Mechanisms of Oxidative Modification of Low Density Lipoproteins under Conditions of Oxidative and Carbonyl Stress
V. Z. Lankin1*, A. K. Tikhaze1, V. I. Kapel'ko1, G. S. Shepel'kova1, K. B. Shumaev2, O. M. Panasenko1, G. G. Konovalova1, and Yu. N. Belenkov1
1Russian Research Cardiology Complex, 3-ya Cherepkovskaya ul. 15a, 121552 Moscow, Russia; fax: (495) 414-6699; E-mail: lankin@cardio.ru2Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; fax: (495) 954-2732; E-mail: inbio@inbio.ras.ru
* To whom correspondence should be addressed.
Received May 4, 2007
Low-molecular-weight aldehydes (glyoxal, methylglyoxal, 3-deoxyglucosone) generated on autooxidation of glucose under conditions of carbonyl stress react much more actively with amino groups of L-lysine and epsilon-amino groups of lysine residues of apoprotein B-100 in human blood plasma low density lipoproteins (LDL) than their structural analogs (malonic dialdehyde (MDA), 4-hydroxynonenal) resulting on free radical oxidation of lipids under conditions of oxidative stress. Glyoxal-modified LDL aggregate in the incubation medium with a significantly higher rate than LDL modified by MDA, and MDA-modified LDL are markedly more poorly absorbed by cultured human macrophages and significantly more slowly eliminated from the rat bloodstream upon intravenous injection. Studies on kinetics of free radical oxidation of rat liver membrane phospholipids have shown that ubiquinol Q10 is the most active lipid-soluble natural antioxidant, and suppression of ubiquinol Q10 biosynthesis by beta-hydroxy-beta-methylglutaryl coenzyme A reductase inhibitors (statins) is accompanied by intensification of lipid peroxidation in rat liver biomembranes and in LDL of human blood plasma. Injection of ubiquinone Q10 protects the human blood plasma LDL against oxidation and prevents oxidative stress-induced damages to rat myocardium. A unified molecular mechanism of atherogenic action of carbonyl-modified LDL in disorders of lipid and carbohydrate metabolism is discussed.
KEY WORDS: low density lipoproteins, lipid hydroperoxides, malonic dialdehyde, glyoxal, methylglyoxal, ubiquinone Q10, oxidative stress, carbonyl stressDOI: 10.1134/S0006297907100069
Abbreviations: apoB) apoprotein B; CHD) coronary heart disease; FITC) fluorescein isothiocyanate; HMG-CoA reductase) beta-hydroxy-beta-methylglutaryl coenzyme A reductase; LDL) low density lipoproteins; MDA) malonic dialdehyde; SOD) superoxide dismutase.
In 1897, A. N. Bach formulated a peroxide theory of biological oxidation
in his paper “On the role of peroxides in slow oxidation
processes” published in the Journal of the Russian
Physicochemical Society of St. Petersburg University. This work,
which ascribed to hydroperoxides a crucial role in metabolism as
intermediates during oxidation of biosubstrates, was really prophetic.
Now the important role of systems of enzymatic generation and
utilization of organic hydroperoxides in the regulation of metabolism
of biologically active substances and provision of normal functioning
of various membrane-bound enzymes has been confirmed by many
experiments and is of no doubt [1-4]. Moreover, from the middle of XX century many works
appeared indicating that disorders in hydroperoxide metabolism can be
important for arising and development of many dangerous diseases, such
as atherosclerosis [5-9],
diabetes mellitus [10-13],
neurodegenerative diseases, etc. [14-18].
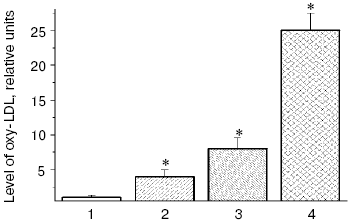
Low density lipoproteins (LDL) are the blood plasma lipoproteins most sensitive to induction of free radical oxidation [6], and we have shown that peroxidation of phospholipids of the outer layer of LDL particles is followed by conformational changes in the basic LDL protein apoprotein B-100 (apoB-100) and its displacement from the hydrophobic zone of the particle into the aqueous phase that can increase the nonreceptor capture of atherogenic LDL by the cells of blood vessel walls [6, 19]. Carbonyl-containing compounds, such as malonic dialdehyde (MDA) and alpha,beta-unsaturated aldehydes (4-hydroxynonenal and 4,5-dihydroxydecenal) [3, 20] generated during oxidative destruction of lipid hydroperoxides can react with amino groups of proteins and produce strong intermolecular cross-links of the Schiff base type [21, 22]. Similarly, aldehydes generated during homolysis of lipid hydroperoxides into oxidized LDL (oxy-LDL) form adducts with epsilon-amino groups of lysine residues of apoB-100 molecules that leads to changes in LDL conformation [6-9, 23, 24]. Modified LDL are recognized by “scavenger receptors” of blood vessel wall macrophages and actively captured by these cells [6-9, 23, 24]. MDA-modified LDL can also be recognized by other receptors of macrophages, which do not compete with scavenger receptors and may be termed “oxy-LDL receptors” [25, 26]. On intensive absorption of oxy-LDL, macrophages transform to lipid-overloaded “foam cells” and secrete a factor stimulating formation of their colonies. This results in clustering of cells with lipid inclusions and arising of primary pre-atherogenic damage to the blood vessel wall as fatty streaks [6-9, 23, 24]. Thus, oxidative stress accompanied by intensification of free radical oxidation of lipids in biomembranes [6] and LDL of blood plasma [6-9, 23, 24] has to promote arising and manifestation of atherosclerosis [5, 6]. Our investigations [27] have shown that the presence in patients with atherosclerosis such risk-factors of this disease as hypercholesterolemia or type II diabetes mellitus stimulates accumulation of oxidized LDL, and diabetic hyperglycemia extremely increases the oxy-LDL level in blood plasma of the patients (Fig. 1).
Patients with diabetes mellitus are prone to development of atherosclerosis, and cardiovascular system diseases are the main cause of death of these patients [12]. Literature data confirm an important role of free radical processes in pathogenesis of the type II diabetes mellitus [10, 30-32], and this induces an idea of common mechanisms of free radical damages in atherosclerosis and diabetes mellitus. Under conditions of diabetic hyperglycemia, autoxidation of glucose is increased, and this can be accompanied by generation of superoxide anion radicals [33, 34]. Therefore, the dose-dependent intensification of free radical oxidation of LDL found by us in vitro in the presence of glucose was not unexpected (Fig. 2), and the glucose levels in the incubation medium were comparable with the blood levels of glucose in patients with type II diabetes mellitus on decompensation of carbohydrate metabolism [36]. A pronounced accumulation of carbonyl compounds occurs in blood of patients with atherosclerosis [6], but in diabetes accumulation of endogenous aldehydes is much higher (so-called “carbonyl stress”) and their production is determined by other mechanisms [10, 30-32]. In particular, low-molecular-weight alpha-oxoaldehydes, such as glyoxal, methylglyoxal, and 3-deoxyglucosone are mainly produced on autoxidation of glucose [20, 37-40]. Similarly to MDA, these carbonyl compounds easily react with amino groups of proteins, producing fluorescent intermolecular links of the Schiff base type [37].Fig. 1. Relative levels of oxy-LDL (the level in control is taken as unity) in blood plasma of patients with coronary heart disease (CHD) and type II diabetes mellitus: 1) control, patients without signs of CHD and type II diabetes mellitus; 2) patients with CHD without hypercholesterolemia; 3) patients with CHD and hypercholesterolemia; 4) patients with type II diabetes mellitus on decompensation of carbohydrate metabolism (HbA1c = 8.1 ± 0.03%). This and the following figures present the levels of lipohydroperoxides in LDL (oxy-LDL) isolated by differential ultracentrifugation in a NaBr density gradient [28] and measured with Fe2+-xylenol orange reagent [29]. * Significant differences compared to the control and preceding group at p <= 0.05.
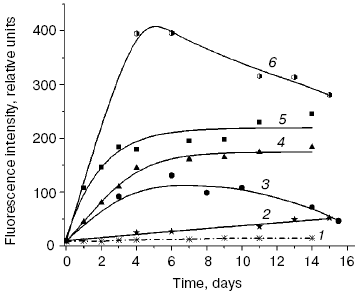
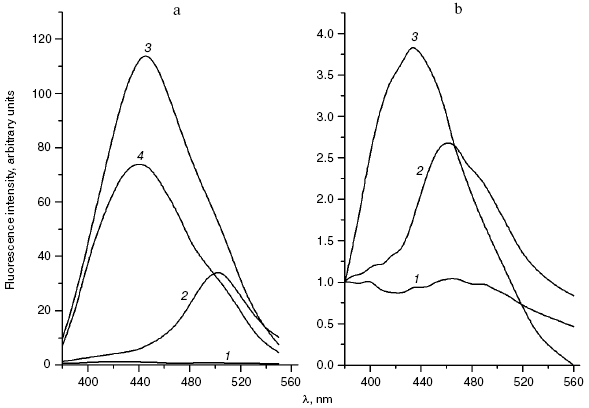
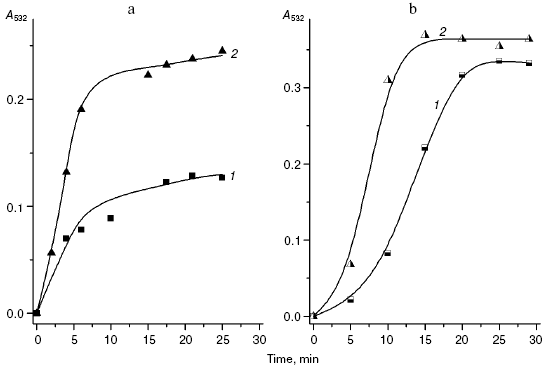
We compared the rates of accumulation of fluorescent products during incubation of L-lysine with glucose and aldehydes produced under conditions of both oxidative (MDA, 4-hydroxynonenal) and carbonyl stress (glyoxal, methylglyoxal, 3-deoxyglucosone) (Fig. 3). The rate of fluorophore accumulation during incubation of L-lysine with alpha-oxoaldehydes generated in the course of glucose autooxidation (glyoxal, methylglyoxal, 3-deoxyglucosone) was significantly higher than during the interaction of L-lysine with aldehydes produced as a result of free radical oxidation of lipids (MDA, 4-hydroxynonenal). Thus, MDA is a less effective modifier of amino acids than alpha-oxoaldehydes, and at the excitation wavelength of 350 nm the fluorescence spectrum maximum of the L-lysine interaction products with MDA is shifted to longer wavelengths compared to the fluorescence maximum of the L-lysine interaction products with the structural analog of MDA, glyoxal (lambdamax is 495 and 425 nm, respectively) (Fig. 4a). These findings are confirmed by data on modification of human blood plasma LDL with low-molecular-weight endogenous aldehydes (Fig. 4b).Fig. 2. Effects of glucose additions on kinetics of Cu2+-initiated free radical oxidation of LDL [35] isolated from blood plasma of a healthy donor: 1) without additions; 2) with 25 µM glucose; 3) with 50 µM glucose.
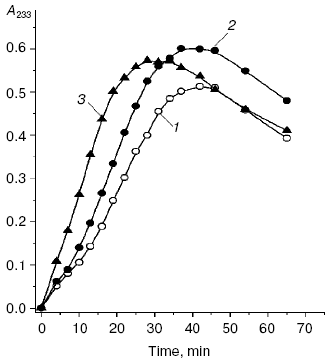
Fig. 3. Changes in fluorescence intensity (excitation wavelength lambdamax = 350 nm) of products of L-lysine (20 mM) interaction with different aldehydes (20 mM) during dark incubation at 37°C in isotonic K,Na-phosphate buffer (pH 7.2) containing 0.02% sodium azide for prevention of bacterial growth: 1) L-lysine + glucose; 2) L-lysine + 4-hydroxynonenal; 3) L-lysine + MDA; 4) L-lysine + glyoxal; 5) L-lysine + methylglyoxal; 6) L-lysine + 3-deoxyglucosone. Measurements were performed as described in [40]; MDA was prepared by acidic hydrolysis of 1,1,3,3-tetraethoxypropane [41]. The figure presents mean results of three determinations; in this and following figures, the fluorescence of a standard solution of dehydrated quinine sulfate (0.01 µg/ml in 0.05 M H2SO4) is taken as unit of fluorescence intensity.
In fact, the quantity of Schiff bases produced on interaction of LDL with glyoxal and methylglyoxal is significantly higher than on LDL interaction with MDA (4-hydroxynonenal did not induce generation of fluorophores during the 24-h incubation with LDL). Similarly to the case of L-lysine, the fluorescence spectrum maximum for MDA-modified LDL is shifted to longer wavelengths, with retention of principal spectral characteristics revealed for the products of aldehyde interaction with L-lysine (for LDL adducts with glyoxal lambdamax = 425 nm, for LDL adducts with MDA lambdamax = 495 nm, at the excitation wavelength of 350 nm).Fig. 4. a) Fluorescence spectra (excitation wavelength lambdamax = 350 nm) of products of L-lysine interaction with glucose (1), MDA (2), methylglyoxal (3), and glyoxal (4); incubation for 24 h, reaction conditions the same as in Fig. 3. b) Fluorescence spectra (excitation wavelength lambdamax = 350 nm) of native LDL from healthy donor blood plasma (1), MDA-modified LDL (2), and glyoxal-modified LDL (3); incubation for 24 h, reaction conditions the same as in Fig. 3. LDL were modified with aldehydes under conditions described in [42] at the ratio of 1 µmol aldehyde per 100 µg apoB, and then excess carbonyls were removed by dialysis.
LDL that has undergone free radical oxidation initiated by copper ions [43] or azoinitiators [44] can form associates similarly to oxidized biomembranes [45], and the aggregated LDL are taken up by cultured vascular walls with an increased rate, i.e. acquire enhanced atherogenicity [46, 47]. Modification by MDA increased the aggregation of LDL; however, the aggregation rate of glyoxal-modified LDL was more than twofold higher than the aggregation rate of MDA-modified LDL (Fig. 5). And although MDA-modified LDL were absorbed by cultured macrophages more effectively than native LDL, glyoxal-modified LDL were absorbed by these cells still more actively than MDA-modified LDL (Fig. 6, a and b). Macrophages were isolated and cultured as described in [48]. Note that similar results presented in Figs. 6a and 6b were obtained by two independent methods of determination of the macrophage-mediated absorption of LDL: by determination of content of intracellular cholesterol [49] and using LDL labeled with fluorescein isothiocyanate (FITC) [50].
Fig. 5. Aggregation rate of healthy donor blood plasma native LDL (1), MDA-modified LDL (2), and glyoxal-modified LDL (3) during incubation in isotonic Na,K-phosphate buffer (pH 7.2) at 37°C. Kinetics of LDL aggregation were measured recording fluctuations of a laser beam (lambda = 780 nm) transmission with a two-channel aggregometer [47]; modified LDL were prepared as described for Fig. 4. * Significant difference compared to control at p <= 0.05.
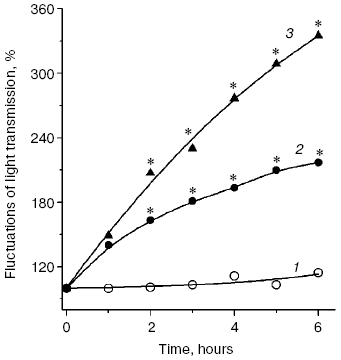
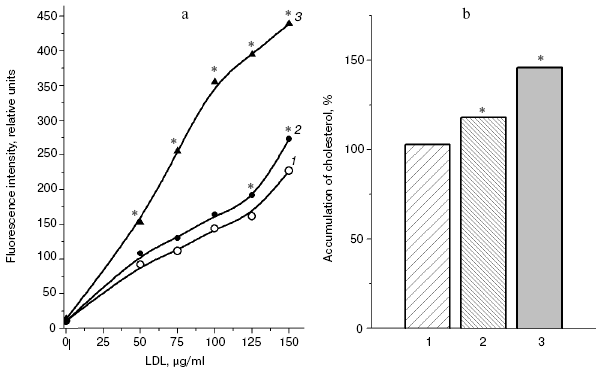
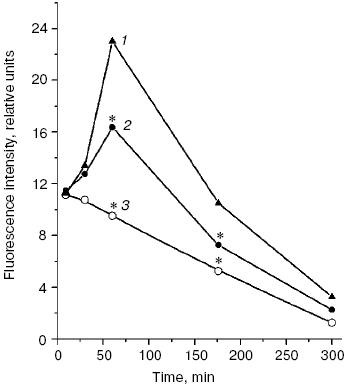
A significant difference in cholesterol accumulation on macrophage absorption of native and modified LDL usually can be revealed at the optimal LDL concentration in the incubation medium (about 100 µg/ml) [51]; thus, in our experiments the pronounced atherogenicity of glyoxal-modified LDL manifested itself more clearly when the concentration-dependent absorption of modified LDL by cultured macrophages was studied (Fig. 6b). The rate of elimination of aldehyde-modified FITC-labeled LDL from the blood flow was studied after injection into the rat femoral vein as described in [52]. Modified LDL were eliminated from the blood flow much faster than native ones (Fig. 7), and the fluorescent label in the animals' blood was sharply increased about 1 h after the injection of FITC-labeled native LDL. The fluorophore peak upon the injection of FITC-labeled MDA-modified LDL was markedly lower, and upon the injection of FITC-labeled glyoxal-modified LDL the fluorophore was monotonously eliminated from the blood flow (Fig. 7). Thus, the MDA-modified LDL were eliminated from the blood flow nearly 1.5-fold faster than native LDL and nearly 1.5-fold more slowly than glyoxal-modified LDL (Fig. 7). Our experimental data suggest significantly higher atherogenicity of alpha-oxoaldehydes (especially glyoxal and methylglyoxal) generated under conditions of carbonyl stress during autooxidation of glucose, as compared to atherogenicity of aldehydes (mainly MDA) generated under conditions of oxidative stress during free radical oxidation of polyene lipids. These findings allowed us for the first time to sufficiently explain reasons for the known rapid progression of atherosclerosis on the background of diabetes mellitus.Fig. 6. a) Uptake by cultured human monocyte macrophages of FITC-labeled LDL isolated from blood plasma of a healthy donor (1), MDA-modified LDL (2), or glyoxal-modified LDL (3) depending on apoB concentration in the incubation medium. LDL were incubated for 7 h with a 14-day-old primary culture of macrophages. Mean results of three experiments are presented. * Significant difference compared to control at p <= 0.05. b) Cholesterol uptake by cultured human macrophages from native LDL (1), MDA-modified LDL (2), or glyoxal-modified LDL (3) at the apoB concentration in the incubation medium of 100 µg/ml. Mean results of three experiments are presented. * Significant difference compared to control at p <= 0.05.
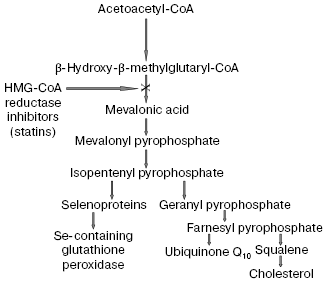
The recognition of an important role of oxidized LDL in pathogenesis of atherosclerosis confirmed in many recent works [53-56] allowed us to consider in a new way the problem of widely used hypolipidemic therapy of coronary heart disease (CHD) by cholesterol-decreasing drugs, statins, which inhibit the activity of a key enzyme beta-hydroxy-beta-methylglutaryl coenzyme A (HMG-CoA) reductase responsible for cholesterol biosynthesis in the body [57]. However, on inhibition of the cholesterol biosynthesis, statins simultaneously suppress synthesis of the most important natural antioxidants (scheme in Fig. 8). In fact, statins inhibit early stages of cholesterol biosynthesis, but metabolites produced during the subsequent reactions are necessary for synthesis of both selenoproteins, including the most important antioxidant enzyme Se-containing glutathione peroxidase [58] and ubiquinone Q10 [57]. It has now been established that ubiquinone Q10 (more accurately, its reduced form ubiquinol Q10) and not vitamin E (alpha-tocopherol), as thought earlier, is a principal natural antioxidant protecting LDL against atherogenic oxidative modification [59, 60]. Vitamin E is not synthesized in the human body, and the demand for it is satisfied only by its intake with food. Although alpha-tocopherol is transferred into peripheral tissues by LDL particles, the oxidizability of LDL cannot be lowered even by introduction of extremely high doses of vitamin E significantly exceeding the daily requirement [6]. Moreover, application of such high doses of vitamin E can even be dangerous [61] because it is associated with a possible accumulation of tocopheroxyl radicals, which can initiate free radical oxidation of LDL and provoke a further development of oxidative stress [62]. The reaction equation presented below shows that alpha-tocopherol (alpha-TOH) can neutralize only one free lipid radical generated in unsaturated lipids of LDL (LO2*), whereas it itself is concurrently oxidized with generation of tocopheroxyl radical (alpha-TO*) [3]:Fig. 7. Rate of elimination from blood flow of FITC-labeled human blood plasma LDL injected into the femoral vein of Wistar rats in the dose of 2 µg apoB per g body weight: 1) native LDL; 2) MDA-modified LDL; 3) glyoxal-modified LDL. Mean results of three experiments are presented. * Significant difference compared to control at p <= 0.05.
LO2* + alpha-TOH → LOOH + alpha-TO*. (1)
However, fully reduced ubiquinone Q10, or ubiquinol Q10 (QH2), which is the only lipid-soluble antioxidant synthesized in the human body, can utilize two lipid radicals concurrently with its consecutive oxidation to semiquinone radical (*QH) and the corresponding quinone (Q), i.e., is more efficient as a trap of radicals than alpha-tocopherol [3]:Fig. 8. Scheme of action mechanism of beta-hydroxy-beta-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins), according to data of [57, 58], with some changes.
LO2* + QH2 → LOOH + *QH,
LO2* + *QH → LOOH + Q. (2)
It is also important that bioregeneration of ubiquinol Q10 in blood plasma LDL can occur as a result of non-enzymatic reduction of its semiquinone radical (*QH) with ascorbic acid (HO-Asc-OH) [3]:
*QH + HO-Asc-OH → HO-Asc-O* + QH2. (3)
Thus, application of the antioxidant ubiquinol Q10, which is synthesized in the body for protection of LDL particles against free radical oxidation, is in a complete agreement with the pragmatism of Nature and is much more advantageous and efficient than application of vitamin E, which is irreversibly spent during LDL transport in the bloodstream and, thus, is unreasonable. Therefore, unsuccessfulness of attempts to prevent oxidative modification of LDL in patients with atherosclerosis using high doses of vitamin E may be explained based on the above-presented considerations, and this has been confirmed by results of recent clinical studies [61, 63, 64]. Suppression of ubiquinone Q10 biosynthesis by statins has to be accompanied by a decrease in its level in LDL of patients, and this has been shown in many clinical investigations [65-69]. Moreover, in elderly patients with atherosclerosis a deficiency of ubiquinone Q10 has been observed [70, 71] which is partly caused by the age-related decrease in its biosynthesis [72, 73]. The intensity of ubiquinone Q10 synthesis in human tissues decreases with age, and, as a result, after 30 years of age its content in human myocardium is 1.5-2.0-fold decreased [72, 74]. Moreover, in patients with atherosclerosis the ratio of ubiquinol Q10/ubiquinone Q10 in LDL is sharply (about twofold) decreased [75, 76], whereas in healthy persons this ratio is 95/5 [77]. And, finally, prescription to patients with atherosclerosis of a diet with a limited amount of animal fats leads to inhibition of the main alimentary sources of ubiquinone Q10 [57], because about 40% of the ubiquinone Q10 pool in the body is compensated due to its delivery with food [57]. Thus, the provision with the most important natural antioxidant, ubiquinol Q10, is initially significantly lowered in patients with atherosclerosis, and a long-term treatment with statins has to still aggravate the deficiency of ubiquinone Q10, and, as a consequence, promote the development of oxidative stress [70, 76, 77] accompanied by accumulation of atherogenic modified LDL.
A side-effect of statins which seems to be associated with intensification of free radical oxidation of membrane phospholipids is a disturbance of liver functions [78, 79], which results in release of hepatic transaminases into blood plasma. Free radical oxidation of biomembranes is associated with disorders in their permeability, which can cause a “leakage” of enzymes from the cell [3, 6]. We have shown that peroral injection of rats with ubiquinone Q10 protects polyene lipids of liver biomembranes against free radical oxidation much more efficiently than injection of an antioxidant vitamin complex consisting of provitamin A, vitamins E and C, and organic selenium [80]. Therefore, suppression of ubiquinone Q10 biosynthesis in tissues with HMG-CoA reductase inhibitors has to be accompanied by intensification of free radical oxidation of membrane phospholipids. In fact, we found [81] that the oxidation degree of liver biomembranes in intact rats treated with simvastatin for four weeks was increased 1.9-fold (p < 0.05) and the oxidation degree of liver biomembranes in rats with hypercholesterolemia treated with lovastatin for four weeks was increased 2.1-fold (p < 0.05) (Fig. 9). It should be noted that the increase in the enzyme levels in blood caused by damage of the liver membrane structures under the influence of HMG-CoA reductase inhibitors could be prevented by injection of patients with ubiquinone Q10, in accordance with results of clinical studies presented in the patent of Merck & Co., Inc., USA (US Patent No. 4929437).
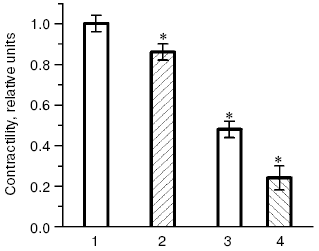
The possibility of unfavorable consequences caused by ubiquinone Q10 deficiency seems to be even more justified, because biological functions of ubiquinone Q10 are far from being limited by its antioxidant effect, and it is also an intermediary electron carrier in the mitochondrial respiratory chain. Therefore, suppression of ubiquinone Q10 biosynthesis on therapy with statins can decrease energy provision of tissues and, what is especially undesirable in CHD, lower energy provision of the myocardium. In our experiments rats were given simvastatin in doses adequate to those prescribed to patients with CHD, and a significant decrease in levels of major energy-rich compounds, ATP and creatine phosphate, was recorded in the heart muscle as early as four weeks after the beginning of injections of the HMG-CoA reductase inhibitor [82, 83]. The decrease in the energy provision of myocardium inevitably has to cause disorders in its functions. Thus, injection of rats for four weeks with another inhibitor of HMG-CoA reductase, atorvastatin [84], was accompanied by a significant decrease in the intensity of the myocardium contractility [85] (Fig. 10). During oxidative stress modeled on the heart isolated from the atorvastatin-treated animals, the myocardium contractility was lowered much more strongly than during oxidative stress modeled on the heart isolated from intact rats (Fig. 10). In another series of experiments [85] preliminary injections of animals with ubiquinone Q10 for six weeks increased its level in the myocardium by 63%, and oxidative stress caused in these rats a significantly lower decrease in the myocardium contractility than in the control groups (not treated with ubiquinone Q10). Oxidative stress also did not suppress activities of the myocardial antioxidant enzymes, SOD and glutathione peroxidase, in the hearts isolated from animals pretreated with ubiquinone Q10 [86]. Moreover, the activities of SOD and glutathione peroxidase strongly correlated with the intensity of myocardial contractility (r = 0.91 or r = 0.94, respectively, at p < 0.05) in cardiomyocytes of the rats pretreated with ubiquinone Q10 [86]. Consequently, the decrease in the energy provision of the heart muscle under the influence of statins can induce functional disorders in the myocardium which are to manifest themselves more sharply under conditions of oxidative stress associated with development of atherosclerosis [5, 6, 87]. A decrease in the ubiquinone Q10 synthesis in myocytes on the therapy statins can lead to dystrophy of muscle tissue and promote manifestations of such side effects of these drugs as myopathy and rhabdomyolysis, the danger of which can occur upon application of the majority of statins, especially in high doses [78, 79]. It should be noted that the story of clinical application of the most promising drug of this class, cerivastatin (“microstatin”, which effectively decreased the cholesterol level in LDL at the daily dose of 0.4 mg) was recently terminated by prohibition of its production because of frequent cases of rhabdomyolysis [88]. Results of clinical studies allowed Merck & Co., Inc. to obtain a patent (US Patent No. 4933165) on application of ubiquinone Q10 for preventing possible cases of myopathy during hypolipidemic therapy of patients with CHD by drugs inhibiting HMG-CoA reductase.Fig. 9. Effect of lovastatin (a) and simvastatin (b) on kinetics of free radical oxidation of liver biomembranes in rats with alimentary hypercholesterolemia (a) and intact rats (b): 1) control; 2) experiment (typical kinetic curves are presented; quantitative data are in the text).
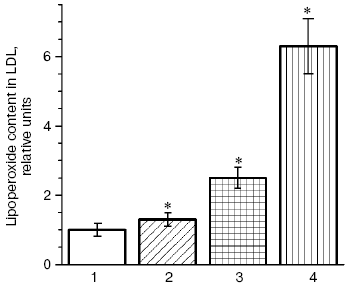
Results of our double blind placebo-controlled clinical studies revealed a significant increase in blood contents of oxy-LDL in patients with CHD and hypercholesterolemia upon long-term treatment with different statins, and in some cases extremely high values were recorded [6, 83, 89-91]. Thus, treatment with pravastatin (40 mg/day for six months), simvastatin (20 mg/day for two months), or cerivastatin (0.4 mg/day for six months) induced a steady increase in the blood level of oxy-LDL by 30%, more than twofold, and more than sixfold, respectively (Fig. 11). It seems doubtless that the observed prooxidant effect of statins is associated with the suppression by these preparations of ubiquinone Q10 biosynthesis, as follows from the scheme presented in Fig. 8. And according to this scheme, which suggests inhibition of selenoprotein biosynthesis by inhibitors of HMG-CoA reductase, we have recorded in patients with CHD and hypercholesterolemia treated with pravastatin (40 mg/day for six months) [6, 83, 89] a sharp decrease (nearly fivefold) in the activity of erythrocytic Se-containing glutathione peroxidase (Fig. 12), which is a key antioxidant enzyme responsible for reduction of toxic lipid hydroperoxides (LOOH) to corresponding alcohols (LOH) on oxidation of reduced glutathione (GSH):Fig. 10. Changes in contractility of isolated rat heart (the product of developed pressure × heart rate) [84] before and after modeling of oxidative stress in intact animals and those treated with atorvastatin [84]; 1, 2) before modeling of oxidative stress; 3, 4) 40 min after addition of 100 µM H2O2 into the perfusate; 1, 3) intact rats; 2, 4) rats treated with atorvastatin (the contractility in intact rats before modeling of oxidative stress is taken as unity). * Significant difference compared to control at p <= 0.05.
LOOH + 2GSH → GSSG + LOH, (4)
which prevents homolysis of lipid hydroperoxides (LOOH) with production of active alkoxyl radicals (LO*) [3]:
LOOH → LO* + OH-. (5)
Fig. 11. Blood level of lipid hydroperoxides in LDL (oxy-LDL) in patients treated with pravastatin (2), simvastatin (3), and cerivastatin (4) for six, two, and six months, respectively, as compared to the corresponding controls; (1) the level of oxy-LDL in patients not treated with statins (control) taken as unity in each study [6, 83, 89-91]. * Significant difference compared to control at p <= 0.05.
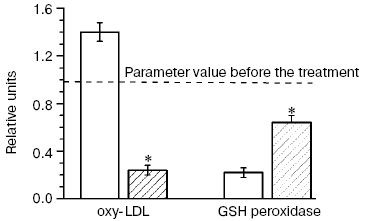
In our double blind placebo-controlled clinical studies [6, 83, 89] patients with CHD and hypercholesterolemia treated with pravastatin (40 mg/day for six months) were concurrently given ubiquinone Q10 (Pharma Nord, Denmark) (60 mg/day for six months), and this not only completely prevented the prooxidant effect of the statin but sharply decreased (more than fourfold) the level of oxy-LDL (Fig. 12). Although the combined treatment of patients with CHD and hypercholesterolemia with pravastatin and ubiquinone Q10 failed to completely normalize the activity of Se-containing glutathione peroxidase in the patients' blood within the observation period, the enzyme activity was decreased nearly threefold less than on monotherapy with pravastatin (Fig. 12). Thus, inhibitors of HMG-CoA reductase possess a pronounced prooxidant effect, and long-term therapy with these drugs can provoke not only disorders in liver function and myopathy, but also induce the manifestation of atherosclerosis because of accumulation of atherogenic oxy-LDL. We have shown (Fig. 1) that such risk factors of atherosclerosis development as hypercholesterolemia and type II diabetes mellitus are also risk factors of arising of oxidative and carbonyl stress [27]. If the supposed scheme of atherosclerosis progression is correct, the principal drug treatment in patients with CHD should be directed to suppress oxidative stress (possibly, by addition of ubiquinone Q10 to hypolipidemic therapy [6, 83, 89]) and carbonyl stress (by normalization of carbohydrate metabolism in diabetes mellitus [92]).Fig. 12. Changes in the blood level of oxy-LDL and activity of glutathione peroxidase in patients treated for six months with pravastatin + coenzyme Q placebo (light columns) or with pravastatin + coenzyme Q (hatched columns); values of the parameters before the treatment (dotted line) are taken as unity [6, 83, 89]. * Significant difference compared to control at p <= 0.05.
REFERENCES
1.Lankin, V. Z., Bondar, T. N., and Tikhaze, A. K.
(1997) Dokl. Akad. Nauk SSSR, 357, 828-831.
2.Lankin, V. Z., Tikhaze, A. K., and Osis, Yu. G.
(2002) Biochemistry (Moscow), 67, 566-574.
3.Lankin, V. Z. (2003) in Free Radicals,
Nitric Oxide, and Inflammation: Molecular,
Biochemical, and Clinical Aspects, NATO Science
Series, Vol. 344 (Tomasi, A., et al., eds.) IOS Press, Amsterdam,
etc., pp. 8-23.
4.Lankin, V. Z., Antonovsky, V. L., and Tikhaze, A.
K. (2004) in Peroxides at the Beginning of the Third Millennium
(Antonovsky, V. L., et al., eds.) Nova Science Publishers Inc., New
York, pp. 85-111.
5.Lankin, V. (1992) Excerpta Med., Int.
Congr. Ser., G98, 385-388.
6.Lankin, V. Z., and Tikhaze, A. K. (2003) in Free
Radicals, Nitric Oxide, and Inflammation:
Molecular, Biochemical, and Clinical Aspects, NATO
Science Series, Vol. 344 (Tomasi, A., et al., eds.) IOS Press,
Amsterdam, etc., pp. 218-231.
7.Steinberg, D., Parthasarathy, S., Carew, T. E.,
Khoo, J. C., and Witztum, J. L. (1989) New Engl. J. Med.,
320, 915-924.
8.Witztum, J. L., and Steinberg, D. (1991) J.
Clin. Invest., 88, 1785-1792.
9.Yla-Herttuala, S. (1994) Drugs Today,
30, 507-514.
10.Oberley, L. W. (1988) Free Rad. Biol.
Med., 5, 113-124.
11.Lankin, V. Z., Korchin, V. I., Konovalova, G. G.,
and Jarkova, R. D. (1994) Free Rad. Biol. Med., 16,
15.
12.Giugliano, D., Ceriello, A., and Paolisso, G.
(1996) Diabetes Care, 19, 257-267.
13.Laight, D. W., Carrier, M. J., and Anggard, E. E.
(2000) Cardiovasc. Res., 47, 457-464.
14.Adams, J. D., and Odunze, I. N. (1991) Free
Rad. Biol. Med., 10, 161-169.
15.Markesbery, W. R. (1997) Free Rad. Biol.
Med., 23, 134-147.
16.Markesbery, W. R., and Carney, J. M. (1999)
Brain Pathol., 9, 133-146.
17.Pratico, D., and Delanty, N. (2000) Am. J.
Med., 109, 577-585.
18.Behl, C., and Moosmann, B. (2002) Free Rad.
Biol. Med., 33, 182-191.
19.Formazyuk, V. E., Osis, Yu. G., Deev, A. I.,
Lankin, V. Z., Vikhert, A. M., and Vladimirov, Yu. A. (1982) Dokl.
Akad. Nauk SSSR, 263, 497-500.
20.Witz, G. (1989) Free Rad. Biol. Med.,
7, 333-349.
21.Tappel, A. L. (1980) Free Rad. Biol.,
4, 1-47.
22.Donato, H. (1981) in Age Pigments (Sohal,
R. S., ed.) Elsevier/North-Holland Biomedical Press, Amsterdam, etc.,
pp. 63-81.
23.Mackness, M. I., and Durrington, P. N. (1995)
Atherosclerosis, 115, 243-253.
24.Goldstein, J. L., Ho, Y. K., Basu, S. K., and
Brown, M. S. (1979) Proc. Natl. Acad. Sci. USA, 76,
333-337.
25.Stanton, L. W., White, R. T., Bryant, C. M.,
Protter, A. A., and Endemann, G. (1992) J. Biol. Chem.,
267, 22446-22451.
26.Endemann, G., Stanton, L. W., Madden, K. S.,
Bryant, C. M., White, R. T., and Protter, A. A. (1993) J. Biol.
Chem., 268, 11811-11816.
27.Lankin, V. Z., Lisina, M. O., Arzamastseva, N.
E., Konovalova, G. G., Nedosugova, L. V., Kaminnyi, A. I., Tikhaze, A.
K., Kukharchuk, V. V., and Belenkov, Yu. N. (2005) Byul. Eksp. Biol.
Med., 140, 41-43.
28.Tertov, V. V., Kaplun, V. V., Dvoryantsev, S. N.,
and Orekhov, A. N. (1995) Biochem. Biophys. Res. Commun.,
214, 608-613.
29.Nourooz-Zadeh, J., Tajaddini-Sarmadi, J., and
Wolff, S. P. (1994) Analyt. Biochem., 220, 403-409.
30.Gillery, P., Monboisse, J. C., Maquart, F. X.,
and Borel, J. P. (1989) Med. Hypotheses, 29, 47-50.
31.Lyons, T. J. (1993) Am. J. Cardiol.,
71, 26B-31B.
32.Tesfamariam, B. (1994) Free Rad. Biol.
Med., 16, 383-391.
33.Wells-Knecht, K. J., Zyzak, D. V., Litchfield, J.
E., Thorpe, S. R., and Baynes, J. W. (1995) Biochemistry,
34, 3702-3709.
34.Yim, H. S., Kang, S. O., Hah, Y. C., Chock, P.
B., and Yim, M. B. (1995) J. Biol. Chem., 270,
28228-28233.
35.Tikhaze, A. K., Lankin, V. Z., Kolycheva, S. V.,
Konovalova, G. G., Shumaev, K. B., Kozachenko, A. I., Gurevich, S. M.,
Zharova, E. A., and Smirnov, L. D. (1998) Byul. Eksp. Biol.
Med., 126, 551-554.
36.Mowri, H.-O., Frei, B., and Keaney, J. F. (2000)
Free Rad. Biol. Med., 29, 814-824.
37.Thornalley, P. J., Langborg, A., and Minhas, H.
S. (1999) Biochem. J., 344, 109-116.
38.Knott, H. M., Brown, B. E., Davies, M. J., and
Deant, R. T. (2003) Eur. J. Biochem., 270, 3572-3582.
39.Beisswenger, P., and Ruggiero-Lopez, D. (2003)
Diabetes Metab., 29, 6S95-6S103.
40.Zhang, X., Ma, Y., Liu, H., de Sa, P. F., Brown,
P. R., and Dain, J. (2004) Analyt. Biochem., 325,
255-259.
41.Requena, J. R., Fu, M. X., Ahmed, M. U., Jenkins,
A. J., Lyons, T. J., Baynes, J. W., and Thorpe, S. R. (1997)
Biochem. J., 322, 317-325.
42.Fogelman, A. M., Shechtrer, I., Seager, J.,
Hokom, M., Child, J. S., and Edvards, P. A. (1980) Proc. Natl. Acad.
Sci. USA, 77, 2214-2218.
43.Hoff, H. F., Whitaker, T. E., and O'Neil, J.
(1992) J. Biol. Chem., 267, 602-609.
44.Kawabe, Y., Cynshi, O., Takashima, Y., Suzuki,
T., Ohba, Y., and Kodama, T. (1994) Arch. Biochem. Biophys.,
310, 489-496.
45.May, H. E., and McCay, P. B. (1968) J. Biol.
Chem., 243, 2288-2305.
46.Tertov, V. V., Orekhov, A. N., Martsenyuk, O. N.,
Perova, N. V., and Smirnov, V. N. (1989) Exp. Mol. Pathol.,
50, 337-347.
47.Tertov, V. V., Sobenin, I. A., Orekhov, A. N.,
Gabbasov, Z. A., Popov, E. G., Yaroslavov, A. A., and Smirnov, V. A.
(1992) Circulation Res., 71, 218-228.
48.Skarlatos, S. I., Rao, R., and Kruth, H. S.
(1992) J. Tissue Culture Meth., 14, 113-118.
49.Orechov, A. N., Tertov, V. V., Novicov, I. D.,
Krushinsky, A. V., Andreeva, E. R., Lankin, V. Z., and Smirnov, V. N.
(1986) Exp. Mol. Pathol., 42, 117-137.
50.Amante, A., Ancona, A., and Forni, L. (1972)
J. Immunol. Meth., 1, 289-301.
51.Jessup, W., Simpson, J. A., and Dean, R. T.
(1993) Atherosclerosis, 99, 107-120.
52.Nieuwenhuizen, W., Emeis, J. J., Vermond, A.,
Kurver, P., and van der Heide, D. (1980) Biochem. Biophys. Res.
Commun., 97, 49-55.
53.Holvoet, P., Vanhaecke, J., Janssens, S., van de
Werf, F., and Collen, D. (1998) Circulation, 98,
1487-1494.
54.Holvoet, P., Mertens, A., Verhamme, P., Bogaerts,
K., Beyens, G., Verhaeghe, R., Collen, D., Muls, E., and van de Werf,
F. (2001) Arterioscler. Thromb. Vasc. Biol., 21,
844-848.
55.Holvoet, P. (2004) Acta Cardiol.,
59, 479-484.
56.Johnston, N., Jernberg, T., Lagerqvist, B.,
Siegbahn, A., and Wallentin, L. (2006) Am. J. Cardiol.,
97, 640-645.
57.Bliznakov, E. G., and Wilkins, D. J. (1998)
Adv. Therapy, 15, 218-228.
58.Moosmann, B., and Behl, C. (2004) Lancet,
363, 892-894.
59.Stocker, R., Bowry, V. W., and Frei, B. (1991)
Proc. Natl. Acad. Sci. USA, 88, 1646-1650.
60.Mohr, D., Bowry, V. W., and Stocker, R. (1992)
Biochim. Biophys. Acta, 1126, 247-254.
61.Hoves, R. M. (2006) Ann. N. Y. Acad. Sci.,
1067, 22-26.
62.Bowry, V. W., Ingold, K. U., and Stocker, R.
(1992) Biochem. J., 288, 341-344.
63.Vivekananthan, D. P. (2003) Lancet,
361, 2017-2023.
64.Lankin, V. Z., Tikhaze, A. K., and Belenkov, Yu.
N. (2004) Kardiologiya, 44, 72-81.
65.Laaksonen, R., Ojala, J.-P., Tikkanen, M. J., and
Himberg, J.-J. (1994) Eur. J. Clin. Pharmacol., 46,
313-317.
66.Mortensen, S., Leth, A., Agner, E., and Rohde, M.
(1997) Mol. Asp. Med., 18 (Suppl.), S137-S144.
67.Langsjoen, P. H., and Langsjoen, A. M. (2003)
Bio Factors, 18, 101-111.
68.Bleske, B. E., Willis, R. A., Anthony, M.,
Casselberry, N., Datwani, M., Uhley, V. E., Secontine, S. G., and Shea,
M. J. (2001) Am. Heart J., 142, E2.
69.Laaksonen, R., Jokelainen, K., Laakso, J., Sahi,
T., Harkonen, M., Tikkanen, M. J., and Himberg, J. J. (1996) Am. J.
Cardiol., 77, 851-854.
70.Kontush, A., Reich, A., Baum, K., Spranger, T.,
Finckh, B., Kohlschutter, A., and Beisiegel, U. (1997)
Atherosclerosis, 129, 119-126.
71.Kaikkonen, J., Nyyssonen, K., and Salonen, J. T.
(1999) Scand. J. Clin. Lab. Invest., 59, 457-466.
72.Kalen, A., Appelkvist, E. L., and Dallner, G.
(1989) Lipids, 24, 579-584.
73.Aejmelaeus, R., Metsa-Ketela, T., Laippala, P.,
Solakivi, T., and Alho, H. (1997) Mol. Asp. Med., 18
(Suppl.), S113-S120.
74.Abete, P., Napoli, C., Santoro, G., Ferrara, N.,
Tritto, J., Chiariello, M., Rengo, F., and Ambrosio, G. (1999) J.
Mol. Cell. Cardiol., 31, 227-236.
75.Mabuchi, H., Higashikata, T., Kawashiri, M.,
Katsuda, S., Mizuno, M., Nohara, A., Inazu, A., Koizumi, J., and
Kobayashi, J. (2005) J. Atheroscler. Thromb., 12,
111-119.
76.Lagendijk, J., Ubbink, J. B., Delport, R.,
Vermaak, W. J., and Human, J. A. (1997) Res. Commun. Mol. Pathol.
Pharmacol., 95, 11-20.
77.Yamashita, S., and Yamamoto, Y. (1997) Analyt.
Biochem., 250, 66-73.
78.McTaggart, F., Buckett, L., Davidson, R.,
Holdgate, G., McCormick, A., Schneck, D., Smith, G., and Warwick, M.
(2001) Am. J. Cardiol., 87, 28B-32B.
79.Alsheikh-Ali, A. A., Ambrose, M. S., Kuvin, J.
T., and Karas, R. H. (2005) Circulation, 111,
3052-3057.
80.Tikhaze, A. K., Konovalova, G. G., Lankin, V. Z.,
Kaminnyi, A. I., Kaminnaya, V. I., Ruuge, E. K., and Kukharchuk, V. V.
(2005) Byul. Eksp. Biol. Med., 140, 181-183.
81.Lankin, V. Z., Ivanova, M. V., Konovalova, G. G.,
Tikhaze, A. K., Kaminnyi, A. I., and Kukharchuk, V. V. (2007) Byul.
Eksp. Biol. Med., 143, 390-393.
82.Pisarenko, O. I., Studneva, I. M., Lankin, V. Z.,
Konovalova, G. G., Tikhaze, A. K., Kaminnaya, V. I., and Belenkov, Yu.
N. (2001) Byul. Eksp. Biol. Med., 132, 956-958.
83.Lankin, V. Z., Tikhaze, A. K., Kukharchuk, V. V.,
Konovalova, G. G., Pisarenko, O. I., Kaminnyi, A. I., Shumaev, K. B.,
and Belenkov, Yu. N. (2003) Mol. Cell. Biochem., 249,
129-140.
84.Lakomkin, V. L., Kapel'ko, V. I., Lankin, V. Z.,
Konovalova, G. G., and Kaminnyi, A. I. (2007) Byul. Eksp. Biol.
Med., 143, 383-385.
85.Lakomkin, V. L., Konovalova, G. G., Kalenikova,
E. V., Zabbarova, I. V., Tikhaze, A. K., Tsyplenkova, V. G., Lankin, V.
Z., Ruuge, E. K., and Kapel'ko, V. I. (2004) Biochemistry
(Moscow), 69, 520-526.
86.Lakomkin, V. L., Konovalova, G. G., Kalenikova,
E. V., Zabbarova, I. V., Kaminnyi, A. I., Tikhaze, A. K., Tsyplenkova,
V. G., Lankin, V. Z., Ruuge, E. K., and Kapel'ko, V. I. (2005)
Biochemistry (Moscow), 70, 79-84.
87.Madamanchi, N. R., Vendrov, A., and Runge, M. S.
(2005) Arterioscler. Thromb. Vasc. Biol., 25, 29-38.
88.Charatan, F. (2001) Br. Med. J.,
323, 359.
89.Lankin, V. Z., Tikhaze, A. K., Kaminnaya, V. I.,
Kaminnyi, A. I., Konovalova, G. G., and Kukharchuk, V. V. (2000)
Byul. Eksp. Biol. Med., 129, 176-179.
90.Lankin, V. Z., Tikhaze, A. K., Konovalova, G. G.,
Tutunov, V. S., Medvedeva, N. V., Kotkina, T. I., Kukharchuk, V. V.,
and Belenkov, Yu. N. (2002) Byul. Eksp. Biol. Med., 134,
39-42.
91.Lankin, V. Z., Konovalova, G. G., Tikhaze, A. K.,
Nezhdanova, I. B., Olfer'ev, A. M., and Kukharchuk, V. V. (2003)
Byul. Eksp. Biol. Med., 136, 42-45.
92.Nedosugova, L. V., Lankin, V. Z., Balabolkin, M.
I., Konovalova, G. G., Lisina, M. O., Antonova, K. V., Tikhaze, A. K.,
and Belenkov, Yu. N. (2003) Byul. Eksp. Biol. Med., 136,
132-134.