxygenation of substrate molecules when they were spatially separated from photosensitizer molecules because the photosensitizers and substrates were adsorbed on different silica gel grains. This experiment showed that photooxygenation in this heterogeneous system was mediated by a gaseous particle [30-32]. It is of interest that about 30 years before Kautsky's papers Raab already suggested in his dissertation that the primary intermediate could be gas [8].
Kautsky noticed also that oxygen quenched fluorescence and delayed fluorescence of dyes absorbed by silica gel. Fluorescence quenching was relatively low efficiency. At the same time, the delayed fluorescence was quenched by very low oxygen concentrations. Kautsky proposed that singlet oxygen can be generated by both fluorescent and metastable states responsible for delayed fluorescence of dyes but the 1O2 generation by the dye metastable states were much more efficient. Kautsky noted also that the latter assumption was consistent with Gaffron's experiments, which showed that the quantum yields of photodynamic reactions practically did not change when the oxygen concentration is changed in a wide range. Gaffron also reported that bacteriochlorophyll is an efficient photosensitizer of thiourea oxygenation though the long wavelength absorption maximum of bacteriochlorophyll corresponded to smaller energy than the 1Sigmag+ level of oxygen. This suggested that the lower 1Deltag level of oxygen was responsible for the photoreaction [32]. However, these conceptions were not recognized by researchers at that time. They were too innovative and strongly differed from the views of contemporaries. Kautsky passed away not knowing that his ideas were fully confirmed in the middle 1960's, 33 years after his first publication in this field [33].

On the other hand, in the 1930's many important discoveries were made in the field of chemistry of free radicals. It was proved that free radicals actively participated in many processes including the polymerization reactions, which were used for synthesis of rubber and different plastic materials [34]. In particular, Backström's papers were published (1934), which dealt with benzophenone-photosensitized oxygenation of alcohols and aldehydes. Backström proposed that excited photosensitizer molecules directly reacted with the substrate molecules without involvement of oxygen, and the primary products of this reaction were free radicals of photosensitizer and substrate molecules. Further reaction of these free radicals with oxygen was suggested to be responsible for development of oxygenation process [35]. The photoreactions of this type Schenck and Terenin called reactions of primary photodehydrogenation.
Simultaneously, Weis [36] and then, Frank [37] proposed the free-radical explanation of Kautsky's experiment. These authors suggested that the photooxygenation occurred due to reactivity of the ·O2- or HO2· radicals formed as a result of oxidation of excited photosensitizer molecules by oxygen:
Sens* + O2 → *Sens+ + *O2- (HO2*).
These ideas opened up one more direction in investigation of oxygen photoactivation and photodynamic action. However, these assumptions seemed hypothetic because at that time reliable methods for detection of short-lived free radicals had not been developed. Such methods appeared later after the discovery by Zavoysky in 1945 of electron paramagnetic resonance [38] and invention of flash-photolysis (Norris and Porter, 1949) [39, 40]. However, these methods became widely available only in the 1960s.
Discovery of the pigment triplet state. In 1933-1935, Jablonski claimed that two excited states of one dye molecule exist: one is the short-lived fluorescence state and the second, is the metastable long-lived phosphorescence state [41]. The famous Jablonski diagram, which is the basis for photochemistry and spectroscopy, was initially introduced as a formal generalization of the experimental data on luminescence of organic chromophores (Fig. 3). The nature of the excited states indicated in this diagram was unknown.
Fig. 3. Simplified Jablonski diagram and the scheme of the mechanism of photosensitized oxygen phosphorescence.
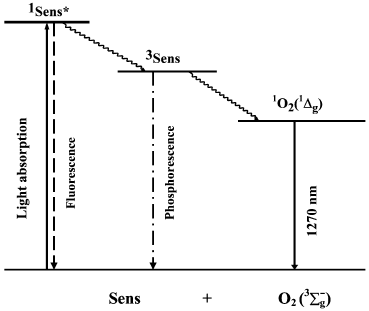
Terenin (in 1943) and Lewis and Kasha (in 1944) formulated a concept, which is presently universally adopted. The fluorescence state is singlet, i.e. its population does not require inversion of the electron spin in dye molecules. The metastable state is triplet, i.e. it has two unpaired electrons. Therefore, deactivation of the triplet state forbidden by spin selection rules proceeds much slower [42-44]. In addition, Terenin indicated that the spin conservation rule (the Wigner rule) allows two mechanisms of 1O2 generation by photoexcited dye molecules:

1Sens*(^v) + 3O2(^^) → 3Sens (^^) + 1O2(^v),
3Sens (^^) + 3O2(vv) → 1Sens (^v) + 1O2(^v),
where 1Sens, 1Sens*, and 3Sens are molecules of pigments-photosensitizers in the ground and excited singlet and triplet states. The first mechanism is possible for relatively small group of photosensitizers whose energy gaps between 1Sens* and 3Sens is more than energies of the singlet states of oxygen (Fig. 4). The second mechanism is possible for much the more abundant group of photosensitizers whose triplet levels are higher than the singlet levels of oxygen. It is of interest that the first mechanism suggests that two molecules of singlet oxygen can be generated by one Sens molecule (Fig. 4) [42, 43]. Terenin also stressed that the triplet states of dyes should be much more efficient than the singlet states in promotion of photodynamic oxygenation reactions because the lifetime of 3Sens is much longer than the lifetimes of 1Sens*.
From moloxide to singlet oxygen. Terenin's mechanisms provided comprehensive explanation of Kautsky's data obtained in the heterogeneous systems. However, Terenin suggested in his first papers that biradical complex of triplet dye molecules with oxygen (moloxide) should be more reactive and play a more important role in photodynamic reactions in homogeneous solutions [42, 43, 45]. The moloxide hypothesis was supported by all recognized photochemists at that time, though their views on the moloxide structure were different [20, 33, 45-48]. The moloxide idea seemed attractive because cyclic peroxides were known to accumulate upon photosensitized oxygenation of many compounds as for instance, aromatic hydrocarbons, certain heterocyclic compounds, and linear and cyclic alkenes [19, 33, 46, 47, 49-52]. In addition, the kinetic features of these photoreactions were consistent with the intermediate moloxide formation [49, 52]. However, different information was also accumulated. For example, Bowen has shown that identical kinetic equations describe mechanisms based on the involvement of moloxide and singlet oxygen [53]. Schenk has shown that the kinetic parameters of moloxide do not depend on dyes [50].Fig. 4. Energy diagram showing mechanisms of generation of singlet oxygen by the singlet and the triplet states of dye molecules (Terenin's mechanisms). Horizontal arrows show that deactivation of the excited states of the dye are accompanied by transition of the oxygen molecule into the singlet state.
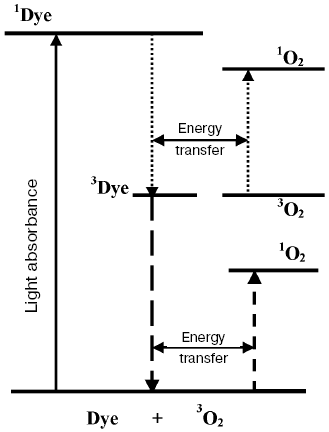
Luminescence measurements allowed further progress of this discussion. In 1947, Kaplan discovered that deactivation of the 1Sigmag+ state of monomeric oxygen molecules in the gas phase is accompanied by luminescence whose spectral maximum corresponded to the oxygen absorption band at 762 nm [54]. Then, photosensitized luminescence was found in gaseous oxygen corresponding to deactivation of the 1Deltag state and transition of oxygen molecules from the 1Sigmag+ to 1Deltag state [55-57] (Fig. 2).
In 1962-1965, Seliger's, Stauff's, Kasha's, and Ogryzlo's groups found that luminescence of monomeric and dimeric singlet oxygen molecules appeared under microwave electrodeless discharge in the stream of gaseous oxygen or in bubbles of oxygen released during the chemical reaction of Cl2 or ClO- with H2O2. Kasha's and Ogryzlo's groups were especially active in investigation of oxygen luminescence. Detailed analysis of their data has been presented in reviews [58, 59].
In 1964, Foote and Wexler added the substrates of the above “oxygen transfer” photoreactions to the chemiluminescence mixture of ClO- with H2O2 and found that oxygenation products formed in this mixture were identical to those formed in photochemical reactions [60, 61]. In a parallel paper submitted 25 days later, Corey and Taylor obtained a similar result using 1O2 generation by microwave discharge [62]. These experiments allowed Foote to claim that moloxide, which was thought to be involved in the solution-phase photodynamic reactions, was in fact the singlet 1Deltag state of oxygen [60, 61]. In the same year, Gollnick and Schenck repeated Foote's experiments using alpha-pinene as an oxidation substrate. They supported Foote's conclusions but also indicated that free radicals could be involved in olefin photooxygenation [52]. It was shown later that the rate constants of 1O2 reactions with certain substrates of the “oxygen transfer” photoreactions in dark chemical systems coincided with the reactions rate constants for “moloxide” in photochemical systems [63]. Thus, the data indicated that the 1Deltag oxygen state was highly reactive [60-62]. At the same time, these papers showed that the substrates of the “oxygen transfer photoreactions” described mostly by Duffraise's and Schenck's groups can be regarded as chemical traps of singlet oxygen, therefore oxygenation of these traps can be used for detection and investigation of singlet oxygen.

Nevertheless, the conclusions of the first of Foote's papers did not exclude certain doubts because they were based on analysis of chemical systems, which contained strong oxidants ClO-, H2O2, and maybe other active particles, besides 1O2. Evans [64], Matheson and Lee [65], and later other researchers have shown that photooxygenation of singlet oxygen traps can be observed without dyes upon direct excitation of oxygen monomols and dimols by red and infrared light in solutions saturated by oxygen at 130 atm (Fig. 2).
Parallel studies of several groups dealt with physical detection methods of photosensitized singlet oxygen formation. In 1968, Snelling discovered luminescence (1268 nm) of the 1Deltag-state of molecular oxygen photosensitized by benzene vapors in the gas phase and showed that this emission appeared owing to energy transfer from excited benzene molecules to oxygen (Fig. 3). At present, the term “phosphorescence” is most frequently used to define this luminescence, because it accompanies the forbidden intersystem transition from the singlet to the triplet state of oxygen molecules. Snelling noticed that at low oxygen pressure 1O2 responsible for this phosphorescence was formed due to energy transfer from triplet benzene molecules to O2. When oxygen pressure increased, the 1O2 phosphorescence, which resulted from energy transfer from the benzene singlet state to O2, was also observed [66]. In 1969, photosensitized ESR signal of singlet oxygen was detected in gas phase experiments [67, 68]. In 1971, photosensitized phosphorescence from the 1Sigmag+ state of oxygen (762 nm) was found in the gas phase [69, 70].
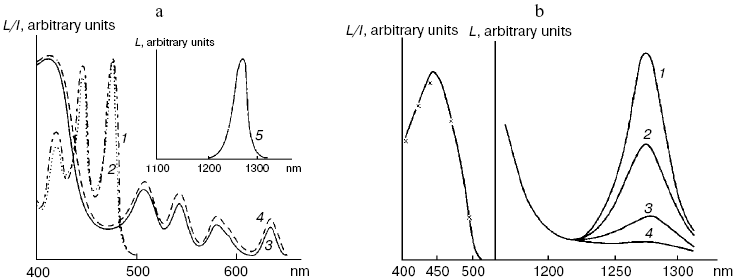
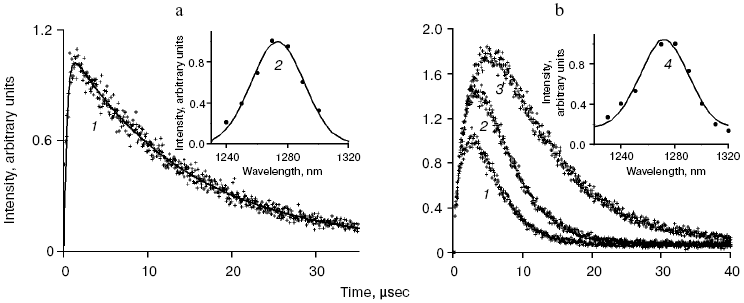
Application of the above physical methods to air-saturated solutions was not successful for some time. The ESR method is still not used. In 1974, Matheson et al. observed luminescence of singlet oxygen dimols (633 nm) under direct laser excitation (1064 nm) of oxygen in the gas phase and 1,1,2-trichlorotrifluoroethane (Freon 113) at about 130 atm oxygen pressure [71]. The dimol light emission was detected if the concentration of oxygen was >=3.9 M that exceeded by three orders the concentration of oxygen in solutions saturated with air at normal atmospheric pressure. In 1976 using sensitive home-made phosphorimeters, the author of the present review discovered dye-photosensitized phosphorescence of 1O2 (1Deltag, 1270 nm) in air-saturated solutions [72]. It was also the first experimental observation of phosphorescence of dissolved 1O2. In this and subsequent papers of 1977-1979, we reported the first application of phosphorescence measurements to analysis of generation and quenching of 1O2 by many biologically important compounds including porphyrins, chlorophylls, bacteriochlorophylls, retinals, flavins, anthracene derivatives, carotenoids, and others [72-81]. The results of the first measurement are shown in Fig. 5. Phosphorescence was observed in air-saturated solvents due to energy transfer from triplet molecules of photosensitizers to oxygen as shown in Fig. 3. In 1979, reliable phosphorescence measurements were performed also by other researchers [82-84]. In papers [82, 83], the phosphorescence kinetic traces were recorded after pulsed laser excitation. Thus, it was finally proved that 1O2 is formed upon photoexcitation of photosensitizer solutions. Since that time, the phosphorescence method of 1O2 detection has been widely applied for photochemical and photobiological studies. Measurements with pulsed laser excitation are especially informative because they combine kinetic and spectral analysis of 1O2 phosphorescence. Figure 6 shows the kinetic traces and spectra of photosensitized 1O2 phosphorescence in ethanol and aqueous solutions of porphyrins, measured in our laboratory using a time-resolved photon counting set-up with pulsed laser excitation [85]. A series of the reviews of this author were devoted to the development of the phosphorescence method and to results of application of this method to the problems of photochemistry, photobiology, and photomedicine [86-92].
Fig. 5. First measurements of photosensitized phosphorescence of singlet oxygen (a) in organic solvent (CCl4) and (b) aqueous solutions. a) Absorption spectra of tetracene and protoporphyrin IX (dimethyl ester) (1, 3) in air-saturated CCl4. Excitation (2, 4) and emission (5) spectra of singlet oxygen phosphorescence in the same solutions [72]. b) Right part: phosphorescence spectra of singlet oxygen in solutions of riboflavin in D2O (1), in mixtures of D2O and H2O containing 5% (2) and 50% H2O (3), and in H2O (4); left part: absorption spectrum of riboflavin in D2O (solid line) and excitation spectrum of singlet oxygen phosphorescence in the same solutions (×) [80].
Mechanisms of energy transfer from excited dye molecules to oxygen. In 1952, Terenin and Ermolaev discovered triplet-triplet energy transfer between dye molecules [93]. According to Dexter, this process results from exchange energy transfer [94]. Singlet oxygen formation was proposed to be due to similar energy transfer between triplet dye and oxygen molecules where oxygen molecules are energy acceptors [95, 96]. This concept is presently generally adopted, though the analyses of quenching of dye fluorescence and the dye triplet states by oxygen show that in many cases the quenching rate constants depend upon the redox potentials of the dye excited states. Hence, it is possible that exciplexes (Sens...O2)* with charge transfer between dye and oxygen are involved in 1O2 generation. Thus, the initial moloxide hypothesis is now transformed into the hypothesis of the exciplex intermediate, whose formation is followed by 1O2 generation [97-99]. It should be noted here that according to Schenck, in 1947, Kautsky already suggested that intermediate formation of the dye-oxygen exciplexes preceded 1O2 formation in photosensitizer solutions [33, 52].Fig. 6. a) Kinetic curve (1) and spectrum (2) of 1O2 phosphorescence in air-saturated ethanolic solution of tetra(p-sulfophenyl)porphyrin (15 µM) after a laser pulse. The kinetic curve is obtained as a result of averaging the signal from 2.4·106 laser pulses. b) Kinetic curves (1-3) and spectrum (4) of 1O2 phosphorescence in air-saturated solutions of tetra(p-sulfophenyl)porphyrin (15 µM, pH 5.8) in water (1, 4) and aqueous solutions of Triton X-100 containing 1% and 80% detergent (2, 3) after laser pulses. The curves were obtained as a result of averaging the signal from ~107 laser pulses. Dots show experimental data, the solid lines are computer approximations. The spectra correspond to the overall phosphorescence intensity in the interval 1-45 µsec after the laser flash [85, 92].
Application of modern research methods showed that Terenin's mechanisms of singlet oxygen generation are valid in dye solutions. It has been proved that both singlet and triplet states of photosensitizer molecules generate 1O2 in the solution-phase. As mentioned above, the energy transfer to oxygen from 1Sens* is possible if the energy gap between the singlet and triplet states of a photosensitizer is more than energy of one of the singlet levels of oxygen (Fig. 4). Many compounds have been found which allow this type of energy transfer. They are aromatic hydrocarbons: tetracene, rubrene, pyrene, chrysene, anthracene derivatives, furan derivatives, and others. This photosensitization mechanism is probably possible for the following biologically important photosensitizers: furocumarins, anthraquinones, retinals, and certain carotenoids. Detailed discussion of experimental information on this subject is presented in recent reviews [98, 99]. However, it should be noted that because of the short lifetimes of the excited singlet states of dyes (not more than 10-20 nsec), high 1O2 quantum yield is possible only at high oxygen pressure. In solutions and biological systems at normal pressure of oxygen, this mechanism of 1O2 generation should be of very low efficiency.
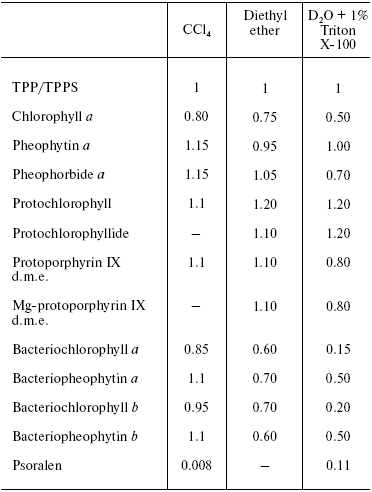
On the contrary, the efficiency of 1O2 generation by the pigment triplet states is known to be very high. In solutions, the 1O2 quantum yields are close to the quantum yields of the photosensitizer triplet states at the oxygen concentrations >=10-5 M. However, two cases are possible: one, if energy of the triplet state (Et) is higher than the 1Sigmag+ level of O2, and the second, if Et is less than energy of the 1Sigmag+ level but higher than energy of the 1Deltag level. In the first case, the 1Sigmag+ state is mostly populated. Population of this state is accompanied by luminescence at 765 and 1930 nm [100-104]. In the second case, only the 1Deltag level is populated. The lifetime of the 1Sigmag+ state in solutions is very short because of efficient 1O2 quenching by solvents that causes rapid energy dissipation and population of the 1Deltag state. Table 1 indicates the lifetimes of the 1Sigmag+ state (tauSigma) in different solvents calculated by this author in reference [86] using the data of Ogryzlo's group dealing with quenching of the 1Sigmag+ state by vapors of different solvents in the gas phase [105]. Calculations showed that tauSigma = 1 nsec in organic solvents whose molecules contain hydrogen atoms, tauSigma ~_75 psec in deuterium oxide and even less in H2O and alcohols. However, in CCl4, the calculated lifetime was about 130 nsec. This value resembles the experimentally measured tauSigma (105-130 nsec) obtained from decays of photosensitized phosphorescence of the 1Sigmag+ state after laser shots [100-104]. Because of the low lifetime and peculiarities of the electronic structure, the 1Sigmag+ state does not show any chemical activity; therefore the role of this state in chemical activation of oxygen is limited by spontaneous generation of the reactive 1Deltag state [86, 100-105].
Table 1. Estimate of the lifetime of the
1Sigmag+ state of singlet
oxygen in different solvents [86]
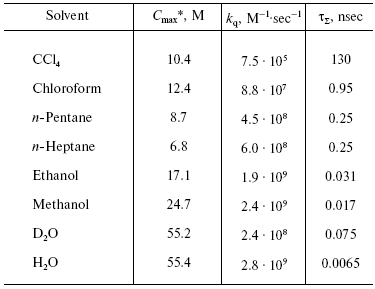
*Cmax is the molar concentrations of
solvents.
The lifetime and reactivity of the 1Deltag state are studied in detail. It is known that owing to physical quenching of the 1Deltag state by solvent molecules, the lifetime of this state varies from 3.1 µsec in water to several tens of milliseconds in CCl4 and other solvents whose molecules do not have hydrogen atoms. In the structures of living cells, the lifetime is decreased to 10-200 nsec due to additional quenching of singlet oxygen by components of biological structures. The major targets are amino acids of proteins (tryptophan, histidine, cysteine, methionine, and phenylalanine), nucleosides (guanosine and thiouridine), unsaturated fatty acids, and other compounds [86, 90, 91, 98].
Thus, energy transfer from the pigment triplet states to oxygen causes oxygen activation due to population of the reactive 1Deltag singlet state:
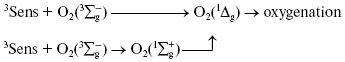
Table 2. Quantum yields of
1O2 (1Deltag)
generation by certain biologically important pigments in solutions
saturated with air at atmospheric pressure
Note: TPP, tetraphenylporphyrin; TPPS, water-soluble analog of TPP,
tetra(p-sulfophenyl)porphyrin. The quantum yields of singlet
oxygen generation by these porphyrins are about 0.7. The excitation
wavelength was 337 nm [87, 89].
Dimols (1O2)2. In solutions of many pigments, photosensitized 1O2 generation is accompanied by luminescence of dimols, (1O2)2, with the main maximum at 703 nm and weaker bands at 635 and 775-780 nm (Fig. 7). Photosensitized dimol luminescence was first observed in our experiments using solutions of protoporphyrin, pheophytins, tetraphenylporphyrin, 2,3,7,8-dibenzopyrene-1,6-quinone (DBPQ), and Pd-tetraphenylporphyrin (Pd-TPP) in CCl4 and C6F6. The best results were obtained in solutions of non-fluorescent pigments DBPQ and Pd-TPP, whose singlet states had much higher energy than dimols [72, 75, 78, 106, 107]. Later, similar dimol light emission was studied also by Chou et al. who used solutions of non-fluorescent compounds phenalenone, 2-acetonaphthone, 1-acetonaphthone, and 1,4-dimethylnaphthalene endoperoxides in CCl4, C6F6, and C6D6 [101, 108]. According to our data, the intensity of dimol luminescence strongly depended on the nature, concentration, and state of photosensitizers in solutions. Most likely, the luminescence, which we detected was emitted by dimol molecules, (1O2)2, which formed contact complexes with pigment molecules. The mechanism of this luminescence is consistent with two kinetic schemes:
Sens + 1O2 → (Sens...O2)* + 1O2 → [(Sens...(O2)2]* → Sens + (O2)2 → hvdim, (1)
1O2 + 1O2 → (1O2)2 + Sens → [(Sens...(O2)2]* → Sens + (O2)2 → hvdim. (2)
According to Scheme (1), collisions of 1O2 molecules with dye molecules lead to formation of a complex (Sens...O2)*. Collision of this complex with the second 1O2 molecule causes formation of the second contact complex, which spontaneously breaks down with formation of dimols (O2)2 and emission of photons corresponding to dimol luminescence. According to our data, dyes increase radiative deactivation of dimols in such complexes. According to Scheme (2), dimols are formed as a result of collisions of two 1O2 molecules. Then, after collision with a dye molecule, dimols are decomposed with light emission. It is noteworthy that in solutions of strongly fluorescent photosensitizers (phthalocyanines, naphthalocyanine, and bacteriopheophytin) whose fluorescent levels had less energy than the singlet oxygen dimols, the dyes accepted energy of two singlet oxygen molecules and emit rather strong delayed fluorescence [106, 107].Fig. 7. Spectrum of photosensitized luminescence of dimols (1O2)2 in air-saturated solutions of 2,3,7,8-dibenzopyrene-1,6-quinone (5 µM) in carbon tetrachloride. Similar spectra were obtained in solutions of this dye in C6F6 and in solutions of Pd-tetraphenylporphyrin (30-100 µM) in CCl4 and C6F6 [106, 107].
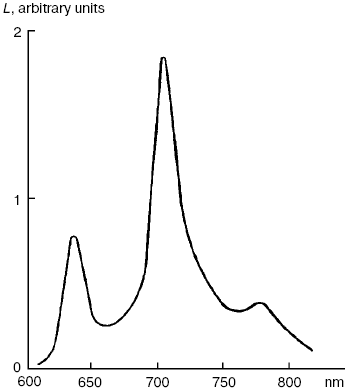
Chou et al. claimed that in their experiments, where much higher fluence rates of excitation radiation were used, photosensitized dimol luminescence accompanied spontaneous deactivation of dimols without collisions with dye molecules [101, 108]. At present, it is difficult to conclude what scheme is correct. This author thinks that the first scheme is more probable. It is not excluded that binding energy in complexes (Sens...O2)* or [(Sens...(1O2)2]* is responsible for the fact that the vibrational (0-1) emission band of dimols at 703 nm is 3-4 times stronger than the (0-0) emission band at 635 nm. It is known that in monomols 1O2, the (0-0) emission band is 50 times stronger than the (0-1) emission band [109, 110]. Using this assumption, one can estimate that the binding energy in the dimol-dye complexes is about 3 kcal/mol. So far, chemical activity of the dimols or their complexes with dyes has not been revealed. Further studies of dimols are needed for better understanding of their nature and mechanisms of their luminescence in solutions.
Activation by supershort high energy laser pulses. The use of femtosecond, picosecond, and powerful nanosecond laser pulses allowed observation of photosensitized singlet oxygen generation upon excitation of photosensitizers in that spectral region where these photosensitizers did not have absorption bands, for instance, at much longer wavelengths than the main absorption maxima of the pigments. It is thought that excitation of pigment molecules is due to summation of energy of two photons because when short powerful laser pulses are used, some pigment molecules are under the influence of the electromagnetic fields of two photons during the time (several femtoseconds) needed for molecular transition from the ground to excited singlet state. In this case, energy of two photons is summed up and excitation of pigment molecules occurs. Phenomenologically, this process resembles the more trivial way of two-photonic molecular excitation as a result of light absorption by weakly pronounced pigment absorption bands. It is suggested that two-photon excitation opens up new opportunities for photomedicine, because one can apply long wavelength dark red or infrared radiation for excitation of photosensitizers [110-114]. The tri-photonic absorption of femtosecond laser pulses has also been reported [114]. In this case, the excitation wavelength can be additionally shifted to longer wavelength. The specificity of this method of oxygen activation consists solely in the mechanism of pigment excitation. Further development of the process is determined by population of the pigment triplet states and energy transfer to oxygen as described above.
Activation by direct oxygen excitation. As mentioned above, Evans and Matheson and Lee have shown that oxygenation of chemical traps of singlet oxygen can be observed upon direct photoexcitation of monomeric and dimeric oxygen molecules dissolved in Freon at high (130 atm) oxygen pressure [64, 65]. Later, similar effects were observed at high oxygen pressure also in other solvents and, in particular, in deuterium oxide [115, 116]. Though these experiments were performed under conditions which were far from normal for biological systems, Ambartzumian suggested that the action spectra of laser radiation reported in Karu's papers [17] indicated the involvement of direct laser oxygen excitation in certain biological effects of laser radiation [117]. This hypothesis, which was later termed “light oxygen effect”, was later discussed by Zakharov and Ivanov [118]. In recent papers, we investigated oxygenation of the 1O2 traps in solutions at normal atmospheric pressure under the action of the laser radiation whose wavelength corresponded to the electronic transitions in oxygen molecules [119-123]. The use was made of the relatively low intensity lasers, which generated radiation at 720-800 nm (500-700 mW) and 1200-1290 nm (30-150 mW). It was shown that these lasers caused oxygenation of chemical traps, 1,3-diphenylisobenzofuran (DPIBF) and tetracene, dissolved in organic solvents or water saturated with air at normal pressure. The rates of these reactions linearly depended on laser energy and increased 5-fold when the solutions were saturated with pure oxygen. Singlet oxygen quenchers strongly inhibited oxygenation of the traps. The maxima of the action spectra of the photooxygenation reactions coincided with the maxima of oxygen absorption bands and corresponded to 1273 nm [120-122] and 765 nm [123] (Fig. 8). These data provided unambiguous evidence that the oxygenation of the traps occurred owing to the activity of the singlet 1Deltag state of oxygen formed as a result of the direct excitation of oxygen molecules by laser radiation:
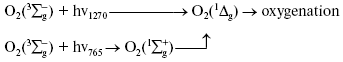
In other words, activation of oxygen by its direct photoexcitation was shown to have appreciable rate in natural conditions.Fig. 8. Action spectra of tetracene oxygenation in air-saturated CCl4 upon direct excitation of oxygen molecules by laser radiation. V is the rate of tetracene photooxygenation, n is the number of photons of laser radiation [121, 124].
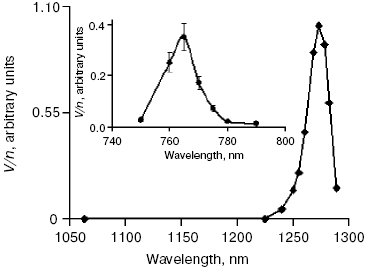
The results of the above measurements were used for estimation of the optical densities (A1270 and A765) and molar absorption coefficients (epsilon1270 and epsilon765) of dissolved oxygen. It was shown that in CCl4 saturated with air at atmospheric pressure A1270 = 7.2·10-6 and epsilon1270 = 0.003 M-1·cm-1; A765 and epsilon765 were estimated to be about 3.5 times less [120, 123]. Dependence of the epsilon1270 values on solvents was investigated. It was found that relative values of epsilon1270 in organic solvents correlated with relative values of the rate constants of 1O2 (1Deltag) radiative deactivation (kr) [121, 122]. In aqueous solutions (H2O and D2O), we used detergents 0.1 M sodium dodecyl sulfate or 0.2% Cremafore 6E for solubilization of DPIBF. It was found that in both cases, the rate of photooxygenation of this trap was 4-10 times more than one can expect from the kr value in water. We explained this effect as a result of heterogeneity of detergent solutions, because kr and the solubility of oxygen is higher in the micellar phase than in water. Therefore, though the volume of the micellar phase is small, contribution of this phase to total generation of singlet oxygen is comparable with the contribution of the water phase [121, 122]. If this assumption is correct, at certain micelle concentrations the micellar phase could generate singlet oxygen with higher intensity than the water phase.
This experiment suggests that hydrophobic structures of living cells are more sensitive to destructive action of IR laser. However, it should be noted that the absorbance and molar absorption coefficient of oxygen are very low. The value of epsilon1270 is about eight orders less than the molar absorption coefficient of the Soret band of porphyrins. We compared the DPIBF photooxygenation rates upon direct and protoporphyrin-sensitized excitation of oxygen dissolved in CCl4 [121]. The absorbance of porphyrin was 0.065 at the excitation wavelength (565 nm). It was shown that the quantum efficiency (the ratio of the photoreaction rate to the intensity of exciting light in photons per second) was 6600 times more in the photosensitized reaction than upon direct oxygen excitation. The maximum efficiency of porphyrin-sensitized photoreaction corresponding to absorption of 100% of the light is 7 times higher. Hence, photosensitized oxygenation was 5 orders more efficient than the photoreaction caused by direct oxygen excitation [121]. It is known that the concentration of free oxygen in living cells should be decreased by respiration by 2-3 orders as compared to the concentration of oxygen in CCl4. Therefore, the efficiency of direct photoactivation of free oxygen in living cells should be less than in CCl4 by 2-3 orders [119].
Thus, it is difficult to expect that direct excitation of free oxygen dissolved in cell structures causes appreciable destructive effects. More likely, IR radiation influences enzyme-bound oxygen molecules whose concentration is much higher. Singlet oxygen formed in this process might trigger expression of antistress genes and apoptosis [124-127] or cause structural changes of biomembranes, which strongly influence their activities [128]. At any rate, our data show that direct excitation of oxygen molecules by IR light is a real, though low efficiency process whose existence should be accounted for in photobiology research.
Free-radical activation mechanisms. In parallel with the above discussion about moloxide and singlet oxygen, oxygen activation mechanisms based on the primary dehydrogenation of substrates by excited dye molecules were also studied. According to Schenck [51, 52], this scheme was first suggested by Backström for the mechanism of the benzophenone-photosensitized oxygenation of alcohols and aldehydes [35].
Similar ideas were discussed in parallel papers of other groups who studied photoreduction of fluorescein derivatives and methylene blue in aerobic solutions in the presence of polyatomic alcohols, organic acids, or phenylhydrazine. Though detailed mechanisms of these reactions could not be proved at that time, the authors suggested that the primary dehydrogenation of the excited dye molecules and further oxidation of photoreduced dyes by oxygen occurred (cited according to reviews [45, 129]).
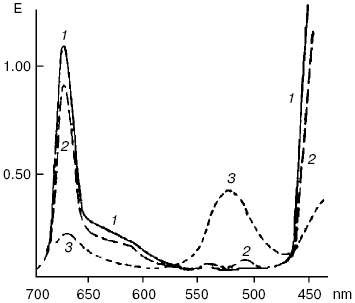
An ability of biologically important pigments--porphyrins, chlorophylls, and bacteriochlorophylls--to the reactions of photodehydrogenation was first experimentally demonstrated by my father, Academician A. A. Krasnovsky, in 1948-1952 [130-133]. The oxidation substrates were ascorbic acid, phenyl hydrazine, cysteine, polyphenols, certain organic acids, reduced NAD (nicotinamide adenine dinucleotide), N-benzylnicotinamide, and cytochromes c. The best results were obtained in solutions of pigments in aqueous pyridine containing up to 50% water and also in ethanol in the presence of organic bases (pyridine, nicotine, ammonium, and others). Illumination of chlorophyll in anaerobic conditions in the presence of ascorbic acid led to the decrease of the main chlorophyll absorption bands and appearance of the pink form with the absorption maximum at 525 nm (Fig. 9). This form was slowly decomposed with restoration of the initial pigment. Restoration was strongly accelerated by oxidants: oxygen, quinones, and others. This was the first fully reversible photoreaction of the main pigment of photosynthesis. It was named the Krasnovsky reaction and attracted attention of many researchers of photosynthesis [131-134]. On the other hand, this was a reversible photoreaction of the pigment which belonged to the most important class of photodynamic dyes. It was shown already in the first studies of this reaction that chlorophyll photoreduction occurred due to activity of the chlorophyll triplet state, and the stable pink form was a secondary photoproduct. The primary short-lived radicals of pigments were later revealed by the methods of electrochemistry, radical polymerization, ESR, and flash-photolysis [131-135].

According to the modern conceptions, the primary step of these reactions is electron (hydrogen) transfer from the substrate molecule (RH) to photoexcited (usually triplet) molecules of photosensitizers. Oxygen oxidizes photoreduced photosensitizer molecules. As a result, dye molecules are restored and superoxide anion-radical is formed:Fig. 9. Reversible photochemical reduction of chlorophyll a (Chl) in pyridine by ascorbic acid (Krasnovsky reaction): 1) absorption spectrum of initial chlorophyll; 2) absorption spectrum of Chl after reverse reaction of reduced Chl with oxygen or other oxidants; 3) approximate spectrum of a labile photoproduct 6 min after the end of illumination (precision of the E values was ± 10%) [130].
Sens* + RH → -Sens* + +R*,
-Sens* + O2 → Sens + -O*2.
Hence, the primary photoreaction causes formation of two free radicals +R* and -O2* [131-135]. These primary radicals initiate a further dark oxygenation process, which strongly depends on the chemical structure of the oxidation substrates. Mechanisms of dark oxygenation processes are a special subject of research. Many year efforts of many groups were required for their analysis. The Bach-Engler peroxide theory [2-4] and then, discovery of free radicals and chain and branching chemical reactions [34, 136] were the initial basis for this research.
According to the modern views, which were established in the 1960s, oxidation of hydrocarbons, alcohols, organic esters, acids, and lipids leads to formation of the primary radicals +R* or their deprotonated forms, which attach oxygen and form reactive peroxy radicals RO2* (see references in [137-141]). The peroxy radicals give rise to formation of peroxides (ROOH, ROOR) and stable degradation products containing keto- and hydroxyl groups. Superoxide radical and its protonated analog HO2* are also oxidizing agents. In addition, they form hydrogen peroxide upon dismutation. Reactions of peroxides with free radicals give rise to a potent oxidant, hydroxyl radical (the Fenton and Haber-Weiss reactions). This process is accelerated by ferric ions:
![]()
The reactions of excited dye molecules with oxygen can also initiate free radical formation (the Weiss-Frank mechanism [36, 37]):
3Sens + O2 → ·Sens+ +
-O2*,
*
In this case, the initial dye molecule can be restored in reaction of the dye cation-radical or its deprotonated form with RH. As a result of the photochemical process, two free radicals *R+ (*R) and -O2· are formed. As shown above, these radicals resemble those which appear upon primary dye photoreduction. Experiments indicate that the probability of dye oxidation by oxygen is much less than the probability of the energy transfer leading to singlet oxygen formation. It should be noted that peroxy radical can also be produced owing to interaction of oxygen with free-radicals formed due to primary dye photooxidation by quinones or other electron acceptors. More detailed discussion of these processes was presented in reviews [86, 145, 146].
Concluding this section, one should note that free radicals and peroxides are also sources of singlet oxygen. The quantum yield of 1O2 upon disproportionation of peroxy radicals RO2* reaches 12% [139, 140, 142-144, 147, 148]. The 1O2 yield upon thermal or catalytic decomposition of endoperoxides, dioxetanes, and dioxyranes reaches 100% [139, 140]. Singlet oxygen is likely generated by ·O2-, HO2·, and *OH radicals [148, 149]:
2H+ + 2·O2- → 1O2(1Deltag) + H2O2,
*OH + *O2- → 1O2(1Deltag) + OH-.
Decomposition of hydrogen peroxides catalyzed by ions of molybdenum, vanadium, and calcium is known to be accompanied by efficient singlet oxygen production [150-153]. Singlet oxygen is formed in peroxidase-catalyzed reactions of hydrogen peroxides with halide anion or indole-3-acetic acid and in other enzymatic reactions [142, 143].
Classification of photoactivation processes. Abundance and diversity of photodynamic reactions hampered their mechanistic analysis. Therefore, the simplified conception formulated in the 1960s by Schenck and his collaborators that photodynamic reactions are based on two different types of primary processes, type I and type II, became most useful [33, 50-52]. This conception in the form proposed by Foote is now universally adopted [60, 145, 146, 154]. In the photoreactions of the type I, the primary stage is interaction of excited photosensitizer molecules with oxygenation substrates and the primary products are free radicals, which activate oxygen and lead to accumulation of peroxy radicals and peroxides. In the photoreactions of the type II, the primary stage is interaction of excited photosensitizer molecules with oxygen. The major product of this interaction is singlet oxygen, though oxidation of photosensitizers by oxygen also occurs.

Type I: Sens* + X → free-radical intermediates + O2 → oxygenation products of RH
Type II: Sens* + O2 → active products + RH → oxygenation products of RH
Here X denotes a compound which is responsible for the primary oxidation or reduction of excited molecules of photosensitizers.
At present, spontaneous change of the sense of this terminology occurs. The term “type I” is often applied to all photoreactions that involve primary formation of free radicals, even if oxygen does not participate in the formation of final products. The term “type II” is usually applied to photoreactions that are due to intermediate singlet oxygen formation. These views reflect some uncertainty in the classic definitions. It is apparent that the classic terminology makes sense only if one deals with photodynamic reactions and believes that the term “photodynamic reaction” is equivalent to the term “photooxygenation reaction”. As shown above, according to the classic terminology, the type of the photodynamic process is determined by nature of the primary photoreaction. Nature of the primary intermediates (free radicals or singlet oxygen) does not influence the classification. However, the primary intermediates of the Weiss-Frank reaction, which according to the classic definition corresponds to the type II, are free radicals that resemble the primary photoproducts of the type I photoreaction.
On the other hand, singlet oxygen and free radicals cannot be a basis for this classification, because they are formed in both the type I and type II reactions. For example, reactions of 1O2 with organic substrates lead to formation of unstable cyclic peroxides, which are then decomposed and cause formation of free radicals. Free radicals are formed also if oxygen accepts electrons from excited molecules of photosensitizers (the Weiss-Frank mechanism) or 1O2 accepts electrons from oxidizing compounds. In these cases, superoxide and peroxy radicals appear.
Hence, the type II reactions are mostly due to the primary formation of singlet oxygen and also suggest less efficient free radical formation. The type I reactions are mostly due to the primary free radical formation. However, as shown above, they are always accompanied by less efficient appearance of singlet oxygen, which is formed during the secondary processes: recombination of peroxy radicals and peroxide decomposition. Thus, the appearance of 1O2 cannot be considered as an unambiguous indication of the type II contribution.
It is noteworthy that the above classification was proposed at that time when elementary mechanisms of photodynamic reactions were unknown and the role of singlet oxygen was under discussion. At present, it is more natural to classify the primary stages of photodynamic reactions accounting mechanisms of oxygen photoactivation. In this case, the type I corresponds to the photodynamic reactions which occur owing to photochemical oxygen activation as a result of primary photosensitized electron transfer. The type II corresponds to the photodynamic reactions which occur due to photophysical oxygen activation as a result of energy transfer from excited photosensitizer molecules to oxygen or direct oxygen excitation followed by singlet oxygen formation.
Thus, the basic ideas of Bach and Engler stimulated mechanistic investigation of both dark and photoinduced reactions of oxygen activation and oxygenation of organic compounds. It is well established at present that, in natural conditions, oxygen photoactivation is determined by photophysical and photochemical mechanisms. Photophysical activation involves energy transfer to oxygen from singlet and triplet pigment molecules or direct photoexcitation of oxygen molecules. As a result, the reactive 1Deltag, state of oxygen molecules is populated, which is responsible for oxygenation of biomolecules. Photochemical activation is due to primary reduction or what is less probable, primary oxidation of photoexcited pigment molecules, which cause appearance of the primary free radicals. These radicals actively interact with oxygen and form reactive organic peroxy radicals and superoxide and hydroxyl radical, which determine oxygenation processes. Photoactivation of oxygen underlies photodynamic action - the phenomenon which is widely spread in nature being a reason for photooxidative stress. It is also involved in regulation of the expression of genes responsible for protective reactions of living cells and organisms. Control of the processes of biological oxygen photoactivation is very important for survival of living organisms, and the methods of artificial stimulation or suppression of oxygen photoactivation are of great interest for photomedicine, for example, in connection with development of the methods of photodynamic therapy of cancer and skin and infectious diseases [13-15, 155-157].
The author is grateful to Professor Helmut Sies for the photograph of Hans Kautsky and his kind permission to publish it.
This work was supported by the Russian Foundation for Basic Research, project No. 07-03-00183.
REFERENCES
1.Shönbein, C. F. (1845) Poggend. Ann.,
65, 196-199.
2.Bach, A. N. (1897) Zh. Rus. Fiz.-Khim.
Ob-va, 29, 373-398.
3.Bach, A. N. (1897) Comp. Rend. Acad. Sci.,
124, 951-967.
4.Engler, C., and Wild, W. (1897) Ber. Dt. Chem.
Ges., 30, 1669-1681.
5.Bach, A. N. (1893) Moniteur Scientifique,
7, 669.
6.Bach, A. N. (1894) Moniteur Scientifique,
8, 241.
7.Mehler, A. H. (1951) Arch. Biochem.
Biophys., 33, 65-89.
8.Raab, O. (1900) Z. Biol. (Muenich),
39, 524-546.
9.Tappeiner, H. (1900) Muench. Med.
Wochenschr., 47, 5-7.
10.Tappeiner, H., and Jodlbauer, A. (1904) Deut.
Arch. Klin. Med., 80, 427-487.
11.Straub, W. (1904) Arch. Exp. Pathol.
Pharmacol., 51, 383-390.
12.Clare, N. T. (1956) in Radiation Biology
(Hollaender, A., ed.) McGraw-Hill Book Company, Inc., New
York-Toronto-London, pp. 693-723.
13.Blum, H. F. (1964) Photodynamic Action and
Diseases Caused by Light, Hafner Publishing Company, N. Y.
14.Spikes, J. D. (1985) in Primary Photoprocesses
in Biology and Medicine (Bensasson, R. V., Jori, G., Land, E. J.,
and Truscott, T. G., eds.) Plenum Publishing Corporation, pp.
209-227.
15.Moan, J., and Peng, Q. (2003) Anticancer
Res., 23, 3591-3600.
16.Vladimirov, Y. A., Osipov, A. N., and Klebanov,
G. I. (2004) Biochemistry (Moscow), 69, 81-90.
17.Karu, T. (2001) Uspekhi Sovrem. Biol.,
121, 110-120.
18.Frizsche, M. (1867) Compt. Rend.,
64, 1035-1037.
19.Moureau, C., Duffraisse, C., and Dean, P. M.
(1926) Comp. Rend., 182, 1584-1587.
20.Schönberg, A. (1935) Liebigs Ann.
Chem. B., 518, S299-302.
21.Mulliken, R. S. (1928) Nature, 122,
505.
22.Mulliken, R. S. (1928) Phys. Rev.,
32, 880-887.
23.Wollaston, W. H. (1802) Phil. Trans.
II, 365-380.
24.Fraunhofer, J. (1814-1815) Denkshriften der
Münch. Akad. Wiss., 5, 193-226.
25.Kirchhoff, G. R. (1862) Untersuchungen
über das Sonenspektrum und die Spektern der chemischen
Elemente, Dummler, Berlin.
26.Ellis, J. M., and Kneser, H. O. (1933) Z.
Physik., 86, 583-591.
27.Herzberg, G. (1934) Nature, 133,
759.
28.Minaev, B. F., and Tikhomirov, V. A. (1984)
Zh. Fiz. Khim., 58, 646-652.
29.Kasha, M. (1985) in Singlet O2,
Vol. 1 (Frimer, A. A., ed.) CRC Press, Boca Raton, Florida, pp.
1-11.
30.Kautsky, H., and de Bruin, H. (1931)
Naturwiss., 19, 1043.
31.Kautsky, H., de Bruin, H., Neuwirtch, R., and
Baumeister, W. (1933) Chem. Ber., 66, 1588-1600.
32.Kautsky, H. (1939) Trans. Farad. Soc.,
35, 216-219.
33.Schenck, G. O. (1970) Ann. N. Y. Acad.
Sci., 171, 67-77.
34.Pryor, W. A. (1979) in Free Radicals in
Biology, Vol. 1 (Pryor, W. A., ed.) [Russian translation], Mir,
Moscow, pp. 13-87.
35.Backström, L. I. (1934) Z. Physik.
Chem., Abt. B, 25, 99-138.
36.Weiss, J. (1935) Naturwiss., 34,
610.
37.Franck, J., and Livingston, R. (1941) J. Chem.
Phys., 9, 184-190.
38.Zavoyskii, E. K. (1945) Zh. Eksp. Teor.
Fiz., 15, 245.
39.Norrish, R. G., and Porter, G. (1949)
Nature, 167, 658-671.
40.Porter, G. (1950) Proc. Roy. Soc.,
A200, 284-300.
41.Jablonski, A. (1935) Z. Physik, 94,
38-46.
42.Terenin, A. N. (1943) Acta Phisicochim.
(USSR), 18, 210-241.
43.Terenin, A. N. (1944) Zh. Fiz. Khim.,
17, 1-12.
44.Lewis, G. N., and Kasha, M. (1944) J. Amer.
Chem. Soc., 66, 2100-2116.
45.Terenin, A. N. (1947) Photochemistry of
Dyes [in Russian], USSR Academy of Sciences Publisher,
Moscow-Leningrad.
46.Schenck, G. O. (1948) Naturwis.,
35, 28-29.
47.Duffraisse, C., and Ecary, S. (1946) Compt.
Rend., 223, 735-737.
48.Rabinowitch, E. I. (1951) Photosynthesis and
Related Process, Vol. 2, Pt. 1, Interscience Publishing Inc., N.
Y.
49.Schenck, G. O. (1951) Z. Electrochem.,
55, 505-611.
50.Schenck, G. O., and Koch, E. (1960) Z.
Electrochem., 64, 170-177.
51.Schenck, G. O. (1963) Ind. Eng. Chem.,
55, 40-43.
52.Gollnick, K., and Schenck, G. O. (1964) Pure
Appl. Chem., 9, 507-525.
53.Bowen, E. J. (1953) Dissc. Farad. Soc.,
14, 143-146.
54.Kaplan, J. (1947) Phys. Rev., 71,
274.
55.Vallance-Jones, A., and Harrison, A. W. (1958)
J. Atmosph. Terr. Phys., 13, 45-60.
56.Noxon, J. F. (1961) Can. J. Phys.,
39, 1110-1119.
57.Noxon, J. F., and Vallance-Jones, A. (1962)
Nature, 196, 157-158.
58.Khan, A. U., and Kasha, M. (1970) J. Am. Chem.
Soc., 92, 3293-3300.
59.Khan, A. U. (1985) in Singlet
O2, Vol. 1 (Frimer, A. A., ed.) CRC Press, Boca Raton,
Florida, pp. 39-176.
60.Foote, C. S., and Wexler, S. (1964) J. Amer.
Chem. Soc., 86, 3879-3880.
61.Foote, C. S., and Wexler, S. (1964) J. Amer.
Chem. Soc., 86, 3880-3881.
62.Corey, E. J., and Taylor, W. C. (1964) J.
Amer. Chem. Soc., 86, 3881-3882.
63.Foote, C. S., Denny, R. W., Weaver, L., Chang,
Y., and Peters, J. (1970) Ann. N. Y. Acad. Sci., 171,
139-148.
64.Evans, D. F. (1969) Chem. Comm.,
367-368.
65.Matheson, I. B. C., and Lee, J. (1970) Chem.
Phys. Lett., 7, 475-476.
66.Snelling, D. R. (1968) Chem. Phys. Lett.,
2, 346-348.
67.Kearns, D. R., Khan, A. U., Duncan, C. K., and
Maki, A. H. (1969) J. Am. Chem. Soc., 91, 1038-1040.
68.Wasserman, E., Kuck, V. J., Delavan, W. M., and
Yager, W. A. (1969) J. Am. Chem. Soc., 91, 1040-1041.
69.Andrews, L. J., and Abrahamson, E. W. (1971)
Chem. Phys. Lett., 10, 113-116.
70.Duncan, K., and Kearns, D. R. (1971) J. Chem.
Phys., 55, 5822-5823.
71.Matheson, I. B. C., Lee, J., Yamanashi, B. S.,
and Wolbrast, M. L. (1974) Chem. Phys. Lett., 27,
355.
72.Krasnovsky, A. A., Jr. (1976) Biofizika,
21, 748-749.
73.Krasnovsky, A. A., Jr. (1977) Biofizika,
22, 927-928.
74.Krasnovsky, A. A., Jr. (1977) Acta Phys. Chem.
(Szeged University), 23, 147-154.
75.Krasnovsky, A. A., Jr. (1978) Izv. AN SSSR.
Ser. Fiz., 41, 343-348.
76.Krasnovsky, A. A., Jr., and Kagan, V. E. (1978)
Dokl. AN SSSR, 242, 229-232.
77.Krasnovsky, A. A., Jr., and Venediktov, E. A.
(1978) Biofizika, 23, 387-389.
78.Krasnovsky, A. A., Jr. (1979) Photochem.
Photobiol., 29, 29-36.
79.Krasnovsky, A. A., Jr., and Kagan, V. E. (1979)
FEBS Lett., 108, 152-154.
80.Krasnovsky, A. A., Jr. (1979) Biofizika,
24, 747-748.
81.Venediktov, E. A., and Krasnovsky, A. A., Jr.
(1979) Izv. Vuzov. Khim. Khim. Tekhnol., 22,
395-398.
82.Salokhiddinov, K. I., Byteva, I. M., and
Dzhagarov, B. M. (1979) Optika Spektr., 47, 881-886.
83.Byteva, I. V., and Gurinovich, G. P. (1979) J.
Luminesc., 21, 17-20.
84.Khan, A. U., and Kasha, M. (1979) Proc. Natl.
Acad. Sci. USA, 76, 6047-6049.
85.Butorina, D. N., Krasnovsky, A. A., Jr., and
Prieszev, A. V. (2003) Biofizika, 48, 201-209.
86.Krasnovsky, A. A., Jr. (1990) in Advances in
Science and Technology. Modern Problems of Laser Physics, Vol. 3
(Akhmanov, S. A., and Chernyaeva, V. B., eds.) [in Russian], VINITI,
Moscow, pp. 63-135.
87.Egorov, S. Yu., and Krasnovsky, A. A., Jr. (1990)
SPIE Proc., 1403, 611-621.
88.Krasnovsky, A. A., Jr. (1991) in Light in
Biology and Medicine, Vol. 2 (Douglas, R. H., Moan, J., and
Ronto, G., eds.) Plenum Press, N. Y.-London, pp. 437-452.
89.Krasnovsky, A. A., Jr. (1993) Proc. SPIE,
1887, 177-186.
90.Krasnovsky, A. A., Jr. (1998) Membr. Cell
Biol., 12, 665-660.
91.Krasnovsky, A. A., Jr. (2001) in Progress in
Porphyrin Chemistry (Golubchikov, O. A., ed.) [in Russian], Vol. 3,
NII Khimii SpbGU Publisher, St. Petersburg, pp. 191-216.
92.Krasnovsky, A. A., Jr. (2004) Biofizika,
49, 305-322.
93.Terenin, A. N., and Ermolaev, V. L. (1952)
Dokl. AN SSSR, 85, 547-550.
94.Dexter, D. L. (1953) J. Chem. Phys.,
21, 8365-850.
95.Terenin, A. N. (1967) Photonics of Dye
Molecules [in Russian], Nauka, Leningrad.
96.Ermolaev, V. L., Bodunov, E. N., Sveshnikova, E.
V., and Shakhverdov, T. A. (1977) Non-radiative Energy Transfer of
Electronic Excitation [in Russian], Nauka, Leningrad.
97.Dzhagarov, B. M., Sagun, E. I., Ganza, V.
A., and Gurinovich, G. P. (1987) Khim. Fiz., 6,
919-928.
98.Schweitzer, C., and Schmidt, R. (2003) Chem.
Rev., 103, 1685-1757.
99.Schmidt, R. (2006) Photochem. Photobiol.,
82, 1161-1177.
100.Schmidt, R., and Bodeshein, M. (1994) J.
Phys. Chem., 98, 2874-2876.
101.Chou, P. T., Wei, G.-T., Lin, C.-H., Wei,
C.-Y., and Chang, C.-H. (1996) J. Am. Chem. Soc., 118,
3031-3032.
102.Bachilo, S. M., Nichiporovich, I. N., and
Losev, A. P. (1998) Zh. Prikl. Spektr., 65, 811-814.
103.Scurlock, R. D., Wang, B., and Ogilby, P. R.
(1996) J. Amer. Chem. Soc., 118, 388-392.
104.Bodesheim, M., and Schmidt, R. J. (1997) J.
Phys. Chem., 101, 5672-5677.
105.Davidson, J., and Ogryzlo, E. A. (1973) in
Chemiluminescence and Bioluminescence (Hercules, D. M., and Lee,
J., eds.) Plenum Press, N. Y., pp. 111-126.
106.Krasnovsky, A. A., Jr., and Neverov, K. V.
(1990) Chem. Phys. Lett., 167, 591-597.
107.Krasnovsky, A. A., Jr., and Neverov, K. V.
(1993) Proc. SPIE, 1890, 56-61.
108.Chou, P.-T., Chen, Y.-C., Wei, C.-Y., and
Lee, M.-Z. (1998) J. Am. Chem. Soc., 120,
4883-4884.
109.Khan, A. U. (1980) Chem. Phys. Lett.,
72, 112-114.
110.Salokhiddinov, K. I., Dzhagarov, B. M., Byteva,
I. M., and Gurinovich, G. P. (1980) Chem. Phys. Lett.,
76, 85-87.
111.Karotki, A., Kruk, M., Drobishev, M., Rebane,
A., Nickel, E., and Spangler, C. W. (2001) IEEE J. Selected Topics
Quantum Electronics, 7, 971-975.
112.Drobizhev, M., Karotki, A., Kruk, M., and
Rebane, A. (2002) Chem. Phys. Lett., 355, 175-182.
113.He, G. S., Lin, T.-C., Dai, J., Prasad, P. N.,
Kannan, R., Dombroskie, A. G., Vaia, R. A., and Tan, L.-S. (2004) J.
Chem. Phys., 120, 5275-5284.
114.He, G., and Prasad, P. S. (2003) Proc.
SPIE, 5211, 1-12.
115.Matheson, I. B. C. (1979) Photochem.
Photobiol., 29, 875-878.
116.Matheson, I. B. C., and Lee, J. (1979)
Photochem. Photobiol., 29, 879-881.
117.Ambartzumian, R. V. (1987) Proc. SPIE,
701, 341-343.
118.Zakharov, S. D., and Ivanov, A. V. (1999)
Kvant. Elektron., 29, 1031-1053.
119.Krasnovsky, A. A., Jr., Drozdova, N. N.,
Ivanov, A. V., and Ambartzumian, R. V. (2003) Biochemistry
(Moscow), 68, 963-966.
120.Krasnovsky, A. A, Jr., and Ambartzumian, R. V.
(2004) Chem. Phys. Lett., 400, 531-535.
121.Krasnovsky, A. A., Jr., Drozdova, N. N.,
Roumbal, Ya. V., Ivanov, A. V., and Ambartzumian, R. V. (2005)
Chinese Opt. Lett., 3S, 1-4.
122.Krasnovsky, A. A., Jr., Roumbal, Ya. V.,
Ivanov, A. V., and Ambartzumian, R. V. (2006) Chem. Phys. Lett.,
430, 260-264.
123.Krasnovsky, A. A., Jr., Kryukov, I. V., and
Sharkov, A. V. (2007) Proc. SPIE, 6535, Q1-Q5.
124.Briviba, K., Klotz, L. O., and Sies, H. (1997)
Biol. Chem., 378, 1259-1265.
125.Klotz, L. O. (2002) Biol. Chem.,
383, 443-456.
126.Klotz, L. O., Kronke, K.-D., and Sies, H.
(2003) Photochem. Photobiol. Sci., 2, 88-94.
127.Antony, R. J., Warczak, K. L., and Donohue, T.
J. (2005) Proc. Natl. Acad. Sci. USA, 102, 6502-6507.
128.Zakharov, S. D., Ivanov, A. V., Volf, E. V.,
Danilov, T. M., et al. (2003) Kvant. Elektron., 33,
149-162.
129.Wurmser, R. (1935) Biological Oxidation and
Reduction [Russian translation], ONTI, Main Editorial Office of
Chemical Literature, Moscow.
130.Krasnovsky, A. A. (1948) Dokl. AN SSSR,
60, 421-424.
131.Krasnovsky, A. A. (1960) Uspekhi Khim.,
29, 736-759.
132.Krasnovsky, A. A. (1960) Ann. Rev. Plant
Physiol., 11, 363-410.
133.Krasnovsky, A. A. (1965) Photochem.
Photobiol., 4, 661-665.
134.Seely, G. R. (1977) in Primary Processes of
Photosynthesis (Barber, J., ed.) Elsevier/North-Holland Biomedical
Press, Amsterdam, pp. 2-52.
135.Chibisov, A. K. (1969) Photochem.
Photobiol., 10, 331-347.
136.Semenov, N. N. (1934) Chain Reactions
[in Russian], Goskhimizdat, Moscow.
137.Russell, G. A. (1975) J. Am. Chem. Soc.,
97, 3871-3877.
138.Vladimirov, Yu. A., Olenev, V. I., Suslova, T.
V., and Cheremisina, Z. V. (1980) Adv. Lipid Res., 17,
173-249.
139.Fedorova, G. F., Trofimov, A. V., Vasil'ev, R.
F., and Veprintsev, T. L. (2007) ARKIVOS, 8, 163-215.
140.Adam, W., Kazakov, D. V., and Kazakov, V. P.
(2005) Chem. Rev., 105, 3371.
141.Krinsky, N. I. (1984) in Oxygen Radicals in
Chemistry and Biology (Bors, W., Sarah, M., and Tait, D., eds.)
Walter de Gruyter Co., Berlin, pp. 453-464.
142.Kanofsky, J. R. (1986) J. Org. Chem.,
51, 3386-3388.
143.Kanofsky, J. R. (1989) Chem.-Biol.
Interactions, 70, 1-28.
144.Kanofsky, J. R. (2000) in Methods in
Enzymology, Vol. 319 (Paker, L., and Sies, H., eds.) pp. 59-67.
145.Foote, C. S. (1976) in Free Radicals in
Biology, Vol. 2 (Pryor, W. A., ed.) Academic Press, N. Y., pp.
85-133.
146.Foote, C. S. (1979) in Singlet Oxygen
(Wasserman, H. H., and Murray, R. W., eds.) Academic Press, N. Y., pp.
139-171.
147.Howard, J. A., and Ingold, K. U. (1968) J.
Am Chem. Soc., 90, 1056-1058.
148.Jason Niu, Q., and Mendenhall, G. D. (1992)
J. Am. Chem. Soc., 114, 165-172.
149.Khan, A. U., and Kasha, M. (1994) Proc.
Natl. Acad. Sci. USA, 91, 12362-12364.
150.Aubry, J. M. (1985) J. Am. Chem. Soc.,
107, 5844-5848.
151.Buhme, K., and Brauer, H.-D. (1992) Inorg.
Chem., 31, 3468-3471.
152.Kovalev, Y. V., Moiseeva, N. I., Minin, V. V.,
Larin, G. M., Krasnovsky, A. A., Gehman, A. E., and Moiseev, I. I.
(2001) Dokl. AN SSSR, 381, 74-77.
153.Nardello, V., Briviba, K., Sies, H., and Aubry,
J. M. (1998) Chem. Comm., 5, 599-600.
154.Foote, C. S. (1991) Photochem.
Photobiol., 54, 659.
155.Chissov, V. I., Sokolov, V. V., and Filonenko,
E. V. (1998) Ros. Khim. Zh., 62, 5-9.
156.Weishaupt, R., Gomer, J., and Dougherty, T. J.
(1993) J. Natl. Cancer Inst., 85, 443-456.
157.Stranadko, E. F., and Ivanov, A. V. (2004)
Biofizika, 49, 380-383.