Effect of Co-evolving Amino Acid Residues on Topology of Phylogenetic Trees
D. Yu. Sherbakov and T. I. Triboy*
Limnological Institute, Siberian Branch of the Russian Academy of Sciences, Ulanbatorskaya ul. 3, P.O. Box 278, 664033 Irkutsk, Russia; fax: (395) 242-5405; E-mail: sherb@lin.irk.ru; ttriboy@mail.ru* To whom correspondence should be addressed.
Received July 30, 2007
The presence in proteins of amino acid residues that change in concert during evolution is associated with keeping constant the protein spatial structure and functions. As in the case with morphological features, correlated substitutions may become the cause of homoplasies--the independent evolution of identical non-homological adaptations. Our data obtained on model phylogenetic trees and corresponding sets of sequences have shown that the presence of correlated substitutions distorts the results of phylogenetic reconstructions. A method for accounting for co-evolving amino acid residues in phylogenetic analysis is proposed. According to this method, only a single site from the group of correlated amino acid positions should remain, whereas other positions should not be used in further phylogenetic analysis. Simulations performed have shown that replacement on the average of 8% of variable positions in a pair of model sequences by coordinately evolving amino acid residues is able to change the tree topology. The removal of such amino acid residues from sequences before phylogenetic analysis restores the correct topology.
KEY WORDS: phylogenetic analysis, correlated substitutions of amino acid residues, homoplasiesDOI: 10.1134/S0006297907120103
Abbreviations: OTU) operational taxonomical unit.
One of the main hypotheses that are the basis of many methods of
phylogenetic analysis is that all features undergo evolution
independently of each other. For polypeptides, this means that the
probability of the substitution fixation in some position of the
sequence should be independent of amino acids situated in all the other
positions. However, this supposition is not always fulfilled. It is
violated not only due to different functional significance of amino
acids in different positions, but because in many cases the potentially
unfavorable effect of some substitutions may be compensated by one or
several substitutions in other positions of the sequence [1, 2]. Such series of mutually
governed substitutions were called coordinated or correlated
substitutions [3]. When the proportion of
coordinated substitutions is relatively high, non-observance of the
rule of feature independence may result in significant errors in
phylogenetic analysis, i.e. in an incorrect topology of a phylogenetic
tree.
A number of works have appeared recently which deal with methods of detection of coordinated substitutions when comparing amino acid sequences. These methods are based on the supposition that, as a rule, the coordinated substitutions should result in non-random alterations of certain physicochemical properties of amino acid residues due to strong requirements caused by the necessity of preservation of the protein structure and function.
Methods based on analysis of information about primary structure of proteins without considering their phylogenetic relationships consist of searching for groups of interacting residues by correlation of amino acid residues in variable positions of sequences [4]. One such method that determines correlated substitutions in aligned protein sequences was incorporated into the CRASP program [5]. Instead of more frequently used matrices of similarity between amino acid residues, this method uses the matrix of similarity between physicochemical properties of amino acids (dissociation constant, the volume of side chain, etc.). As in the analogous method of Fleishman [6], the most precise results are obtained when the topology of a phylogenetic tree binding the proteins is known. However, the presence of co-evolving residues may result in an incorrect topology, which in turn, may lead to incorrect identification of correlated residues; that is why the source of a phylogenetic model should be dissimilar data, able to help in building the tree of species.
From the point of view of phylogenetics, correlated substitutions of amino acids is one of most probable sources of homoplasies, the independent evolution of identical non-homologous adaptations in response to identical conditions [7]. This circumstance is in not considered by phylogenetic programs.
In this work, we suggest applying to molecular data (amino acid sequences) the method of accounting for correlated substitutions similar to that proposed by Marques and Gnaspini [8] for morphological features responsible for homoplasies. The essence of this method is that in the group of features that evolved during convergent evolution only one remains, whereas the rest are removed.
Our approach includes a series of successive procedures with the analyzed amino acid sequences of proteins. We suppose that this approach will allow us to avoid the use of amino acid residues that evolved during convergent evolution and production of incorrect phylogenetic reconstructions.
We show using computer simulations that the account of correlations significantly decreases the risk of obtaining incorrect topologies when in the course of evolution restrictions by physicochemical properties are superimposed on some amino acid positions.
METHODS OF INVESTIGATION
Simulation of sequences containing co-evolving sites. Sets of monophyletic sequences were obtained using the pseq-gen program [9] according to the tree binding a variable number of operational taxonomical units (OTUs). The tree length was chosen so that about 50% of positions in sequences contained substitutions compared to the original. The substitution probabilities were preset by the JTT matrix [10].
Correlated substitutions were introduced by addition to the sequence end of a short fragment (10 amino acid residues) containing at the beginning of the sequence correlated positions for the first ten homologous amino acid residues of variable charge. Correlation between amino acid residues was prescribed as follows:
- the first 10 variable amino acid positions (columns), where the charge of amino acid residue is changed, were found in the alignment;
- depending on the amino acid residue found in this position, another residue was added to C-terminus provided that Asp will be the correlating residue for Arg, Glu for Lys, and any non-charged amino acid residue for Ala.
For sequences generated using the pseq-gen program and the same sequences but with added correlated residues, phylogenetic analysis was carried out using the PHYML program [11], and the resulting trees were compared with those used by pseq-gen for simulations. The comparison was performed using the Robinson-Foulds distance [12] realized in the treedist program (the package of phylogenetic programs PHYLIP v.4.0 [13]).
Simulation of homoplasy-containing sequences. Sequences were generated under the pseq-gen program by the prescribed tree consisting of four OTUs. The length of the tree branches corresponded to number of substitutions between sequences. Maximally distant OTUs differed by substitutions in 50% of the positions. The following simulation conditions were prescribed for pseq-gen: sequence length 1000 amino acid residues and substitution matrix JTT. Overall, 10 sets of four sequences in each were simulated. Proteins were aligned using the Clustalw v.1.4 program [14] and phylogenetic analysis was carried out by two methods realized in the PHYLIP program package: the method of Maximum Likelihood [15] (program PROML) and in addition, the matrix of distances between OTUs was obtained under the PROTDIST program; the matrix was used to restore the trees by Neighbor-joining method [16] (program NEIGHBOR). The resulting phylograms were compared visually with the tree used for sequence generation.
Homoplasies were introduced by hand into simulated sequences in the sequence editor BioEdit [17] and then phylogenetic analysis for modified sequences was carried out (PROML program).
After obtaining sets of homoplasy-containing sequences and extraction of corresponding phylogenetic trees for them, columns with introduced homoplasies were removed from aligned sequence in the BioEdit editor and phylogenetic analysis was carried out again (PROML program).
The pI and charge values at pH 7.0 were found for initial sequences and those carrying homoplasies (Protein Calculator v.3.3 program [18]).
The change in the sequence charge before and after introduction of homoplasies was compared with the same physicochemical parameters obtained on sequences of serum albumin isolated from eight organisms (identification numbers of sequences in the protein databases are shown in parentheses): Homo sapiens (CAA00606.1), Bos taurus (AAA51411.1), Sus scrofa (NP_001005208.1), Felis catus (NP_001009961.1), Canis familiaris (NP_001003026.1), Microtus fortis cala (AAW79113.1), Rattus norvegicus (NP_599153.1), Gallus gallus (NP_990592.1). The charge value for albumin was calculated using the Protein Calculator v.3.3 program for unfolded protein at pH 7.0.
RESULTS AND DISCUSSION
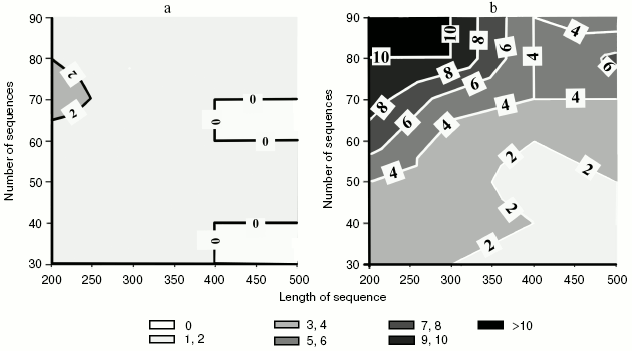
Effect of co-evolving positions on phylogenetic analysis. The supposition concerning the effect of correlated substitutions on phylogenetic analysis was tested on model trees that prescribed evolution of hypothetical sequences. Sets of dichotomous trees of random topology were obtained for different numbers of operational taxonomical units (30-90 OTUs). We supposed that these trees are in principle the “standards” because they indicate the real arrangement of hypothetical OTUs. Different length amino acid sequences (200-500 amino acid residues) were modeled in accordance with the order of each tree branching. Such “proteins” evidently do not carry any correlated substitutions.
We suppose that in this case the tree topology determined by standard methods of phylogenetic analysis should coincide with the topology of model trees for which these sequences were obtained. Simulations were repeated ten times for each combination of parameters (the amount and length of sequences in a set) and in each case the similarity in topology of restored and original trees was determined (figure, panel (a)).
The same algorithm of investigation was used for sets containing sequences with added correlated amino acid residues. Results of comparison are given in the figure (panel (b)). It is clear that the addition of 10 amino acids in accordance with the rules suggested for coordinated evolution generates a tree restored by phylogenetic analysis and distinct from the original one. As expected, these distinctions are most pronounced in the region of a large number of relatively short sequences.
Effect of homoplasies on phylogenetic analysis. We have considered above the effect of correlated substitutions on the topology of resulting trees. In this work, in addition to correlated substitutions, we have simulated homoplasies as a result of coordinated evolution by introduction of substitutions into positions of variable charge.a) Data obtained for sequences without correlated substitutions. b) Sequences contained sites correlated in physicochemical properties. Figures on isolines correspond to mean Robinson-Foulds topological distances between calculated and model trees
To simplify the experiment, we have used sets of four OTUs. The aligned sequences were free of inserts and deletions. Homoplasies were introduced in any of four sequences and we have tried, at the minimum of substitution positions, to “draw together” two OTUs, not bound by a node in common on the original tree. As a rule, a single amino acid residue was replaced in the same alignment position of one of four sequences. The main condition in the case of homoplasy simulation was that the residue that was supposed to replace another in one sequence would be of the same type (or charge) as the residue in the same site in the other, supposed to be combined in one node in common on the tree. At the same time, the residues should differ from those in the two other sequences. Unlike the previous experiment, in this one the condition of residue substitutions depending on their charge is not observed: in some cases, in order to combine two OTUs in a node in common, the charged amino acid in one of them was replaced by a neutral one or vice versa. Introduction of homoplasies produced sets of modified sequences with the tree topology different from the original.
Phylogenetic analysis using Maximum Likelihood and Neighbor-joining methods was carried out on the basis of sets free of the model sequence homoplasies simulated in the pseq-gen program. Results of analysis have shown that the built trees were identical to the original one used for the sequence evolution in pseq-gen. Thus we have supposed that in our case the choice of phylogenetic method has no effect on the drawn topology and the whole following phylogenetic analysis was carried out using the method of Maximum Likelihood.
The introduction of homoplasies into model sequences according to the procedure described in “Methods” altered the tree topology. The number of introduced substitutions necessary to change the tree strongly varied in different sets (2.6-22.7% homoplasies) of variable positions. On the average for all sets, it was necessary to introduce homoplasies into 8% of variable amino acid residues in each pair of sequences (a pair of original and modified sequences is implied) in order to change the tree topology.
The described method of introduction of homoplasies makes highly probable generation of an “incompetent” protein (if the simulated sequences had structure and functions). We have calculated the difference in sequence charges before and after introduction of homoplasies and thus checked how strong changes in the properties of the modified sequence were. It was supposed to compare the results with the difference of the same physicochemical properties of serum albumin of different organisms. In such a way, we have established the limits in which the physicochemical parameters under study may vary in homologous natural proteins.
To calculate the charge of model “proteins” and albumin of eight vertebrate species, the Protein Calculator v.3.3 program was used. The results were placed in a table and the absolute difference was calculated as:
a = |m - i|,
where m is the charge of the modified sequence and i is the charge of the original sequence. Since it is difficult to judge the extent of charge alteration of the modified sequence, we also calculated relative difference (in %) as the ratio of absolute difference to the high value modulus of the studied physicochemical parameter in the pair of “original-modified” sequences. The same calculations were carried out for albumin, where the absolute and relative differences were obtained by comparing in pairs the properties of all sequences.
The calculated charges as well as relative difference between them are given in the table for model sequences. As is seen in the table, in six out of forty pairs of model sequences (or 15% of the pairs) the introduction of homoplasies has changed the sign of the charge. It was assumed that “proteins” in such pairs lost their native structure and parameters of their physicochemical properties were not used in future calculations. Besides, the difference between charges of sequence pairs varied over wide limits up to 96.15% (protein A in set No. 7). On the average, charges under sequence comparison by pairs differed for 30.38 and 35.21% in model proteins and albumin, respectively. Based on data obtained on model proteins one can say that about 8% of variable positions, in which convergent evolution (homoplasies) took place in charged protein positions, are able to influence the accuracy of phylogenetic analysis.
Charge calculated by the Protein Calculator v.3.3 program for original
sequences and those carrying homoplasies
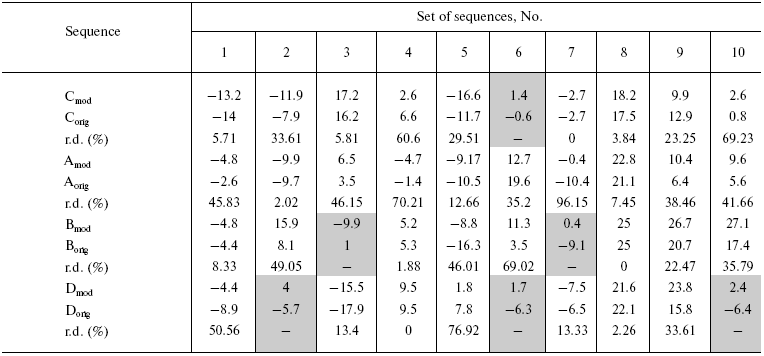
Note: orig, original sequence; mod, modified sequence; r.d.,
relative difference between charges of original and modified sequences
(%); sequences in corresponding sets where the charge sign was changed
after introduction of homoplasies into original sequence are
highlighted with gray.
The removal of correlated substitutions (homoplasies) from model sequences carrying them resulted in restoration of the tree topology according to which original sequences underwent evolution. This means that the removal of correlated substitutions from real proteins before phylogenetic analysis gives more chances to obtain trees that are indicative of a real evolution process that took place among taxons under analysis.
Analysis of a large volume of information concerning amino acid sequences of orthologous proteins shows that very often strict epistatic interactions are established between pairs or groups of individual amino acid positions which are not enrolled in the concept of adaptation density, according to which most sites are considered independently of each other [19]. After mutation in one such position, a compensatory mutation in an associated one can be selectively advantageous, even if it was harmful in the wild-type protein. Meanwhile, concrete mechanisms that would be able to carry out such coordinated evolutionary events at the population level are still unknown.
There are now a number of works showing nonrandom fixation of amino acid substitutions in proteins. Analysis of compensatory substitutions was carried out by Kondrashov et al. in the course of investigation of pathogenetic missense mutations in human protein sequences [20]. They studied 32 mutant and normal human proteins and their normal orthologs in mammals. According to their results, about 10% of substitutions, by which proteins of other species differ from their human orthologs, are compensated pathogenetic mutations.
Simulations on model proteins have shown that a smaller number of correlated substitutions are enough to change topology of the resulting phylogenetic trees.
When co-evolving amino acid positions exist in sequences, groups of co-evolving amino acid residues should be removed from sequences to obtain phylogeny close to reality, and after that to begin phylogenetic analysis.
This means that combination of phylogenetic analysis with approaches enabling determination of groups coordinately evolving positions in amino acid sequences of orthologous proteins will make it possible to avoid possible mistakes. However, this requires development of new algorithms for analysis of these sequences.
The mechanisms able to contribute to emergence of multiple coordinated amino acid substitutions in populations are still not investigated. The problem is that any trajectory of step-by-step substitutions between two really existing protein sequences (like horse and human b-hemoglobins [20]) passes through states of strongly reduced viability. Nevertheless, such events not only are observed but they are rather widespread.
Authors express their sincere gratitude to V. V. Aleshin (Belozersky Institute of Physico-Chemical Biology, Moscow State University) for valuable critical comments and are also grateful to Yu. S. Bukin for consultations during preparation of material for this article.
This work was carried out within the framework of a joint Russian-Holland scientific project “Information System Simulation and Analysis of Complex Histories of Evolution (SACHE)” supported by a grant from The Netherlands Organization on Scientific Investigations (Ref. 047.016.013).
REFERENCES
1.Oosawa, K., and Simon, M. (1986) Proc. Natl.
Acad. Sci. USA, 83, 6930-6934.
2.Hughes, A. L., and Yeager, M. (1997) J. Mol.
Evol., 44, 675-682.
3.Pazos, F., Helmer-Citterich, M., Ausiello, G., and
Valencia, A. (1997) J. Mol. Biol., 271, 511-523.
4.Gobel, U., Sander, C., Schneider, R., and Valencia,
A. (1994) PROTEINS: Structure, Function, and
Genetics, 18, 309-317.
5.Afonnikov, D. A., and Kolchanov, N. A. (2004)
Nucleic Acids Res., 32, 64-68.
6.Fleishman, S. J., Yifrach, O., and Ben-Tal, N.
(2004) J. Mol. Biol., 340, 307-318.
7.Harris, S. R., Wilkinson, M., and Marques, A. C.
(2003) Cladistics, 19, 128-130.
8.Marques, A. C., and Gnaspini, P. (2001)
Cladistics, 17, 371-381.
9.Rambaut, A., and Grassly, N. C. (1997)
Comput. Appl. Biosci., 13, 235-238.
10.Jones, D. T., Taylor, W. R., and Thornton, J. M.
(1992) Comput. Appl. Biosci., 8, 275-282.
11.Guindon, S., and Gascuel, O. (2003) Syst.
Biol., 52, 696-704.
12.Robinson, D., and Foulds, L. (1981)
Math. Biosci., 53, 131-147.
13.Felsenstein, J. (1996) PHYLIP (phylogeny
inference package). v.4.0. Seattle: University of Washington,
Department of Genetics, USA.
14.Thompson, J. D., Higgins, D. G., and Gibson, T.
J. (1994) Nucleic Acids Res., 22, 4673-4680.
15.Felsenstein, J. (1981) J. Mol. Evol.,
17, 368-376.
16.Saitou, N., and Nei, M. (1987) Mol. Biol.
Evol., 4, 406-425.
17.Hall, T. A. (1999) Nucleic Acids. Symp.
Ser., 41, 95-98.
18.http://www.scripps.edu/~cdputnam/protcalc.html
19.Pal, C., Papp, B., and Lercher, M. J. (2006)
Nature, 7, 337-348.
20.Kondrashov, A. S., Sunyaev, Sh., and Kondrashov,
F. (2002) Proc. Natl. Acad. Sci. USA, 99,
14878-14883.