REVIEW: “Blue” Laccases
O. V. Morozova, G. P. Shumakovich, M. A. Gorbacheva, S. V. Shleev, and A. I. Yaropolov*
Bach Institute of Biochemistry, Russian Academy of Sciences, Leninskii pr. 33, 119071 Moscow, Russia; fax: (495) 954-2732; E-mail: yaropolov@inbi.ras.ru* To whom correspondence should be addressed.
Received April 28, 2007
This review concerns copper-containing oxidases--laccases. Principal biochemical and electrochemical properties of laccases isolated from different sources are described, as well as their structure and mechanism of catalysis. Possible applications of laccases in different fields of biotechnology are discussed.
KEY WORDS: laccase, molecular features, structure, bioelectrocatalysis, biotechnologyDOI: 10.1134/S0006297907100112
Abbreviations: ABTS) 2,2´-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt; DET) direct electron transfer; LMS) laccase mediator systems; NHE) normal hydrogen electrode.
Laccase (p-diphenol:oxygen oxidoreductase, EC 1.10.3.2) is a
copper-containing oxidase that catalyzes reduction of molecular oxygen
to water, bypassing a stage of hydrogen peroxide production [1-3].
Laccases are the most important components of the lignolytic complex of wood-destroying white rot fungi responsible for decomposition (but in some cases also synthesis) of lignin, which is one of the most widely distributed natural polymers. The role of laccase in these processes is not known in detail. These enzymes are also very interesting for studies on their structure and catalytic mechanisms. This interest is reasoned by structure of the laccase active site, which includes four copper ions of three types, and their coordinated interaction during catalysis of one-electron oxidation of donor substrates results in reduction of molecular oxygen to water. The substrate specificity of laccases is also very interesting because of their ability to catalyze oxidation of a wide variety of organic (especially aromatic) and inorganic compounds. Organic compounds are oxidized through a free radical mechanism, which is insufficiently studied for the majority of reactions.
DISTRIBUTION IN NATURE AND PHYSIOLOGICAL FUNCTIONS
Laccase was first isolated from sap of the lacquer tree Rhus vernicifera late in the XIXth century [4]. It is the best-studied laccase of plant origin [5-9]. Data on other higher plant laccases are scarce. Laccases from Rhus succedanea [5, 8], Acer pseudoplatanus [10], Pinus taeda [11, 12], Populus euramericana [13], Liriodendron tulipifera [14], Nicotiana tobacco [15], Lolium perenne [16], and Zea mays [17] are characterized partially.
The majority of laccases described in literature were isolated from higher fungi. Laccases have been found in many agents of soft rot, in the most of white rot-causing polypores, in geophilous saprophytic and phytopathogenic fungi (ascomycetes or their hyphomycetous anamorphas) and also in many agarics, including cultured edible mushrooms: the champignon Agaricus bisporus, oyster mushroom Pleurotus ostreatus, and the rice mushroom Lentinula edodes. There are other laccase producers, in particular actively wood-rotting fungi, such as Trametes (Coriolus) versicolor, T. hirsuta (C. hirsutus), T. ochracea, T. villosa, T. gallica, Cerrena maxima, Coriolopsis polyzona, Lentinus tigrinus, Pleurotus eryngii, etc.
Laccases are also found in saprophytic compost-inhabiting ascomycetes Myceliophthora thermophila [18] and Chaetomium thermophilum [19] and in the geophilous hyphomycete Mycelia sterlia INBI 2-26 [20]; they seem to be involved in humification. Laccases or laccase-encoding genes are identified in the grape pathogenic hyphomycete Botrytis cinerea [21] and the wheat and barley root rot agent Gaeumannomyces graminis var. tritici [22]. Interaction of the soft rot agent B. cinerea with host plants has been studied in [21], and extracellular laccase isoforms synthesized by this fungus have been shown to inactivate compounds produced by the host plant and toxic for this fungus.
Up to now functions of both plant and fungal laccases are not defined unambiguously [10, 23-27]. In plants, laccases are involved in synthesis of lignin and catalyze free radical polymerization of lignin structural units: p-coumaryl, coniferyl, and sinapyl alcohols. Fungal laccases are involved in degradation of lignin, pathogenesis and detoxification, and also in development and morphogenesis of fungi [24, 26, 27]. Formation of fungal carposomes is associated with laccase-catalyzed synthesis of extracellular pigments, which act as connecting elements on linking of fungal cell wall components. Laccase-lacking mutants of the oyster mushroom Pleurotis florida failed to form carposomes, as differentiated from revertants with the normal activity of the enzyme [28]. The laccase activity strictly depended on the phase of development of the fungus A. bisporus: the laccase activity sharply decreased upon the formation of carposomes [29].
The role of laccases during delignification is not clear in detail. Laccases can catalyze in vitro degradation of lignin, but the resulting low-molecular-weight components can repolymerize. Therefore, it was suggested that the main function of laccases is not the oxidation of lignin but influence on polymerization of their oxidation products. Enzymatic decomposition of lignin results in some toxic products that are dangerous for the fungal mycelium. Laccase detoxifies these low-molecular-weight phenolic components converting them to polymers nontoxic for fungal hyphae [30].
Findings of laccase activity in bacteria have been recently reported [31-37]. Some of these laccases can function in the presence of high concentrations of chloride and copper ions at neutral pH values. The enzyme isolated from Sinorhizobium meliloti is a dimeric protein with pI 6.2 consisting of two similar 45-kD subunits [38], whereas laccase from Pseudomonas putida is a monomeric 59-kD protein stable at pH 7.0 [39]. Both enzymes can oxidize syringaldazine.
LOCALIZATION IN THE CELL
Considering the substrate specificity, the enzymes involved in lignin degradation are exclusively extracellular. This is true for lignin peroxidase and Mn-peroxidase, but the situation with laccase is different. The majority of known fungal laccases are indeed extracellular proteins, but intracellular laccases of white rot fungi are also described. The majority of white rot fungi are shown to produce both intracellular and extracellular laccases [40], but the greater part of the enzyme (95%) is located outside the cell. Trace intracellular laccase activity was detected in the fungi A. bisporus, but 88% of the activity was found in the supernatant [29]. Both extracellular and intracellular laccases were found in Phanerochaete chrysosporium [41] and Suillus granulatus [42]. This localization of laccases seems to be associated with their physiological functions. Intracellular fungal laccases can be involved in transformation of low-molecular-weight phenolic compounds produced in the cell. Laccases located in the cell walls and spores can be involved in synthesis of melanin and other substances protecting the cell walls [43, 44].
MOLECULAR FEATURES OF LACCASES
To the present, more than a hundred laccases have been isolated and characterized. Based on the literature, general characteristics of these enzymes can be given with the reservation that the majority of laccases were isolated from wood-destroying white rot fungi, whereas plant laccases are much less studied.
Laccases are glycoproteins with molecular weight of 50-130 kD (Table 1). The carbohydrate moiety of the majority of laccases consists of mannose, N-acetylglucosamine, and galactose and constitutes about 45% of the protein mass in laccases of plant origin. Fungal laccases have lower carbohydrate contents (10-20%). In laccases from the basidiomycete P. eryngii the carbohydrate moieties are 1 and 7% [70], whereas in laccases from C. fulvocinerea [50] and B. cinerea [48] carbohydrate contents are 32 and 49%, respectively, i.e. are high for fungal laccases. Many researchers think the carbohydrate moiety of the molecule is responsible for the stability of the protein globule: deglycosylation of laccase from Ganoderma lucidum completely inhibited the activity [87]. The carbohydrate moiety has also been supposed to protect the enzyme molecule against proteolysis and inactivation by free radicals [24, 88].
Table 1. Some characteristics of laccases
isolated from different sources
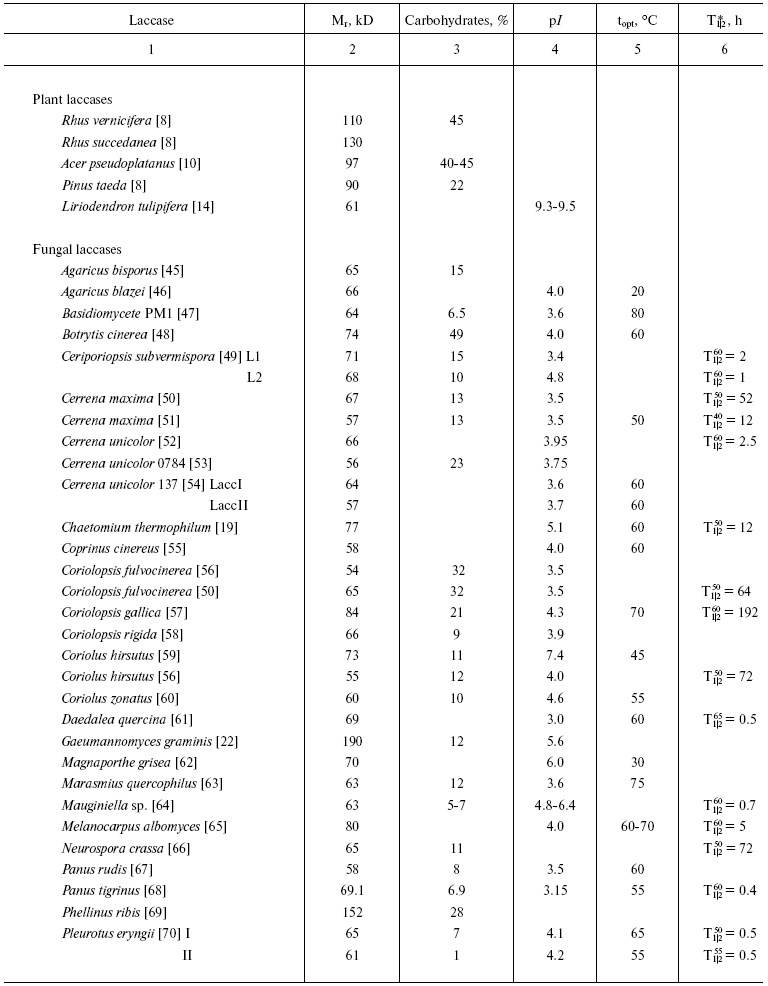
*Half-inactivation time of enzyme at the temperature
indicated.
Most fungi produce several isoforms of laccases. Thus, the fungus P. ostreatus secretes eight different laccase isozymes, six of which have been isolated and characterized (Table 1) [71-73]. The predominant isoform (POXC) is a 59-kD protein with pI 2.9; three isozymes (POXA2, POXB1, and POXB2) have the same molecular weight of about 67 kD, with pI 4.1 for two of them and pI 2.9 for the third. Two other isoforms of this protein (POXA1b and POXA1w) have molecular weight of 61 kD and pI 6.7-6.9, and two more isoforms (POXA3a and POXA3b) are heterodimers (pI 4.1-4.3) consisting of two subunits: of 61 kD and 16 or 18 kD. Formation of P. ostreatus isoenzymes is induced by copper ions in the culture medium and regulated on the level of gene transcription [73, 89]. Although laccase POXC is the predominant isozyme, the highest level of mRNA has been found for isoform POXA1b, which is mainly an intracellular or cell wall-bound protein [89].
The fungus T. pubescens produces two laccase isoforms with the same molecular weight of 67 kD and pI values of 5.1 and 5.3 [81], the fungus C. subvermispora produces four isoenzymes (68-71 kD, pI values from 3.4 to 4.7) [49], and the flat polypore G. lucidum produces five laccase isoforms with pI values of 3.0, 4.25, 4.5, 4.8, and 5.1 and molecular weights from 40 to 66 kD [87, 90]. The multiplicity of laccase isoforms seems to be associated with a variety of processes that they are involved in.
The number of isozymes found depends on conditions of cultivation, in particular on the presence of an inducer in the medium [44, 73, 91, 92]. The fungus P. pulmonarius produces three laccase isoforms, two of which (lcc1 and lcc2) are constitutive and the isoform lcc3 can be detected only when the fungus is cultured in the presence of inducers [91], whereas the fungus M. quercophilus strain 17 produces three constitutive and four inducible forms of the enzyme [93].
Although most of fungal laccases are monomeric proteins, enzymes consisting of several subunits are also known. Thus, laccases from the wood-rotting fungi P. ribis [69], P. pulmonarius [94], and T. villosa [83], the mycorrhyzal fungus Cantharellus cibarius [95], and the ascomycete R. solani [77] consist of two similar subunits with molecular weights typical for monomeric laccases. The above-mentioned fungus P. ostreatus produces two heteromeric laccase isoforms (POXA3a and POXA3b) [73]. Oligomeric laccases have been isolated from some ascomycetes. Thus, gel filtration shows the molecular weight of laccase from Monocillium indicum to be 100 kD, and SDS-electrophoresis reveals in it three subunits: 24, 56, and 72 kD [96]. The fungus G. gramminis produces an enzyme consisting of three 60-kD subunits [22], and a laccase from the ascomycete Podospora anserina is a tetramer consisting of 80-kD subunits [97].
STRUCTURE OF THE ACTIVE SITE OF LACCASES
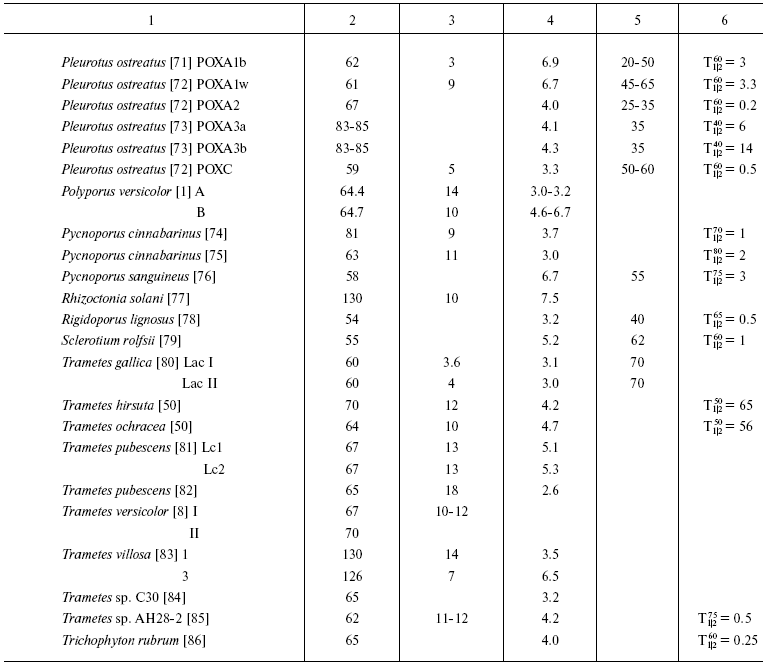
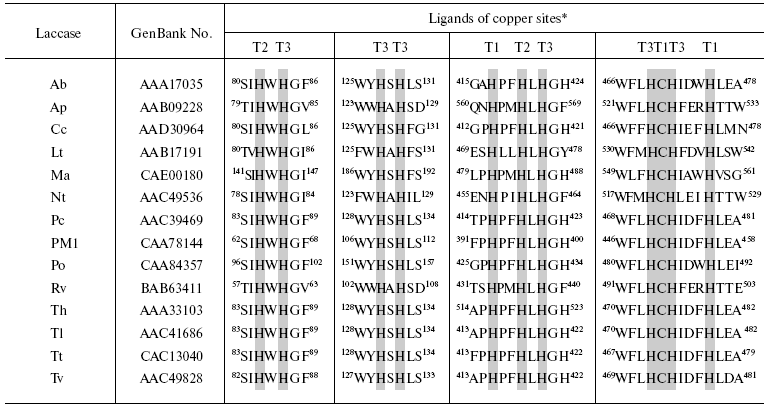
The active site of laccases contains four copper ions: a mononuclear “blue” copper ion (T1 site) and a three-nuclear copper cluster (T2/T3 site) consisting of one T2 copper ion and two T3 copper ions (Fig. 1). The distance between the T2 and T3 sites of the enzyme is 4 Å and the T1 copper ion is located at the distance of about 12 Å from them [98-100].
Copper ions of the laccase active site are classified by their spectral and EPR characteristics [8, 101, 102]. The T1 site of laccases imparts a light blue color to the enzyme solutions and is characterized by a distinctly pronounced band of optic absorption at the wavelength of 600 nm (epsilon ~ 5000 M-1·cm-1) and also by a weak parallel superfine splitting in the EPR spectra (g|| = 2.30, A|| = (40-95)·10-4 cm-1) [8, 103, 104]. The T1 site has as ligands two histidine imidazoles and the sulfhydryl group of cysteine, which form a trigonal structure. This copper ion can be replaced by mercury or cobalt ions [105-107].Fig. 1. Scheme of T1 (Cu1) and T2/T3 (Cu4/Cu2-Cu3) copper sites of laccase CotA from Bacillus subtilis, with indicated distances between the most important atoms [99].
The mononuclear T2 site of the enzyme, which is invisible in electron absorption spectra, displays ultrafine splitting in EPR spectra (g|| = 2.24, A|| = (140-200)·10-4 cm-1), which is typical for copper ions in tetragonal complexes [8, 103, 104]. The T2 site can be selectively removed from the enzyme molecule, and this is accompanied by a significant loss in the enzyme activity [108]. The T3 site of laccases is a binuclear copper site with copper ions paired antiferromagnetically through a hydroxide bridge that makes this site diamagnetic and prevents its detection in the EPR spectra. This site can be identified by the presence of a shoulder at 330 nm in the UV region of the spectrum [8, 109]. Eight imidazoles of histidine are ligands of the T2/T3 cluster [8, 110]. Figure 1 presents a scheme of the active site of laccase CotA from B. subtilis and positions of ligands of the T1, T2, and T3 sites, with indications of the interatomic distances [98].
Note that laccases with a differently structured active site are also described in the literature. The above-mentioned fungus P. ostreatus produces two laccase isoforms with the same molecular weight of 67 kD and virtually identical protein moieties which differ from other laccases only by the absence of activity toward guaiacol [71, 72]. The isoform POXA1b is induced by copper ions, has an absorption maximum at 605 nm, and contains four copper ions in the active site [71]. The other isoform (POXA1w) has no absorption maximum in the blue region of the spectrum [72] but has an absorption maximum at 400 nm, and its active site includes one copper ion, one iron ion, and two zinc ions [72]. The active site of laccase from Phlebia radiata is shown to contain two copper ions (T1 and T2 sites) and a prosthetic group PQQ (pyrroquinolinequinone), which seems to act as a T3 site [111]. The fungus P. ribis produces only one laccase which is a dimer consisting of two 76-kD subunits containing one copper ion, one manganese ion, and two zinc ions [69]. Enzymes lacking the maximum at 600 nm in the absorption spectrum are usually called laccase-like enzymes because they have the catalytic activity inherent in typical “blue” laccases. As a rule, it is not established whether water is the end product of dioxygen reduction by these enzymes.
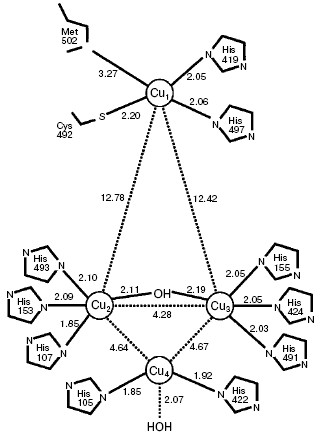
The three-dimensional structures have been determined for fungal laccases from C. cinereus (with the T2 copper removed) [112, 113], M. albomyces [114, 115], T. versicolor [116, 117], R. lignosus [100], and C. maxima [118], as well as the structure of laccase CotA from endospores of B. subtilis [99, 119]. Crystals have been prepared and a primary structural analysis performed for fungal laccases from P. cinnabarinus [120], C. hirsutus [121], and P. tigrinus [122]. All these laccases have pronounced structural homology, but there are also some differences, in particular in the organization of loops and formation of the substrate-binding “pocket”. Figure 2 presents the three-dimensional structure of laccase from C. maxima [118].
Laccase molecules are usually monomers consisting of three consecutively connected cupredoxin-like domains twisted in a tight globule. The size of the protein globule of laccases from C. cinnereus and T. versicolor is 70 × 50 × 45 Å and 65 × 55 × 45 Å, respectively [112, 117]. The T1 copper site of laccases is located in the third domain, and the three-nuclear cluster T2/T3 is located between the first and third domains and has amino acid ligands in each of them [112, 117, 118]. Amino acid residues of the second and third domains are involved in formation of the substrate-binding pocket (the binding site of electron donor substrate). The locations of disulfide bridges have been determined for the molecule of laccase from T. versicolor: one of them (8Cys-Cys488) connects the C-end with the first domain and the other (117Cys-Cys205) connects the first and second domains [117]. The structure of the laccase molecule from M. albomyces [114] is stabilized by three disulfide bridges: 4Cys-Cys12 in the first domain, 114Cys-Cys540 which binds the first and third domains, and 298Cys-Cys332 in the second domain close to the substrate-binding pocket. In the molecule of laccase from C. maxima, the alpha-helix on the C-end is stabilized by the disulfide bridge 85Cys-Cys488, whereas another disulfide bridge (117Cys-Cys205) connects the first and second domains [118].Fig. 2. Three-dimensional structure of laccase from Cerrena maxima [118].
Structures of laccases isolated from different sources are very much alike. On comparing sequences of more than 100 laccases, four conservative regions were revealed which are specific for all laccases [123, 124]. One cysteine and ten histidine residues form a ligand environment of copper ions of the laccase active site and are present in these four conservative amino acid sequences (Table 2).
Table 2. Comparison of amino acid sequences
of four different laccase regions that contain ligands of the active
site copper ions
*Ligands of the active site copper ions represented by histidine and
cysteine residues that are conservative for all laccases are shown in
gray color. Ab, A. bisporus lcc1 [125];
Ap, A. pseudoplatanus [10]; Cc, C.
cinereus lcc1 [126]; Lt, L. tulipifera
lac2-1 [14]; Ma, M. albomyces lac1 [127]; Nt, N. tabacum [15]; Pc, P. cinnabarinus lcc3-1 [128]; PM1, basidiomycete PM1 [129]; Po, P. ostreatus pox2 [130]; Rv, R. vernicifera [131]; Th, T. hirsuta [132]; Tl, T. villosa lcc1 [83]; Tt, T. trogii lcc1 [133]; Tv, T. versicolor lcc1 [134].
FACTORS DETERMINING REDOX POTENTIAL OF THE LACCASE T1 SITE
Standard redox potentials of three copper sites of laccases (T1, T2, T3) are key characteristics of copper-containing oxidases including laccases. Based on the T1 site redox potential, all copper-containing oxidases are subdivided onto high-, medium-, and low-potential enzymes (Table 3) [128, 140-142].
Table 3. Potentials of the T1 site of some
oxidases

While the T1 site potential is known for many laccases (Table 3), potentials of the T2 site are determined only for the plant laccase from R. vernicifera (390 mV) [102] and fungal laccase from T. hirsuta (400 mV) [143], and the T3 site potential is determined for laccases from R. vernicifera (460 mV) [102] and T. versicolor (785 mV) [137]. Fluoride ion has a strong influence on redox potential of the T3 site copper ion and slightly affects the potential of copper ions of the T1 and T2 sites [137-143]. In the presence of fluoride ion the potential of the T3 site of laccase from T. versicolor is 210 mV lower because of a strong interaction of fluoride ion with the T2/T3 cluster [137].
The T1 site potential of copper-containing enzymes was earlier thought to depend on the ligand environment of the type 1 copper ion and amino acid residues forming the substrate-binding pocket [132, 144]. High-potential laccases have a phenylalanine residue as an axial ligand of the T1 site copper; in medium- and low-potential laccases this role is played by leucine and methionine residue, respectively. However, results of direct mutagenesis of laccases from M. thermophila (Mt), R. solani (Rs), and T. villosa (Tv) [136, 144] have shown that the ligand environment of the T1 copper ion does not have a very strong influence on its potential. Mutants in the axial ligand of the T1 site (Mt-L513F, Rs-L470F, Tv-F463L, Tv-F463M) and triple mutants of laccases (Mt-V509L/S510E/G511, Rs-L466V/E467S/A468G) from M. thermophila, R. solani, and T. villosa were obtained, and their spectroscopic and catalytic features were determined. The substitution of leucine by phenylalanine in the axial ligand position in laccases from M. thermophila and R. solani had a slight influence on the T1 site potential and kinetics of substrate oxidation. The triple mutation also slightly changed the T1 copper ion potential, but strongly influenced catalytic properties of the enzyme (the kcat value was markedly decreased on oxidation of syringaldazine and ABTS (2,2´-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt) and fluoride ion-caused inhibition and changed the pH profile of the activity [144]. The substitution of axial phenylalanine residue by leucine in laccase from T. villosa decreased the potential of the copper ion by 50 mV, and the substitution by methionine decreased it by 110 mV. But the potential of the T. villosa-F463M mutant (680 mV) remained much higher than the T1 site potential of laccase from R. vernicifera (430 mV). However, the mutation T. villosa-F463M increased fivefold the Km value for syringaldazine and 38-fold for ABTS, along with a simultaneous 1.7-fold increase in the kcat value. It seemed that this mutation in laccase from T. villosa changed the state of the T1 site copper ion and affected the substrate-binding pocket, increasing the Km value [136]. Thus the axial ligand of the T1 copper ion was shown to have no significant effect on redox potential of the T1 site of laccases.
It was recently suggested that an increase in the length of the Cu-N bond should affect the T1 site redox potential because of a decreased contribution of the free electron pair of the nitrogen atom [117]. The short alpha-helix (residues 455-461) which contains the ligand His458 of the T1 copper in the high-potential laccase from T. versicolor is farther from the copper ion compared to its location in the medium-potential laccase from C. cinereus, and this is likely to be caused by formation of a hydrogen bond between Glu460 and Ser113 of the first domain. Owing to the formation of this hydrogen bond, the whole His458-containing helix is shifted towards the first domain and, thus, increases the distance between the atoms Cu-N. In the laccase from C. cinereus the position of Glu460 is occupied by methionine, which is unable to produce a hydrogen bond. Possibly, just production of this hydrogen bond (Ser-Glu) markedly influences the T1 copper ion potential. Comparison of available amino acid sequences of laccases and their redox potentials confirms this hypothesis: laccases which contain glutamic acid and serine in positions corresponding to Glu460 and Ser113 in laccase from T. versicolor are high-potential ones [117, 140].
The T1 site of the enzyme is the primary acceptor of electrons from reducing substrates. Laccases can directly oxidize only compounds with ionization potential not higher or slightly higher than the redox potential of the T1 site copper ion of the enzyme [145]. The potential of the enzyme T1 site also determines the efficiency of catalysis on oxidation of the majority of laccase substrates, and this makes laccases with the high-potential T1 site promising for biotechnology.
The range of compounds oxidized with involvement of laccases can be enlarged using so-called redox mediators, which are laccase substrates producing high-potential intermediates as a result of enzymatic oxidation. Under diffusion-controlled conditions, these intermediates can chemically (not enzymatically) react with other compounds, which cannot be oxidized with involvement only of laccases. The oxidized mediator is reduced to the initial state by the compound subjected to oxidation, and thus a closed cycle is created [146, 147].
CATALYTIC PROPERTIES OF LACCASES AND MECHANISM OF
CATALYSIS
Reduction of dioxygen. Notwithstanding many works concerning blue copper-containing oxidases including laccases, there is no general opinion about the electron transfer pathway inside the protein globule and the mechanism of dioxygen reduction in the molecule. The T1 site of laccases is thought to accept electrons from reducing substrates, and then they are transferred onto the three-nuclear T2/T3 cluster where molecular oxygen is activated and reduced to water [8, 148, 149].
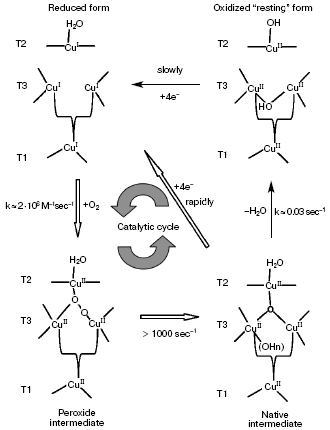
Interaction of a completely reduced laccase with molecular oxygen results in different forms of the enzyme. Two well-studied forms are termed peroxide intermediate and native intermediate [3, 104, 149, 150]. The native intermediate plays an important role in the catalytic cycle of laccase [1]. During reaction with 17O2, this intermediate acts as an oxygen radical. By EPR, all four copper ions in the native intermediate were shown to be oxidized [98]. The structure of this intermediate is different from that of the “resting” enzyme with the T2 site copper ion not connected with ions of the T3 site.
Figure 3 shows a four-electron reduction of a dioxygen molecule to water with involvement of laccase [151]. The completely reduced three-nuclear T2/T3 copper cluster of laccase interacts with molecular oxygen producing the peroxide intermediate. On using laccase with the T1 copper ion substituted by the redox-inactive mercury ion Hg2+, a peroxide bridge is generated between the reduced T2 and oxidized T3 sites [98, 152]. This intermediate changes to a compound similar to the native intermediate of laccase with four copper ions in the active site. Production of the peroxide intermediate (in T1-Hg-laccase) and the native intermediate (in the native enzyme) depends on the dioxygen concentration, and the second order rate constants of these reactions are comparable. The peroxide intermediate was supposed to contain the ion O22-, with one of its oxygen atoms bound with the T2 and T3 copper ions and the other atom coordinated with another copper ion of T3 [150, 151].
Splitting of the peroxide O-O bond on transition from the peroxide to native intermediate includes a mechanism of proton and electron transfer which is not clear in detail. The activation energy of this process is found to be ~9 kcal/mol [98], and this is in agreement with a mechanism that includes a one-electron reducing decomposition of peroxide.Fig. 3. Catalytic cycle of laccase showing the mechanism of reduction and oxidation of the enzyme copper sites [151].
In turn, the native intermediate can slowly transform to a completely oxidized form, which is often termed “resting” laccase. The T1 site of this enzyme form can be reduced by a substrate, but the rate of electron transfer onto the three-nuclear copper cluster is too low to be significant for catalysis [3, 8].
Oxidation of reducing substrates. Types of substrates oxidized by laccases. Laccases catalyze oxidation of a wide variety of organic compounds including phenols, methoxy-substituted phenols, aminophenols, diamines, and also some inorganic ions such as [Mo(CN)8]4-, [Fe(CN)6]4-, [Os(CN)6]4-, and [W(CN)8]4- [1, 2, 8, 24, 153, 154]. In some works laccases were shown to oxidize Mn2+ in the presence of different chelators [155-157].
Values of Km and kcat of different laccases widely vary for the same substrate. The majority of laccases combine a high affinity for ABTS and syringaldazine with a high catalytic constant, whereas guaiacol and 2,6-dimethoxyphenol are oxidized markedly slower and the corresponding Michaelis constants are significantly higher. Low values of Km are specific for hydroquinone and sinapic and syringic acids, whereas relatively high values of Km are found for p-substituted phenols, vanillic acid, and vanillic aldehyde. Laccases capable of oxidizing polycyclic hydrocarbons or pentachlorophenols are described, but catalytic constants have been calculated only for some of them, e.g. laccase from T. versicolor [26]. Note that it is difficult to compare catalytic characteristics of laccases isolated from different sources because of different experimental conditions in different experiments.
In work [50] laccases isolated from culture fluids of the fungi T. hirsuta, T. ochracea, C. fulvocinerea, and C. maxima were comprehensively studied under the same conditions, and kinetic parameters of reactions catalyzed by these laccases on oxidation of some organic substrates and potassium ferrocyanide were determined. The Km values of laccases from T. hirsuta, T. ochracea, C. fulvocinerea, and C. maxima for pyrocatechin were 142, 110, 85, and 120 µM; for guaiacol 63, 90, 70, and 160 µM; for sinapic acid 24, 11, 21, and 24 µM; and for potassium ferrocyanide 180, 96, 170, and 115 µM, respectively. On oxidation of organic substrates, laccase from T. hirsuta had the highest catalytic constants among the enzymes. But laccase from C. maxima displayed the highest value of kcat on oxidation of an inorganic substrate, K4Fe(CN)6. The lowest values of Km for all four enzymes have been recorded on oxidation of sinapic acid, which is a model lignin compound. It seems that the substituent (-CH=CH-COOH) in the para-position with respect to the OH-group of the benzene ring has a negative mezomeric effect that enhances the efficiency of catalysis by nearly an order of magnitude.
Some fungi produce a number of laccase isoforms with similar values of Km and kcat for different substrates. Thus, the basidiomycete T. gallica produces two enzymes (Lac I and Lac II), and the Km values of Lac I and Lac II on oxidation of ABTS, 2,6-dimethoxyphenol, and guaiacol are 1.2, 420, and 405 µM and 0.9, 410, and 400 µM, respectively [80]. Two laccase forms (Lc1 and Lc2) were isolated from culture fluid of the basidiomycete T. pubescens with similar values of kcat for such substrates as hydroquinone, pyrocatechin, ABTS, and K4Fe(CN)6 [81]. The above-mentioned fungus P. ostreatus produces a number of isoenzymes, with very different values of kcat: from 230 µM for POXC to 14,000 µM for POXA3 with 2.6-dimethoxyphenol used as the donor substrate. Moreover, only two enzymes (POXA2 and POXC) could oxidize guaiacol [72, 73].
The optimum pH for the majority of fungal laccases is in the range from 3.5 to 5.0 when organic donors of hydrogen atoms are used as substrates, and the pH-dependence curve is bell-shaped [9, 51, 55, 59, 61, 77, 88, 158, 159]. Such a profile of the laccase activity on oxidation of phenolic compounds is caused by two effects. On one hand, with increase in pH of the solution ionization potential of phenolic compounds decreases as a result of production of phenolate anion (DeltaE/DeltapH = 59 mV at 25°C) that has to enhance the rate of the enzymatic reaction. On the other hand, with increase in pH of the solution the rate of laccase-catalyzed enzymatic reactions decreases at the cost of OH--ion binding with the T2/T3 site of the enzyme [145].
However, during oxidation of substrates which are electron donors (e.g. K4Fe(CN)6, ABTS) the activity of laccases monotonously decreases with changes in the solution pH from 2.5 to 7.0 [49, 61, 68, 77, 159]. This occurs because protons are not involved in oxidation of such substrates and the influence of pH on redox potential of such compounds is minimal. In this case, the decrease in the enzyme activity is associated with binding of the hydroxide ion with the T2/T3 site of the enzyme.
The pH optimum of laccase from lacquer tree for substrates that are donors of hydrogen atoms was different from that of fungal laccases: laccase from Rhus vernicifera displayed maximal activity in neutral and weakly alkaline solutions. For electron donor substrates, the pH profile of the activity was similar to that of fungal laccases [5, 6, 9].
The temperature optimum of the majority of enzymes is in the limits of 50-70°C (Table 1), although the activity of laccase from G. lucidium is maximal at 25°C [87, 90]. The half-inactivation time of laccases varies from a few minutes at 50°C for laccase from B. cinnerea to 3 h at 75°C for laccase from Pycnoporus sanguineus [76].
The activity of laccases is inhibited by various organic and inorganic compounds [2, 160]. Small inorganic anions, such as fluoride, chloride, azide, and hydroxide anions bind with the T2/T3 site of laccases, and this prevents the electron transfer from the T1 site of the enzyme onto the T2/T3 cluster and inhibits the enzymatic activity [154, 160]. Such compounds as dithiothreitol and thioglycolic and ethyldithiocarbamic acids also are inhibitors of laccases [19, 48, 96]. However, in the work [161] some substances described as laccase inhibitors (dithiothreitol, thioglycolic and ethyldithiocarbamic acids, cysteine, and sodium azide) were tested in reactions of ABTS and 2,6-dimethoxyphenol oxidation using laccase from T. versicolor. Only azide ion was a true inhibitor of laccase when either substrate was used. All the other compounds did not inhibit the laccase activity, but the earlier described inhibitory effects were caused by reduction of the reaction products, such as ABTS+* and quinone, or by subsequent nonenzymatic interactions during oxidation of the substrate, which resulted in an apparent decrease in the laccase activity.
ELECTROCATALYTIC PROPERTIES OF LACCASES
Bioelectrocatalysis is a term for reactions accelerating electrochemical processes. Bioelectrocatalysis is can be a direct and or a mediated coupling of enzymatic catalysis and electrochemical reaction. The concept of “bioelectrocatalysis” was introduced in the 1970s in a description of a direct mediator-free biocatalysis by redox enzymes. This phenomenon was later termed direct electron transfer (DET) [162, 163]. Direct mediator-free electrocatalysis is based on the possibility of immediate electron exchange between the active site of the enzyme and an electrode in the absence of mediator [162, 164]. Note that during this reaction the electrode acts as a “substrate” of the enzyme. Bioelectrocatalysis by the DET mechanism was shown for the first time for laccase from T. versicolor [162]. A carbonaceous electrode with laccase immobilized on its surface was the electron donor, and dioxygen was electroreduced to water through a four-electron mechanism. The mechanism of this reaction is similar to that of an enzymatic reaction with involvement of low-molecular-weight compounds as electron donors.
The normal oxygen potential of 1.23 V for the pair O2/H2O can be obtained only under special conditions, with a preliminarily prepared platinum electrode and in especially pure solutions [165]. A stationary potential established in the oxygen atmosphere on carbonaceous electrodes is far from the equilibrium oxygen potential and is ~0.6-0.7 V. Addition into solution of small amounts of laccase caused a displacement of the electrode potential to positive values and accelerated oxygen electroreduction within the potential range of 0.6-1.2 V.
Stationary potentials of electrodes from different materials (gold, pyrographite, glass-carbon, and soot) were studied in the presence and absence of laccase under aerobic and anaerobic conditions [162]. In the presence of laccase, high values of stationary potentials were established only in the oxygen atmosphere. The stationary potential value and currents at this potential increased only with increase in the amount of adsorbed enzyme. The maximal value of potential +1.207 V (versus normal hydrogen electrode (NHE)) in the same solution (when pH values of the operating and reference electrodes were equal) was close to the equilibrium oxygen potential and established on soot electrodes, which were preliminarily kept in laccase solution for 24 h. The enzyme adsorption of soot electrodes was virtually irreversible.
The enzymatic nature of electrocatalysis was proved by a specific inhibition of electrocatalytic effects by fluoride or azide ions and enzyme inactivation by heating [140, 166]. Using a disk-ring electrode, it was shown that in the potential range of 0.6-1.2 V, where oxygen was reduced on the electrode with adsorbed laccase, no hydrogen peroxide was produced as an intermediate product of the reaction [163].
The rate of oxygen electroreduction on laccase-modified electrodes depended on the electrode material, mode of its preparation, and partial oxygen pressure in the system [140, 141, 163, 167].
Electroreduction of molecular oxygen on electrodes made of highly oriented pyrolytic graphite with adsorbed laccase from T. versicolor was studied in [168]. The starting potential for laccase to catalyze electroreduction of O2 (about 735 mV versus NHE) was close to the redox potential of the enzyme T1 site (780 mV versus NHE).
Notwithstanding multiple publications concerning direct (mediator-free) electron transfer, the molecular mechanism of DET between electrode and laccase is not clear in detail. The phenomenon of bioelectrocatalysis was used as a basis for creating biosensors and biofuels [140, 169-179].
POSSIBLE APPLICATIONS OF LACCASES IN BIOTECHNOLOGY
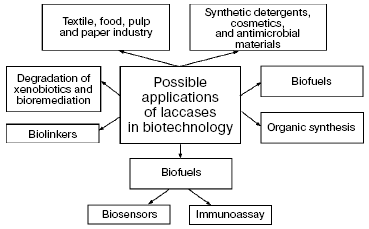
Catalytic and electrocatalytic properties of laccases pose the possibility of their wide application in pulp and paper, textile, and cosmetic industries, for detoxification and decoloration of sewage, in organic synthesis, for degradation of xenobiotics and bioremediation, to create antimicrobial compositions, in production of wood-fiber plates, wood blocks, and cardboard without using toxic linkers, for production of detergents, and in elaboration of biosensors and cathodes of biofuels [2, 23, 30, 154, 180-187] (Fig. 4).
Application of laccases in practice is based on two mutually related lines: a search for enzymes with new physicochemical properties and also for highly effective intensifiers and promoters of the action of these enzymes. All approaches and prospects of application of laccases in biotechnology are based on their ability to produce free radicals during oxidation of different substrates.Fig. 4. Possible applications of laccases in biotechnology.
There are attempts at using redox enzymes, including laccases, for delignification of lignocellulose and modification of lignocellulose materials to impart new features to them [180, 181, 188]. At present delignification of paper pulp with laccase-mediator systems (LMS) is realized on the level of pilot setups. Application of crude laccase preparations combined with different mediators for bleaching flax fiber seems promising, especially in Russia [189].
Laccases are also very promising for application in food industry for stabilization and improvement of quality of different drinks and deoxygenation of perishable products containing plant oils. Prospects of laccase applications in food industry are described in reviews [23, 183, 186, 190, 191].
The phenomenon of direct electron transfer from an electrode to laccase molecules was used for creation of a biofuel cathode [172-174]. Based on this phenomenon, microbiofuels were created during recent years using glucose oxidase (anode) and laccase or bilirubin oxidase (cathode) [192, 193].
Amperometric biosensors based on laccases have been described for analysis of different phenolic compounds (chloro-substituted phenols, catecholamines, lignin, tea tannins) and also ascorbic acid [175, 177, 178, 194-197]. In such biosensors a Clark-type oxygen or a carbonaceous electrode is used. These biosensors function in a flow-injection regimen or in a system with stirring. To enlarge the range of phenolic compounds to be analyzed, a biosensor has been created on the base of co-immobilized laccase and tyrosinase [169].
Laccase from the fungus C. hirsutis (T. hirsuta) can be used as a marker enzyme for analysis of the pesticide 2,4-D [198]. As differentiated from peroxidase, which has hydrogen peroxide as another substrate, the second substrate of laccase is air oxygen, and this makes the analytic procedure much easier and allows the standard equipment for ELISA to be used. On the base of DET a pseudo-free of reagents immunosensor has been elaborated, with laccase as a marker enzyme [171]. Laccase-based highly sensitive systems are proposed for analysis of narcotic-containing compounds and biogenic amines [199-201].
Bioremediation includes all procedures directed for biotransformation of a pollution-changed environment in order to recover its initial state. For this purpose, laccases (and LMS) are very promising owing to their wide substrate specificity and ability to degrade many phenolic compounds [154, 184, 202-205]. Oxidation of toxic substrates by laccase with production of insoluble products and the subsequent separation of the precipitate is a promising approach for purification of industrial sewage.
Organophosphorus compounds (in particular, various insecticides and neuroparalytic poisons) are very toxic and cannot be sufficiently hydrolyzed nonenzymatically. Purified laccase from the fungus P. ostreatus in the presence of ABTS completely and at high rate oxidatively destroyed neuroparalytic VX gases and the insecticide diisopropylamiton [206].
Laccase can be also used in fine organic synthesis, e.g. for oxidation of hydroxyl groups of sugar derivatives [207, 208], vinblastin synthesis from vindolin [2], free radical polymerization on polysulfostyrene matrix resulting in electroconducting water-soluble polyaniline [209].
Thus, laccases are very interesting for both fundamental studies on their structure and catalytic mechanism and application in biotechnology.
REFERENCES
1.Reinhammar, B. (1984) in Copper Proteins and
Copper Enzymes, Vol. 3 (Lontie, R., ed.) CRC Press, Boca Raton, pp.
1-35.
2.Yaropolov, A. I., Skorobogat'ko, O. V., Vartanov,
S. S., and Varfolomeev, S. D. (1994) Appl. Biochem. Biotechnol.,
49, 257-280.
3.Lee, S.-K., George, S. D., Antholine, W. E.,
Hedman, B., Hodgson, K. O., and Solomon, E. I. (2002) J. Am. Chem.
Soc., 124, 6180-6193.
4.Yoshida, H. (1883) J. Chem. Soc., 43,
472-486.
5.Omura, T. (1961) J. Biochem. (Tokyo),
50, 264-272.
6.Holwerda, R. A., and Gray, H. B. (1975) J. Am.
Chem. Soc., 97, 6036-6041.
7.Morpurgo, L., Graziani, M. T., Finazzi-Agro, A.,
Rotilio, G., and Mondovµ, B. (1980) Biochem. J.,
187, 361-366.
8.Solomon, E. I., Sundaram, U. M., and Machonkin, T.
E. (1996) Chem. Rev., 96, 2563-2606.
9.Shiba, T., Xiao, L., Miyakoshi, T., and Chen, C.-L.
(2000) J. Mol. Catal. B: Enzym., 10, 605-615.
10.Sterjiades, R., Dean, J. F. D., and Eriksson,
K.-E. L. (1992) Plant Physiol., 99, 1162-1168.
11.Bao, W., O'Malley, D. M., Whetten, R., and
Sederoff, R. R. (1993) Science, 260, 672-674.
12.Sato, Y., Wuli, B., Sederoff, R., and Whetten, R.
(2001) J. Plant Res., 114, 147-155.
13.Ranocha, P., McDougall, G., Hawkins, S.,
Sterjiades, R., Borderies, G., Stewart, D., Cabanes-Macheteau, M.,
Boudet, A.-M., and Goffner, D. (1999) Eur. J. Biochem.,
259, 485-495.
14.LaFayette, P. R., Eriksson, K.-E. L., and Dean,
J. F. D. (1999) Plant Mol. Biol., 40, 23-35.
15.Kiefer-Meyer, M. C., Gomord, V., O'Connell, A.,
Halpin, C., and Faye, L. (1996) Gene, 178, 205-207.
16.Gavnholt, B., Larsen, K., and Rasmussen, S. K.
(2002) Plant Sci., 162, 873-885.
17.Caparros-Ruiz, D., Fornale, S., Civardi, L.,
Puigdomènech, P., and Rigau, J. (2006) Plant Sci.,
171, 217-225.
18.Berka, R. M., Schneider, P., Golightly, E. J.,
Brown, S. H., Madden, M., Brown, K. M., Halkier, T., Mondorf, K., and
Xu, F. (1997) Appl. Environ. Microbiol., 63,
3151-3157.
19.Chefetz, B., Chen, Y., and Hadar, Y. (1998)
Appl. Environ. Microbiol., 64, 3175-3179.
20.Vasil'chenko, L. G., Koroleva, O. V., Stepanova,
E. V., Landesman, E. O., and Rabinovich, M. L. (2000) Prikl.
Biokhim. Mikrobiol., 36, 412-421.
21.Bar-Nun, N., and Mayer, A. M. (1989)
Phytochemistry, 28, 1369-1371.
22.Edens, W. A., Goins, T. Q., Dooley, D., and
Henson, J. M. (1999) Appl. Environ. Microbiol., 65,
3071-3074.
23.Mayer, A. M., and Staples, R. C. (2002)
Phytochemistry, 60, 551-565.
24.Thurston, C. F. (1994) Microbiology,
140, 19-26.
25.Gavnholt, B., and Larsen, K. (2002)
Physiol. Plant., 116, 273-280.
26.Baldrian, P. (2006) FEMS Microbiol. Rev.,
30, 215-242.
27.Leonowicz, A., Cho, N.-S., Luterek, J.,
Wilkolazka, A., Wojtas-Wasilewska, M., Matuszewska, A., Hofrichter, M.,
Wesenberg, D., and Rogalski, J. (2001) J. Basic Microbiol.,
41, 185-227.
28.Das, N., Sengupta, S., and Mukherjee, M. (1997)
Appl. Environ. Microbiol., 63, 4120-4122.
29.Wood, D. A. (1980) J. Gen. Microbiol.,
117, 339-345.
30.Bolobova, A, V., Askadskii, A. A.,
Kondrashchenko, V. I., and Rabinovich, M. L. (2002) Theoretical
Principles of Technology of Wood Composites. Book II. Enzymes,
Models, Processes [in Russian], Nauka, Moscow.
31.Alexandre, G., and Zhulin, I. B. (2000) Trends
Biotechnol., 18, 41-42.
32.Diamantidis, G., Effosse, A., Potier, P., and
Bally, R. (2000) Soil Biol. Biochem., 32, 919-927.
33.Hullo, M.-F., Moszer, I., Danchin, A., and
Martin-Verstraete, I. (2001) J. Bacteriol., 183,
5426-5430.
34.Martins, L. O., Soares, C. M., Pereira, M. M.,
Teixeira, M., Costa, T., Jones, G. H., and Henriques, A. O. (2002)
J. Biol. Chem., 277, 18849-18859.
35.Claus, H. (2003) Arch. Microbiol.,
179, 145-150.
36.Enguita, F. J., Marçal, D., Martins, L.
O., Grenha, R., Henriques, A. O., Lindley, P. F., and Carrondo, M. A.
(2004) J. Biol. Chem., 279, 23472-23476.
37.Ruijssenaars, H. J., and Hartmans, S. (2004)
Appl. Microbiol. Biotechnol., 65, 177-182.
38.Rosconi, F., Fraguas, L. F., Martinez-Drets, G.,
and Castro-Sowinski, S. (2005) Enzyme Microb. Technol.,
36, 800-807.
39.McMahon, A. M., Doyle, E. M., Brooks, S., and
O'Connor, K. E. (2007) Enzyme Microb. Technol., 40,
1435-1441.
40.Blaich, R., and Esser, K. (1975) Arch.
Microbiol., 103, 271-277.
41.Dittmer, J. K., Patel, N. J., Dhawale, S. W., and
Dhawale, S. S. (1997) FEMS Microbiol. Lett., 149,
65-70.
42.Gunther, Th., Perner, B., and Gramss, G. (1998)
J. Basic Microbiol., 38, 197-206.
43.Eggert, C., Temp, U., Dean, J. F. D., and
Eriksson, K.-E. L. (1995) FEBS Lett., 376, 202-206.
44.Galhaup, C., and Haltrich, D. (2001) Appl.
Microbiol. Biotechnol., 56, 225-232.
45.Perry, C. R., Matcham, S. E., Wood, D. A., and
Thurston, C. F. (1993) J. Gen. Microbiol., 139,
171-178.
46.Ullrich, R., le Huong, M., Dung, N. L., and
Hofrichter, M. (2005) Appl. Microbiol. Biotechnol., 67,
357-363.
47.Coll, P. M., Fernández-Abalos, J. M.,
Villanueva, J. R., Santamaría, R., and Perez, P. (1993) Appl.
Environ. Microbiol., 59, 2607-2613.
48.Slomczynski, D., Nakas, J. P., and Tanenbaum, S.
W. (1995) Appl. Microbiol. Biotechnol., 61, 907-912.
49.Fukushima, Y., and Kirk, T. K. (1995) Appl.
Environ. Microbiol., 61, 872-876.
50.Shleev, S. V., Morozova, O. V., Nikitina, O. V.,
Gorshina, E. S., Rusinova, T. V., Serezhenkov, V. A., Burbaev, D. S.,
Gazaryan, I. G., and Yaropolov, A. I. (2004) Biochimie,
86, 693-703.
51.Koroleva, O. V., Yavmetdinov, I. S., Shleev, S.
V., Stepanova, E. V., and Gavrilova, V. P. (2001) Biochemistry
(Moscow), 66, 618-622.
52.Bekker, E. G., Petrova, S. D., Ermolova, O. V.,
Elisashwili, V. I., and Sinitsyn, A. P. (1990) Biokhimiya,
55, 2019-2024.
53.Stepanova, E. V., Pegasova, T. V., Gavrilova, V.
P., Landesman, E. O., and Koroleva, O. V. (2003) Prikl. Biokhim.
Mikrobiol., 39, 427-434.
54.Michniewicz, A., Ullrich, R., Ledakowicz, S., and
Hofrichter, M. (2006) Appl. Microbiol. Biotechnol., 69,
682-688.
55.Schneider, P., Caspersen, M. B., Mondorf, K.,
Halkier, T., Skov, L. K., Ostergaard, P. R., Brown, K. M., Brown, S.
H., and Xu, F. (1999) Enzyme Microb. Technol., 25,
471-545.
56.Smirnov, S. A., Koroleva, O. V., Gavrilova, V.
P., Belova, A. B., and Klyachko, N. L. (2001) Biochemistry
(Moscow), 66, 774-779.
57.Calvo, A. M., Copa-Patino, J. L., Alonso, O., and
Gonzalez, A. E. (1998) Arch. Microbiol., 171, 31-36.
58.Saparrat, M. C. N., Guillen, F., Arambarri, A.
M., Martinez, A. T., and Martinez, M. J. (2002) Appl. Environ.
Microbiol., 68, 1534-1540.
59.Shin, K.-S., and Lee, Y.-J. (2000) Arch.
Biochem. Biophys., 384, 109-115.
60.Koroljova, O., Stepanova, E., Gavrilova, V.,
Biniukov, V., Jaropolov, A., Varfolomeyev, S., Scheller, F., Makower,
A., and Otto, A. (1999) Appl. Biochem. Biotech., 76,
115-128.
61.Baldrian, P. (2004) Appl. Microbiol.
Biotechnol., 63, 560-563.
62.Iyer, G., and Chattoo, B. B. (2003) FEMS
Microbiol. Lett., 227, 121-126.
63.Dedeyan, B., Klonowska, A., Tagger, S., Tron, T.,
Iacazio, G., Gil, G., and Le Petit, J. (2000) Appl. Environ.
Microbiol., 66, 925-929.
64.Froehner, S. C., and Eriksson, K.-E. (1974) J.
Bacteriol., 120, 458-465.
65.Kiiskinen, L.-L., Viikari, L., and Kruus, K.
(2002) Appl. Microbiol. Biotechnol., 59, 198-204.
66.Froehner, S. C., and Eriksson, K.-E. (1974) J.
Bacteriol., 120, 458-465.
67.Zhang, M., Wu, F., Wei, Z., Xiao, Y., and Gong,
W. (2006) Enzyme Microb. Technol., 39, 92-97.
68.Quaratino, D., Federici, F., Petruccioli, M.,
Fenice, M., and D'Annibale, A. (2007) Antonie Van Leeuwenhoek,
91, 57-69.
69.Min, K.-L., Kim, Y.-H., Kim, Y.
W., Jung, H. S., and Hah, Y. C. (2001) Arch. Biochem.
Biophys., 392, 279-286.
70.Munoz, C., Guillen, F., Martinez, A. T., and
Martinez, M. J. (1997) Appl. Environ. Microbiol., 63,
2166-2174.
71.Giardina, P., Palmieri, G., Scaloni, A.,
Fontanella, B., Faraco, V., Cennamo, G., and Sannia, G. (1999)
Biochem. J., 341, 655-663.
72.Palmieri, G., Giardina, P., Bianco, C., Scaloni,
A., Capasso, A., and Sannia, G. (1997) J. Biol. Chem.,
272, 31301-31307.
73.Palmieri, G., Cennamo, G., Faraco, V., Amoresano,
A., Sannia, G., and Giardina, P. (2003) Enzyme Microb. Technol.,
33, 135-325.
74.Eggert, C., Temp, U., and Eriksson, K. E. (1996)
Appl. Environ. Microbiol., 62, 1151-1158.
75.Schliephake, K., Mainwaring, D. E., Lonergan, G.
T., Jones, I. K., and Baker, W. L. (2000) Enzyme Microb.
Technol., 27, 100-107.
76.Litthauer, D., van Vuuren, M. J., van Tonder, A.,
and Wolfaardt, F. W. (2007) Enzyme Microb. Technol., 40,
563-568.
77.Wahleithner, J. A., Xu, F., Brown, K. M., Brown,
S. H., Golightly, E. J., Halkier, T., Kauppinen, S., Pederson, A., and
Schneider, P. (1996) Curr. Genet., 29, 395-403.
78.Cambria, M. T., Cambria, A., Ragusa, S., and
Rizzarelli, E. (2000) Protein Exp. Purif., 18,
141-147.
79.Ryan, S., Schnitzhofer, W., Tzanov, T.,
Cavaco-Paulo, A., and Gubitz, G. M. (2003) Enzyme Microb.
Technol., 33, 766-774.
80.Dong, J. L., and Zhang, Y. Z. (2004) Prep.
Biochem. Biotech., 34, 179-194.
81.Nikitina, O. V., Shleev, S. V., Gorshina, E. S.,
Rusinova, T. V., Serezhenkov, V. A., Burbaev, D. Sh., Belovolova, L.
V., and Yaropolov, A. I. (2005) Biochemistry (Moscow),
70, 1274-1279.
82.Galhaup, C., Goller, S., Peterbauer, C. K.,
Strauss, J., and Haltrich, D. (2002) Microbiology, 148,
2159-2169.
83.Yaver, D. S., Xu, F., Golightly, E. J., Brown, K.
M., Brown, S. H., Rey, M. W., Schneider, P., Halkier, T., Mondorf, K.,
and Dalboge, H. (1996) Appl. Environ. Microbiol., 62,
834-841.
84.Klonowska, A., Gaudin, C., Fournel, A., Asso, M.,
Le Petit, J., Giorgi, M., and Tron, T. (2002) Eur. J. Biochem.,
269, 6119-6125.
85.Xiao, Y. Z., Tu, X. M., Wang, J., Zhang, M.,
Cheng, Q., Zeng, W. Y., and Shi, Y. Y. (2003) Appl. Microbiol.
Biotechnol., 60, 700-707.
86.Jung, H., Xu, F., and Li, K. (2002) Enzyme
Microb. Technol., 30, 161-168.
87.Ko, E.-M., Leem, Y.-E., and Choi, H. T. (2001)
Appl. Microbiol. Biotechnol., 57, 98-102.
88.Yoshitake, A., Katayama, Y., Nakamura, M.,
Iimura, Y., Kawai, S., and Morohoshi, N. (1993) J. Gen.
Microbiol., 139, 179-185.
89.Palmieri, G., Giardina, P., Bianco, C.,
Fontanella, B., and Sannia, G. (2000) Appl. Environ. Microbiol.,
66, 920-924.
90.Kumari, H. L., and Sirsi, M. (1972) Arch.
Microbiol., 84, 350-357.
91.De Souza, C. G. M., Tychanowicz, G. K., de Souza,
D. F., and Peralta, R. M. (2004) J. Basic Microbiol., 44,
129-136.
92.Dong, J. L., Zhang, Y. W., Zhang, R. H., Huang,
W. Z., and Zhang, Y. Z. (2005) J. Basic Microbiol., 45,
190-198.
93.Farnet, A. M., Criquet, S., Tagger, S., Gil, G.,
and Le Petit, J. (2000) Can. J. Microbiol., 46,
189-194.
94.De Souza, C. G. M., and Peralta, R. M. (2003)
J. Basic Microbiol., 43, 278-286.
95.Ng, T. B., and Wang, H. X. (2004) Biochem.
Biophys. Res. Commun., 313, 37-41.
96.Thakker, G. D., Evans, C. S., and Rao, K. K.
(1992) Appl. Microbiol. Biotechnol., 37, 321-323.
97.Molitoris, H. P., and Esser, K. (1970) Arch.
Microbiol., 72, 267-296.
98.Palmer, A. E., Lee, S. K., and Solomon, E. I.
(2001) J. Am. Chem. Soc., 123, 6591-6599.
99.Enguita, F. J., Martins, L. O., Henriques, A. O.,
and Carrondo, M. A. (2003) J. Biol. Chem., 278,
19416-19425.
100.Garavaglia, S., Cambria, M. T., Miglio, M.,
Ragusa, S., Iacobazzi, V., Palmieri, F., D'Ambrosio, C., Scaloni, A.,
and Rizzi, M. (2004) J. Mol. Biol., 342, 1519-1531.
101.Malmstrom, B. G. (1982) Annu. Rev.
Biochem., 51, 21-59.
102.Reinhammar, B. R., and Vanngard, T. I. (1971)
Eur. J. Biochem., 18, 463-468.
103.Solomon, E. I., Baldwin, M. J., and Lowery, M.
D. (1992) Chem. Rev., 92, 521-542.
104.Quintanar, L., Yoon, J., Aznar, C. P., Palmer,
A. E., Andersson, K. K., Britt, R. D., and Solomon, E. I. (2005) J.
Am. Chem. Soc., 127, 13832-13845.
105.Larrabee, J. A., and Spiro, T. G. (1979)
Biochem. Biophys. Res. Commun., 88, 753-760.
106.Morie-Bebel, M. M., Morris, M. C., Menzie, J.
L., and McMillin, D. R. (1984) J. Am. Chem. Soc., 106,
3677-3678.
107.Li, J.-B., and McMillin, D. R. (1990) Inorg.
Chim. Acta, 167, 119-122.
108.Malkin, R., Malmstrom, B. G., and Vanngard, T.
(1969) Eur. J. Biochem., 7, 253-259.
109.Solomon, E. I., Tuczek, F., Root, D. E., and
Brown, C. A. (1994) Chem. Rev., 94, 827-856.
110.Messerschmidt, A., and Huber, R. (1990) Eur.
J. Biochem., 187, 341-352.
111.Karhunen, E., Niku-Paavola, M.-L., Viikari, L.,
Haltia, T., van der Meer, R. A., and Duine, J. A. (1990) FEBS
Lett., 267, 6-8.
112.Ducros, V., Brzozowski, A. M., Wilson, K. S.,
Brown, S. H., Ostergaard, P., Schneider, P., Yaver, D. S., Pedersen, A.
H., and Davies, G. J. (1998) Nat. Struct. Biol., 5,
310-316.
113.Ducros, V., Brzozowski, A. M., Wilson, K. S.,
Ostergaard, P., Schneider, P., Svendson, A., and Davies, G. J. (2001)
Acta Crystallogr., 57, 333-336.
114.Hakulinen, N., Kiiskinen, L. L., Kruus, K.,
Saloheimo, M., Paananen, A., Koivula, A., and Rouvinen, J. (2002)
Nat. Struct. Biol., 9, 601-605.
115.Hakulinen, N., Kruus, K., Koivula, A., and
Rouvinen, J. (2006) Biochem. Biophys. Res. Commun., 350,
929-934.
116.Bertrand, T., Jolivalt, C., Caminade, E., Joly,
N., Mougin, C., and Briozzo, P. (2002) Acta Crystallogr.,
58, 319-321.
117.Piontek, K., Antorini, M., and Choinowski, T.
(2002) J. Biol. Chem., 277, 37663-37669.
118.Lyashenko, A. V., Zhukhlistova, N. E.,
Gabdoulkhakov, A. G., Zhukova, Y. N., Voelter, W., Zaitsev, V. N.,
Bento, I., Stepanova, E. V., Kachalova, G. S., Koroleva, O. V.,
Cherkashyn, E. A., Tishkov, V. I., Lamzin, V. S., Schirwitz, K.,
Morgunova, E. Y., Betzel, C., Lindley, P. F., and Mikhailov, A. M.
(2006) Acta Crystallogr., 62, 954-957.
119.Enguita, F. J., Matias, P. M., Martins, L. O.,
Placido, D., Henriques, A. O., and Carrondo, M. A. (2002) Acta
Crystallogr., 58, 1490-1493.
120.Antorini, M., Herpoel-Gimbert, I., Choinowski,
T., Sigoillot, J. C., Asther, M., Winterhalter, K., and Piontek, K.
(2002) Biochim. Biophys. Acta, 1594, 109-114.
121.Pegasova, T. V., Zwart, P., Koroleva, O. V.,
Stepanova, E. V., Rebrikov, D. V., and Lamzin, V. S. (2003) Acta
Crystallogr., 59, 1459-1461.
122.Ferraroni, M., Duchi, I., Myasoedova, N. M.,
Leontievsky, A. A., Golovleva, L. A., Scozzafava, A., and Briganti, F.
(2005) Acta Crystallogr., 61, 205-207.
123.Kumar, S. V. S., Phale, P. S., Durani, S., and
Wangikar, P. P. (2003) Biotechnol. Bioeng., 83,
386-394.
124.Claus, H. (2004) Micron, 35,
93-96.
125.Perry, C. R., Smith, M., Britnell, C. H., Wood,
D. A., and Thurston, C. F. (1993) J. Gen. Microbiol.,
139, 1209-1218.
126.Yaver, D. S., Overjero, M. D. C., Xu, F.,
Nelson, B. A., Brown, K. M., Halkier, T., Bernauer, S., Brown, S. H.,
and Kauppinen, S. (1999) Appl. Environ. Microbiol., 65,
4943-4948.
127.Kiiskinen, L. L., and Saloheimo, M. (2004)
Appl. Environ. Microbiol., 70, 137-144.
128.Eggert, C., LaFayette, P. R., Temp, U.,
Eriksson, K.-E. L., and Dean, J. F. D. (1998) Appl. Environ.
Microbiol., 64, 1766-1772.
129.Coll, P. M., Tabernero, C., Santamaría,
R., and Perez, P. (1993) Appl. Environ. Microbiol., 59,
4129-4135.
130.Giardina, P., Aurilia, V., Cannio, R.,
Marzullo, L., Amoresano, A., Siciliano, R., Pucci, P., and Sannia, G.
(1996) Eur. J. Biochem., 235, 508-515.
131.Nitta, K., Kataoka, K., and Sakurai, T. (2002)
J. Inorg. Biochem., 91, 125-131.
132.Kojima, Y., Tsukuda, Y., Kawai, Y., Tsukamoto,
A., Sugiura, J., Sakaino, M., and Kita, Y. (1990) J. Biol.
Chem., 265, 15224-15230.
133.Colao, M. Ch., Garzillo, A. M., Buonocore, V.,
Schiesser, A., and Ruzzi, M. (2003) Appl. Microbiol.
Biotechnol., 63, 153-158.
134.Ong, E., Pollock, W. B., and Smith, M. (1997)
Gene, 196, 113-119.
135.Garzillo, A. M., Colao, M. C., Buonocore, V.,
Oliva, R., Falcigno, L., Saviano, M., Santoro, A. M., Zappala, R.,
Bonomo, R. P., Bianco, C., Giardina, P., Palmieri, G., and Sannia, G.
(2001) J. Protein Chem., 20, 191-201.
136.Xu, F., Palmer, A. E., Yaver, D. S., Berka, R.
M., Gambetta, G. A., Brown, S. H., and Solomon, E. I. (1999) J.
Biol. Chem., 274, 12372-12375.
137.Reinhammar, B. R. M. (1972) Biochim.
Biophys. Acta, 275, 245-259.
138.Shleev, S., Nikitina, O., Christenson, A.,
Reimann, C. T., Yaropolov, A. I., and Ruzgas, T. (2007) Bioorg.
Chem., 35, 35-49.
139.Klonowska, A., Gaudin, C., Asso, M., Fournel,
A., Reglier, M., and Tron, T. (2005) Enzyme Microb. Technol.,
36, 34-41.
140.Shleev, S., Tkac, J., Christenson, A., Ruzgas,
T., Yaropolov, A. I., Whittaker, J. W., and Gorton, L. (2005)
Biosens. Bioelectron., 20, 2517-2554.
141.Christenson, A., Dimcheva, N., Ferapontova, E.
E., Gorton, L., Ruzgas, T., Stoica, L., Shleev, S., Yaropolov, A. I.,
Haltrich, D., Thorneley, R. N. F., and Aust, S. D. (2004)
Electroanalysis, 16, 1074-1092.
142.Ferapontova, E. E., Shleev, S., Ruzgas, T.,
Stoica, L., Christenson, A., Tkac, J., Yaropolov, A. I., and Gorton, L.
(2005) in Electrochemistry of Nucleic Acids and Proteins: Towards
Electrochemical Sensors for Genomic and Proteomics (Palecek, E.,
Scheller, F., and Wang, J., eds.) Elsevier, Amsterdam, pp. 517-598.
143.Shleev, S., Christenson, A., Serezhenkov, V.,
Burbaev, D., Yaropolov, A., Gorton, L., and Ruzgas, T. (2005)
Biochem. J., 385, 745-754.
144.Xu, F., Berka, R. M., Wahleithner, J. A.,
Nelson, B. A., Shuster, J. R., Brown, S. H., Palmer, A. E., and
Solomon, E. I. (1998) Biochem. J., 334, 63-70.
145.Xu, F. (1997) J. Biol. Chem.,
272, 924-928.
146.Bourbonnais, R., Leech, D., and Paice, M. G.
(1998) Biochim. Biophys. Acta, 1379, 381-390.
147.Fabbrini, M., Galli, C., and Gentili, P. (2002)
J. Mol. Catal. B: Enzymes, 16, 231-240.
148.Messerschmidt, A., Ladenstein, R., Huber, R.,
Bolognesi, M., Avigliano, L., Petruzzelli, R., Rossi, A., and
Finazzi-Agro, A. (1992) J. Mol. Biol., 224, 179-205.
140.Bento, I., Martins, L. O., Lopes, G. G.,
Carrondo, M. A., and Lindley, P. F. (2005) Dalton Trans.,
21, 3507-3513.
150.Rulisek, L., Solomon, E. I., and Ryde, U.
(2005) Inorg. Chem., 44, 5612-5628.
151.Shleev, S., Reimann, C. T., Serezhenkov, V.,
Burbaev, D., Yaropolov, A. I., Gorton, L., and Ruzgas, T. (2006)
Biochimie, 88, 1275-1285.
152.Shin, W., Sundaram, U. M., Cole, J. L., Zhang,
H. H., Hedman, B., Hodgson, K. O., and Solomon, E. I. (1996) J. Am.
Chem. Soc., 118, 3202-3215.
153.Sakurai, T. (1992) Biochem. J.,
284, 681-685.
154.Gianfreda, L., Xu, F., and Bollag, J.-M. (1999)
Bioremediation J., 3, 1-26.
155.Hofer, C., and Schlosser, D. (1999)
FEBS Lett., 451, 186-190.
156.Schlosser, D., and Hofer, C. (2002) Appl.
Environ. Microbiol., 68, 3514-3521.
157.Nikitina, O. V., Shleev, S. V., Gorshina, E.
S., Rusinova, T. V., and Yaropolov, A. I. (2005) Vestnik MGU,
Ser. 2. Khim., 46, 267-273.
158.Shin, K. S., and Kim, C.-J. (1998)
Biotechnol. Tech., 12, 101-104.
159.Shleev, S., Jarosz-Wilkolazka, A., Khalunina,
A., Morozova, O., Yaropolov, A., Ruzgas, T., and Gorton, L. (2005)
Bioelectrochemistry, 67, 115-124.
160.Xu, F. (1996) Biochemistry