REVIEW: A Biochemical Approach to the Problem of Aging: “Megaproject” on Membrane-Penetrating Ions. The First Results and Prospects
V. P. Skulachev1,2
1Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-0338; E-mail: skulach@belozersky.msu.ru2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
Received September 4, 2007
Antioxidants specifically addressed to mitochondria have been studied for their ability to decelerate aging of organisms. For this purpose, a project has been established with participation of several research groups from Belozersky Institute of Physico-Chemical Biology and some other Russian research institutes as well as two groups from the USA and Sweden, with support by the “Mitotechnology” company founded by “RAInKo” company (O. V. Deripaska and Moscow State University). This paper summarizes the first results of the project and estimates its prospects. Within the framework of the project, antioxidants of a new type (SkQ) were synthesized comprising plastoquinone (an antioxidant moiety), a penetrating cation, and decane or pentane linker. Using planar bilayer phospholipid membranes, we selected SkQ derivatives with the highest penetrating ability, namely plastoquinonyl-decyl-triphenylphosphonium (SkQ1), plastoquinonyl-decyl-rhodamine 19 (SkQR1), and methylplastoquinonyl-decyl-triphenylphosphonium (SkQ3). Anti- and prooxidant properties of these substances and also of ubiquinone and ubiquinonyl-decyl-triphenylphosphonium (MitoQ) were tested on isolated mitochondria. Micromolar concentrations of cationic quinones are found to be very strong prooxidants, but in lower (sub-micromolar) concentrations they display antioxidant activity. The antioxidant activity decreases in the series SkQ1 = SkQR1 > SkQ3 > MitoQ, so the window between the anti- and prooxidant effects is smallest for MitoQ. SkQ1 is rapidly reduced by complexes I and II of the mitochondrial respiratory chain, i.e. it is a rechargeable antioxidant. Extremely low concentrations of SkQ1 and SkQR1 completely arrest the H2O2-induced apoptosis in human fibroblasts and HeLa cells (for SkQ1 C1/2 = 1·10-9 M). Higher concentrations of SkQ are required to block necrosis initiated by reactive oxygen species (ROS). In mice, SkQ1 decelerates the development of three types of accelerated aging (progeria) and also of normal aging, and this effect is especially demonstrative at early stages of aging. The same pattern is shown in invertebrates (drosophila and daphnia). In mammals, the effect of SkQs on aging is accompanied by inhibition of development of such age-related diseases as osteoporosis, involution of thymus, cataract, retinopathy, etc. SkQ1 manifests a strong therapeutic action on some already developed retinopathies, in particular, congenital retinal dysplasia. With drops containing 250 nM SkQ1, vision is recovered in 50 of 66 animals who became blind because of retinopathy. SkQ1-containing drops instilled in the early stage of the disease prevent the loss of sight in rabbits with experimental uveitis and restore vision to animals that had already become blind. A favorable effect is also achieved in experimental glaucoma in rabbits. Moreover, the pretreatment of rats with 0.2 nmol SkQ1 per kg body weight significantly decreases the H2O2-induced arrhythmia of the isolated heart. SkQ1 strongly reduces the damaged area in myocardial infarction or stroke and prevents the death of animals from kidney infarction. In p53-/- mice, SkQ1 decreases the ROS level in the spleen cells and inhibits appearance of lymphomas which are the main cause of death of such animals. Thus, it seems reasonable to perform clinical testing of SkQ preparations as promising drugs for treatment of age-related and some other severe diseases of human and animals.
KEY WORDS: aging, mitochondria, reactive oxygen species, SkQs, antioxidantsDOI: 10.1134/S0006297907120139
Abbreviations: Deltaiota) transmembrane electric potential; BLM) planar bilayer phospholipid membrane; C12TPP) dodecyl triphenylphosphonium; DMQ) demethoxyMitoQ; MitoQ) compound of ubiquinone and decyl triphenylphosphonium; ROS) reactive oxygen species; SkQ) compounds of plastoquinone or methylplastoquinone and decyl (or amyl) triphenylphosphonium, methylcarninite, or tributylammonium; SkQ1) compound of plastoquinone and decyl triphenylphosphonium (other SkQ derivatives are shown in Fig. 3).
PREREQUISITES OF THE PROJECT: A HYPOTHESIS ABOUT
INTRAMITOCHONDRIAL ROS AS A COMPONENT OF THE AGING PROGRAM
The finding that in many cases cell death is a result of realization of programs encoded in the cell genome is one of the most important discoveries of recent decades. The programs in question represent different types of apoptosis and necrosis or their combinations (reviews [1, 2]). Similar programs are found in bacteria [3] and unicellular eukaryotes [4]. Based on these phenomena, it has been concluded that at least unicellular organisms possess mechanisms for self-elimination. Such phenomena that we have coined “phenoptosis” [1] are also inherent in multicellular organisms, but their molecular mechanisms are still to be elucidated [2, 5, 6].
The proof of the genetically programmed death of individuals has markedly reinforced positions of a few gerontologists who, following the great biologists of the second half of the XIX century--Darwin [7], Wallace, and Weissmann [8]--are inclined to consider aging of an organism as a final stage of ontogenesis and not a simple consequence of accumulation of accidental errors.
If aging is programmed, a reasonable question arises what the biological role of this program is. I think that this program accelerates evolution. Natural selection cannot recognize the appearance of a small potentially useful change unessential for survival of a young and strong animal. However, this change can be decisive for an organism weakened by aging. Take such an example. Two young hares, a clever one and a silly one, on meeting a fox have nearly similar chances to escape from the enemy because they can run faster. But with age the clever hare will get an advantage compared to the silly one because the speed of the hares' running will decrease as a result of an age-related disease known as sarcopenia (diminution of cell number in muscles). And now the clever hare who will immediately take to his heels on seeing the fox will have much better chance to save his life than the silly hare who lingers at the start. The clever hare will continue procreating leverets, and the hare population will become wiser [2].
Humans stopped adapting themselves to the environment long ago. Instead of adapting, they try to change the environment to their benefit. If we want to fly, we build aircraft rather than waiting until wings will grow on our back. Therefore, mechanisms specialized for acceleration of evolution are atavistic for humans and should be abolished if these mechanisms, such as aging, are obviously harmful for an individual. And in accordance with this logic, we may speak about the switching off of the aging program as a most important problem of medicine of the future. But what does the aging program consist of?
The free radical hypothesis by Harman [9] is the most popular concept on the mechanism of aging. He supposed that oxidation of biopolymers by reactive oxygen species (ROS) plays a leading role in weakening of vital functions with age. This hypothesis has been supported by the finding that the oxidation level of DNA, proteins, and lipids increases with aging. This situation can be a consequence of an increase in ROS production or a decrease in the antioxidant defense in old age, or simply a result of the prolonged damaging action of ROS accumulated proportionally to the organism's age [1, 2, 5, 6]. In the framework of the theory of programmed aging, Harman's hypothesis has to be supplemented by an assumption that an increase in the ROS-induced damages with time is controlled by the organism, similarly to any other change occurring during ontogenesis.
The cell contains a set of enzymes that convert O2 to the primary form of ROS, superoxide (O2-·) or its derivative hydrogen peroxide (H2O2) [2, 10]. However, the activity of all these enzymes is significantly lower than that of enzymes of respiratory chain in the inner mitochondrial membrane. In humans, the area occupied by this membrane is as large as 14,000 m2, and respiratory chain complexes represent its major constituent. Mitochondria of the adult human take up about 400 liters of O2 per day and convert it to water (H2O) by four-electron reduction. However, if even 0.1% of this amount of O2 is reduced through the chemically simpler one-electron pathway, this will produce 0.4 liter of O2-·, which greatly exceeds the abilities of all other mechanisms of ROS generation taken together.
In the respiratory chain O2-· is generated mainly in complexes I and III. The highest rate of superoxide generation in mitochondria not inhibited with any toxin is observed in complex I during the reverse transfer of electrons from succinate to NAD+ at the maximum proton potential (i.e. in the absence of ADP). This rate equal to 1 nmol O2/mg protein per minute is nearly fivefold higher than the rate of O2-· production in complex III under the same conditions. It is 10% of the respiration rate without ADP (in state 4) and about 1.5% of the maximal respiration rate in the presence of ADP (in state 3). Even if this rate is lowered because of partial inhibition by NADH [11], a rather large amount of generated superoxide may be dangerous for the cell due to the high toxicity of O2-· and products formed from O2-·. In fact, we carry in our mitochondria a potential generator of strong toxins, which can easily kill our cells and us together with them. This catastrophe will be caused not only by the direct toxic action of ROS, but as a consequence of triggering apoptosis and necrosis which are induced by ROS.
In 2007 Lambert et al. [12] reported that the lifetime of mammals and birds is longer the lower is the rate of H2O2 generation by heart mitochondria during the reverse transfer of electrons from succinate through complex I (animals of 12 species of very different systematic position were studied: from mice to cattle and baboons and from quails to pigeons). No correlation was found when H2O2 generation was measured under conditions of the forward electron transfer (through both complex I and complex III). The longest-living African rodent, the naked mole-rat, was found to be the only exception to this rule. This creature the size of a mouse has maximal lifespan of about 28 years instead of the mouse's 2.5-4 years. Mole-rat mortality does not depend on age [13], as if the aging program was absent in this animal. The program seemed to be switched off after ROS because both the rate of ROS generation at the reverse transfer of electrons [12] and the level of biopolymer oxidation [14, 15] in mole-rats are higher than in mice. But the latter finding is not surprising on taking into account that activities of superoxide dismutase and catalase in these two species are not very different but the third antioxidant enzyme, glutathione peroxidase, is 70-fold less active in mole-rats than in mice [16]. The key for understanding this paradoxical situation seems to be presented by the observation that even very high concentrations of H2O2 failed to induce apoptosis in the mole-rat's cells [17]. In this respect, the mole-rat is reminiscent of long-living mouse having mutations in genes encoding the protein p66shc [18], the enzyme of CoQ synthesis mClk1 [19], or elongation factor 2 kinase [20], whose cells are also resistant to apoptogenic action of hydrogen peroxide [18, 20] or the prooxidant menadione [19].
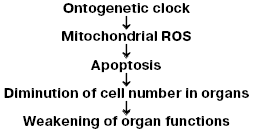
All these observations are in good agreement with our hypothesis on the steps of the aging program (Fig. 1) suggesting a chain of events from “biological clock” (which control stages of ontogenesis at one or another time), through generation of mitochondrial ROS, to diminution in the cell number in organs as a result of apoptosis triggered by these ROS, and, finally, to weakening of the organism's vital functions [2].
Based on this scheme, it can be asserted that protection of cells against mitochondrial ROS has to arrest (or at least decelerate) aging. As to the treatment of aging with antioxidants, the literature is rather vast and ambiguous: from the statement of Ames and colleagues [21, 22] that such a drug against age is already found to conclusions of Howes about a complete fruitlessness of this approach and, thus, erroneousness of Harman's hypothesis [23].Fig. 1. Hypothetical mechanism of aging mediated by mitochondrial ROS [2].
We think that there are two essential omissions in the studies concerning the treatment of aging with antioxidants. 1. If it is necessary to neutralize just intramitochondrial ROS generated by complex I protruding into the mitochondrial matrix, antioxidants addressed to mitochondria are required. But no attempts to use such antioxidants in gerontology are described in the literature. The statement of B. N. Ames [22] that the action of tret-butylhydroxylamine used by him addressed to mitochondria is speculative, and the rather high effective concentration (10-4 M) of this substance seems to indicate that the Ames' hypothesis is erroneous. In our experiments effective concentrations of a mitochondria-addressed geroprotector SkQ1 are much lower (5·10-10-10-5 M, see below). 2. Commonly used antioxidants, even if they principally can reach, like vitamin E, the mitochondrial membranes are natural substances, and their excess can be cleaved by cellular enzymes if their excessive presence is undesirable. We are dealing with just such a situation when vitamin E is used to arrest aging. The organism seeks to realize the aging program, and the coming vitamin E is cleaved by cytochrome P450 which is induced in the liver by excess vitamin E. In fact, the organism disposes systems for defense against both oxygen and antioxidants. The matter is that ROS are responsible for some biological functions indispensable for a proper life. They are directly involved in the struggle with bacteria and viruses and also indirectly help to overcome in this struggle because they are required, in particular, for functioning of the immune system, where ROS-triggered apoptosis plays a certain role [2]. Other useful functions of ROS are also described. In this respect, an experiment of Goldstein is demonstrative. Mice and rats were grown in air deprived of superoxide, and in three weeks the animals died [24].
A “superantioxidant” intended to block the aging program and thus to prolong a healthy human life has to meet the following requirements: 1) the antioxidant must not remove ROS completely, but only their excess generated inside mitochondria at the reverse transfer of electrons in the respiratory chain; 2) the antioxidant must not be inactivated by enzymes of the organism which is by all means seeking to terminate its ontogenesis by switching on the aging program.
These problems might be solved based on a phenomenon observed by us jointly with the group of E. A. Liberman already in 1969-1970 [25-28]. The case in point is the discovery of membrane-penetrating ions, i.e. hydrophobic synthetic compounds that can easily penetrate across the mitochondrial membrane in spite of their ionized group. Triphenylmethylphosphonium is a typical representative of such ions. In this cation, the positive charge on the phosphorus atom is strongly delocalized by three phenyl residues. As a result, water dipoles cannot be held on the cation and do not form an aqueous shell preventing its penetration into the hydrophobic regions of the membrane. The interior of mitochondria is the only cell compartment negatively charged relative to its environment (i.e. the cytosol); therefore, on entering the cell, penetrating cations will be specifically accumulated within mitochondria. This accumulation can be described by the Nernst equation (a 10-fold gradient at Deltaiota = 60 mV). If in the energized mitochondria Deltaiota is ~180 mV [29], the penetrating cation concentration in the mitochondrial matrix has to be 1,000 times higher than in the cytosol.
This consideration allowed us to suppose that penetrating cations can be used by mitochondria as “electric locomotive molecules” for accumulation of uncharged substances attached to these cations [30]. Such an idea allowed us to explain the role of cationic group of carnitine in the transport of fatty acid residues into mitochondria [29, 30]. On being electrophoretically concentrated in the inner leaflet of the phospholipid bilayer of the inner mitochondrial membrane, fatty acid acyls and carnitine would be able to act as antioxidants interrupting chain reactions of peroxidation of phospholipids and proteins which constitute this leaflet. This hypothesis has been confirmed by data obtained in our group by Yu. N. Antonenko and A. A. Pashkovskaya: palmitoyl carnitine prevented photooxidation of gramicidin incorporated into a plane bilayer phospholipid membrane (BLM).
At the border of the two centuries, the principle of “electric locomotive molecule” was successfully used by M. P. Murphy for the addressed delivery into mitochondria of such antioxidants as vitamin E [31] and ubiquinone [32]. The so-called MitoQ composed of ubiquinone and decyl triphenylphosphonium cation seemed to be especially promising. MitoQ is obviously more advantageous than palmitoyl carnitine or cationic derivative of vitamin E because its reduced form oxidized during its functioning as antioxidant can be regenerated by accepting electrons from the respiratory chain. In other words, MitoQ is a rechargeable antioxidant [33]. In fact, in the experiments of Murphy et al., MitoQ was accumulated and regenerated within mitochondria and protected them and also cell cultures against oxidative stress [31-37].
We have confirmed the data of Murphy et al. on the antioxidant activity of MitoQ. However, it was shown that its activity rapidly changed to prooxidant with an increase in the MitoQ concentration. Thus, for rat heart mitochondria C1/2 of the MitoQ antioxidant effect and prooxidant effect was 0.3 and 0.5 µM, respectively (experiments by M. Yu. Vyssokikh and other researchers of our group). Addition of 1.5-2.5 µM MitoQ caused such a strong generation of hydrogen peroxide by mitochondria oxidizing NAD-dependent substrates that its rate became higher than all recorded values described in the literature and approximated the respiration rate in state 4 [38]. Thus, the use of MitoQ in practice as an antioxidant is rather risky. Therefore, we decided to start a search for antioxidants stronger than MitoQ and with a broader window between the anti- and prooxidant effects.
STARTING THE PROJECT
The project of using penetrating ions in practice was started in 2003. It was initiated owing to grants from the Charitable Foundation “Paritet” (now “Volnoe Delo”) created by O. V. Deripaska. Oleg Vladimirovich Deripaska is a 1993 graduate of the Physical Faculty of Moscow State University, and is now one of the richest businessmen in Russia. He is a member of the Moscow State University Board of Trustees and significantly helps his Alma Mater. In early 2003 I shared with him a plan of creating a “superoxidant” drug with the ambitious hope of stopping aging. I did not promise the personal immortality to the foundation's creator, but remarked that even if we fail to beat old age, at least we will overcome hangnails. O. V. burned with the idea that aging was a harmful atavism which would be well to be cancelled, and he thus granted us $120,000 per year for five years.
Upon obtaining the money, we stood before the usual dilemma of what to buy: a fish or a fishing-rod. We preferred the rod and ordered synthesis of demethoxyMitoQ (DMQ), because the nematode Caenorhabditis elegans can live sevenfold longer if it contains a deletion in the gene encoding the demethoxyCoQ conversion to ubiquinone and also the insulin receptor gene [39]. The substance was synthesized and displayed an increased difference between the anti- and prooxidant concentrations compared to MitoQ, but unfortunately this difference was less pronounced than we would have liked.
Then we paid attention to plastoquinone, an electron carrier operating instead of ubiquinone in photosynthetic electron transport chains in chloroplasts of plants as well as in cyanobacteria. During evolution, ubiquinone involved in the mitochondrial respiratory chain was substituted by plastoquinone in the chloroplast chain of the same plant cell, possibly, just because of the better antioxidant properties of plastoquinone, as shown in chemical experiments on model systems [40, 41]. In fact, a chloroplast generating oxygen is under conditions of much stronger oxidative stress than mitochondria, which take up this oxygen. As discriminated from ubiquinone, plastoquinone has methyl groups instead of methoxy groups, and its methyl group is substituted by hydrogen. Whereas the anti- and prooxidant concentrations of MitoQ differ less than twofold (300 and 500 nM), this difference for a plastoquinone derivative of decyl triphenylphosphonium named SkQ1 is increased 32-fold (25 and 800 nM) [38].
This result showed that we found a uniquely effective antioxidant with action specifically addressed to mitochondria and not complicated by a side prooxidant effect. In this connection, I suggested to Deripaska to change the grant to an investment project aimed at creation of a new type of drugs and biotechnology products, i.e. SkQ derivatives. The proposal was accepted, and the investment project was started in the spring of 2005.
Joint-stock company “Mitotechnology” with limited responsibility was organized (Deripaska's company “RAInKo” and Moscow State University as promoters, A. A. Grigorenko as General Director) supported by O. V. The budget in 2005 was $1.7 million, in 2006 - $4.6 million, and in 2007 it increased to $6.8 million. To escape becoming a victim of Parkinson's Law (“Success in research leads to such an increase in its financing that it becomes impossible to continue the work”), we appealed to consulting company “Ikontri” (General Director V. V. Perekhvatov), which helped us in choosing the optimum strategy for organization of the work. The “Ikontri” researchers developed a unique program for managing the project.
A Supervisory Board of Moscow State University was created headed by the Rector Professor V. A. Sadovnichii (the Board members are: MSU Higher Business School Faculty Dean Professor O. S. Vikhansky, Biological Faculty Dean Professor M. P. Kirpichnikov, and Fundamental Medicine Faculty Dean Professor V. A. Tkachuk). The support by the MSU Rector and Deans became an important condition for successful development of the project.
The equipment (sometimes very expensive) was bought, laboratories were repaired, and new researchers were invited; some of them returned from abroad upon receiving in Moscow flats for the time of their participation in the project. The project rapidly left the limits of the Belozersky Institute of Physico-Chemical Biology where the work was performed in the framework of the initial grant. At present, the project includes groups from MSU Faculties of Biology, Chemistry, Physics, Mechanics and Mathematics, Fundamental Medicine, Bioengineering and Bioinformatics; Russian Academy of Sciences Institutes of Bioorganic Chemistry, Molecular Genetics, Developmental Biology in Moscow; Moscow Cancer Research Center; Moscow Cardiology Research Center; Helmholtz Institute of Eye Diseases; Institute of Poliomyelitis, Veterinary Academy; Institutes of Oncology and of Cytology in St. Petersburg; Novosibirsk Institute of Cytology and Genetics; Johnson Medical Institute (USA); Venner-Gren Institute (Stockholm). At present, 260 persons are participating in the project: research workers, postgraduates, technicians, and students. The first conference considering the experimental results of the project took place in March 2007, and in late August there was a round table discussion on the concept of programmed aging as the theoretical basis of the project.
THE FIRST RESULTS OF THE PROJECT: SkQs AS TOOLS AGAINST OXIDATIVE
STRESS, PROMISING FOR TREATMENT OF AGE-RELATED (AND NOT ONLY
AGE-RELATED) DISEASES
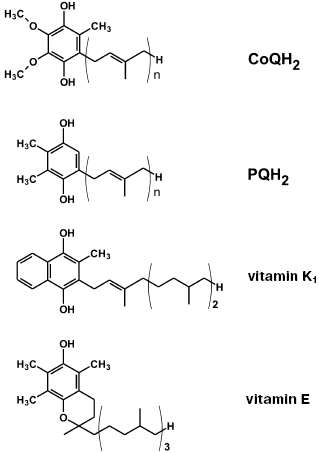
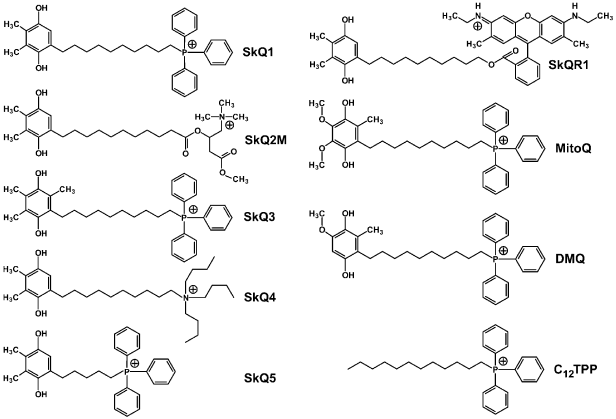
On searching for the best antioxidant among cationic derivatives of quinones, a number of substances were synthesized mainly by our chemists G. A. Korshunova, N. V. Sumbatyan, and L. S. Yaguzhinsky. It was taken into account that (1), in contrast to ubiquinone (CoQ), plastoquinone and another chloroplast electron carrier vitamin K1, as well as a “professional antioxidant” vitamin E, contain no methoxy groups, and (2) vitamin K1, vitamin E, and CoQ have in the fifth position of the quinone ring a methyl group, which is absent in plastoquinone (Fig. 2). As a result, the majority of our quinone derivatives contained either plastoquinone (SkQ1, SkQ2M, SkQ4, SkQ5, SkQR1) or methylplastoquinone (SkQ3). For comparison, MitoQ and DMQ containing, respectively, ubiquinone and demethoxyubiquinone, were also synthesized.
As cations, in the majority of cases alkyl triphenylphosphonium derivatives were used with the alkyl residue linking the cation with the quinone being either decyl (SkQ1, SkQ3, SkQ4, DMQ, and MitoQ) or pentyl (SkQ5). In some cases a different cation was used. Instead of phosphonium, compounds with an ionized nitrogen atom were used: methylcarnitine (SkQ2M), tributylammonium (SkQ4), or Rhodamine 19 (SkQR1, Fig. 3).Fig. 2. Some hydrogen carriers and antioxidants of quinone nature.
All the prepared compounds were first tested for their ability to penetrate across model membranes. I. I. Severina showed that SkQR1 and SkQ3 display the best penetrability across a BLM. The gradient of their concentrations generated Nernst diffusion potential with the “plus” sign in the compartment with the lower concentration of the cation in the range 5·10-6-5·10-5 M (SkQR1) or 5·10-6-5·10-4 M (SkQ3) (SkQR1 at concentrations higher than 5·10-5 M slightly damaged the BLM). The corresponding concentrations of SkQ1, MitoQ, and DMQ were in a higher range (5·10-5-5·10-4 M) indicating a slightly lower permeability of the BLM for these compounds. The BLM permeability for SkQ2M, SkQ4, and SkQ5 was much lower, so that the Nernst potential values could not be reached even in the presence of tetraphenylborate increasing the BLM cationic permeability [38]. Based on these findings, SkQ1, SkQR1, and SkQ3 were chosen for further work.Fig. 3. Some substances that were synthesized and used in the project (the reduced forms are presented) [38].
Anti- and prooxidant properties of these substances were studied in our group by M. Yu. Vyssokikh et al. in comparison with those of MitoQ, DMQ, and C12TPP (dodecyl triphenylphosphonium, i.e. an SkQ analog but lacking quinone). To measure the antioxidant activity of these compounds, rat heart mitochondria were energized by oxidizing succinate and incubated under conditions of the Fenton reaction when OH* radicals are generated from hydrogen peroxide produced by the mitochondria, the reaction being catalyzed by Fe2+ (ascorbic acid was used as a reductant for iron ions). Under these conditions, mitochondrial phospholipids (first of all cardiolipin) were peroxidized, and this peroxidation was followed by accumulation of malonic dialdehyde. All substances tested, except C12TPP, inhibited this accumulation, but their effective concentrations were very different. SkQ1 and SkQR1 were the most active. The antioxidant activity of other quinones decreased in the following series: DMQ > SkQ3 > MitoQ. The activity of decylplastoquinone was nearly 100-fold and that of MitoQ was more than 30-fold lower than the activity of SkQ1.
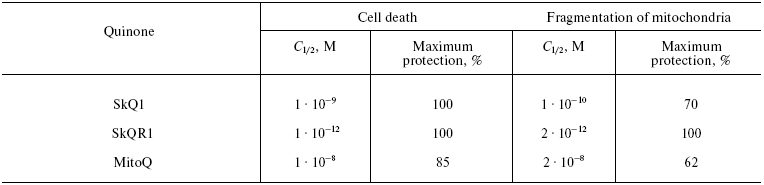
Table 1. Effect of SkQ1, SkQR1, and MitoQ on
(i) H2O2-induced cell death from apoptosis and
(ii) fragmentation of mitochondria in human fibroblasts
The prooxidant activity was tested (i) by the rate of non-enzymatic one-electron reduction of O2 to O2-· mediated by hydroquinone forms of the corresponding compounds in water solutions and (ii) by the stimulation of H2O2 production in rat heart mitochondria oxidizing glutamate and malate in state 4. An important result of the first test was the several times slower reduction of O2 by SkQ1H2 than by MitoQH2 (data by M. Yu. Vyssokikh and E. K. Ruuge). In the second test, these compounds displayed no sharp difference. However, it should be noted that the prooxidant effect of MitoQ on the mitochondria increased at slightly lower concentrations than that of SkQ1 and SkQ3.
The further experiments on cells and animals were performed with SkQ1 and SkQR1, which manifested the best antioxidant properties and a good penetrability when tested, respectively, on mitochondria and BLM. Due to a high quantum yield of the SkQR1 fluorescence, its fate could be followed in the cells and the organism. In HeLa cells, SkQR1 specifically stained only the mitochondria and on intraperitoneal injection into animals it was mainly accumulated in the kidneys and liver (data of B. V. Chernyak's and D. B. Zorov's groups).
Data of Yu. M. Vassiliev, B. V. Chernyak et al., showed that extremely low concentrations of SkQ1 and SkQR1 prevented the H2O2-induced death of human cells in culture. Fragmentation of long mitochondria into small ones (the “thread-grain transition” [42]) in fibroblasts and HeLa cells, induced by H2O2 and other apoptogens, was shown to be the most sensitive parameter. This transition could be prevented by 2-h preincubation with SkQR1, SkQ1, and MitoQ at the C1/2 value, respectively, of 2·10-12, 1·10-10, and 2·10-8 M. SkQ1 very effectively prevented apoptosis of these cells (C1/2 = 1·10-9 M, for MitoQ C1/2 = 1·10-8 M; the antioxidants were added seven days before the addition of H2O2). As shown in Table 1, MitoQ was not only an order of magnitude less active, but even in the optimal concentration, it failed to completely prevent the fragmentation of mitochondria and cell death, whereas SkQR1 and SkQ1 did. This failure of MitoQ could not be overcome by increasing its concentration. At concentrations higher than 1·10-7 M, MitoQ, similarly to SkQ derivatives, did not inhibit but actually strengthened the apoptogenic effect of hydrogen peroxide.
The efficiency of very low concentrations of SkQ1 and SkQR1 in experiments on cells is explained by two factors. First, the coefficient of their distribution between the hydrophobic and aqueous phases is very high: in the octanol-water system, it is 13,000 for SkQ1. Then, we have gained three orders more due to Deltaiota (~180 mV) on the inner mitochondrial membrane and one order more from Deltaiota on the outer cell membrane (~60 mV). In total, the SkQ1 concentration gradient between the extracellular medium and inner half-membrane layer of the inner mitochondrial membrane occurs to be really immense: 13,000·1000·10 = 1.3·108. This means that at the SkQ1 concentration in the medium of 1·10-10 M (Table 1), its concentration in the inner half-membrane layer will be 1·10-10·1.3·108 = 1.3·10-2 M. On the way to the inner mitochondrial membrane, SkQ1 crosses the outer cell membrane, endoplasmic reticulum membranes, and the mitochondrial outer membrane, accumulating within them in accordance with the lipid/water distribution coefficient. That is why a prolonged preincubation is required to provide for the protective effect of picomolar concentrations of SkQ1 on the intracellular mitochondria. The equilibrium concentration in the system of medium/intracellular mitochondria will require less time the higher is the penetrability of our cations. Therefore, in experiments on the cells, SkQR1 was markedly more efficient than SkQ1.
The very high efficiency of SkQs is also explained by their being rechargeable antioxidants. In fact, SkQ1 can be easily reduced by both complexes I and II of the respiratory chain [38].
And one more reason has to be taken into account, namely, the very mechanism of the effect of SkQs on H2O2-induced apoptosis. In our experiments (1-4)·10-4 M H2O2 used for triggering apoptosis induced a dramatic increase in the generation of endogenous ROS by mitochondria, and this seemed to be a signal for the cell to commit suicide. Upon saturating the inner half-membrane layer of a mitochondrion, SkQ1 interrupts in the very beginning the chain reactions of peroxidation of fatty acid residues of phospholipids. SkQs should be highly available for hydrophobic regions of the membrane where these residues are located. Such availability depends on the orientation of SkQs within the membrane: the charged phosphonium group has to be in water, and the flexible decane linker terminated with plastoquinone has to be in the membrane depth. The SkQ molecule can move along the membrane and the plastoquinone head can oscillate pendulum-like between the membrane core and surface.
In line with the above-described reasoning, the effective antioxidant concentrations of SkQ1 on the isolated mitochondria under conditions of external generation of OH* radicals occurred to be higher than on the cells (C1/2 = 2.5·10-8 M, data by M. Yu. Vyssokikh [38], instead of 1·10-9 M on the cells, see Table 1). Still greater amounts of SkQ1 were required to arrest necrosis of cells induced by generation of radicals by a photosensitizer (MitoTracker Red) under illumination (C1/2 = 3.5·10-7 M, preincubation for 1 h with SkQ1). In this case, MitoQ was also less effective than SkQ1 (data of D.S. Izyumov and B.V. Chernyak [38]). It should be noted that highly efficient prevention by cationic quinones of endogenous oxidative stress in cell cultures was for the first time observed by Murphy et al. on fibroblasts from patients with Friedreich's ataxia [37]. This disease is associated with disorders in production of the mitochondrial protein frataxin, which results in an increase in the content of iron ions in the matrix and in oxidative stress. Such fibroblasts can survive in culture only in the presence of antioxidants, in particular, decylubiquinone (C1/2 = 3·10-8 M). MitoQ was much more effective than decylubiquinone (C1/2 = 5·10-10 M), and an uncoupler increased this value to 1·10-8 M. An excess of MitoQ (5·10-7 M and more) could not protect the cells against oxidative stress, whereas an excess of decylubiquinone (1·10-4 M) was as efficient as its lower quantities.
On the next stage of our project, the antioxidant effect of SkQ1 was tested on isolated rat heart. V. I. Kapelko and V. L. Lakomkin (Moscow Research Center of Cardiology) caused heart arrhythmia by addition of H2O2 into the perfusate. The arrhythmia was significantly less pronounced if the hearts had been isolated from rats pretreated for two weeks with SkQ1 (2·10-10 mol/kg body weight) [43].
Then effect of SkQ1 on the in vivo development of heart pathology was studied. O. I. Pisarenko et al. (Moscow Cardiology Research Center) used SkQ1 for a similar pretreatment of rats before the artificial induction of myocardial infarction. SkQ1 (2.5·10-7 mol/kg) decreased by 40% the damaged zone of the heart muscle [43]. In our institute D. B. Zorov and E. Yu. Plotnikov removed one kidney of a rat and some time later induced infarction of the remained kidney by interrupting the blood flow for 90 min. This resulted in the death of 80% of the experimental rats five-six days after the ischemia. However a single intraperitoneal injection of 0.5-1 µmol SkQ1 or SkQR1 per kg body weight the day before ischemia decreased the mortality to 20% [43]. A similar pretreatment with SkQR1 sharply (threefold) decreased the zone of brain necrosis in rats with artificial stroke (data by D. B. Zorov and N. K. Isaev). Injected in this manner, SkQ1 was inefficient, possibly because of its lower penetrability [43].
The majority of in vivo studies in the framework of the project were designed to answer the main question of whether SkQs could serve as geroprotectors. Effects of SkQ1 were studied in three models of accelerated aging (progeria) and in normal aging of rodents, Drosophila, and Daphnia. Positive effects of SkQ1 were observed in X-ray-induced progeria of mice (A. G. Ryazanov et al., Johnson Medical Institute, USA) and in progeria caused by a point mutation in the gene of mitochondrial DNA polymerase (I. Shabalina, B. Cannon, and J. Niedergard, Venner-Gren Institute, Sweden). In the second case, mice with mutated mitochondrial DNA polymerase with damaged proof-reading domain were used (in the mutant mice aspartate was substituted by alanine in the exonuclease site of the enzyme [44]). Ninety percent of mutant mice not treated with SkQ1 died by the 280th day of the life with all signs of premature aging (body, weight loss, osteoporosis causing lordokyphosis, decrease in body temperature and inability to maintain it in the cold, alopecia, torpor, etc.). Only 10% of mice in the group of the mice treated with SkQ1 (2.5 µmol/kg with water) died during this period. Most signs of aging in these animals were poorly pronounced or even absent [43].
Many experiments were conducted on rats of the OXYS strain with progeria caused by permanent oxidative stress. The selection of this strain was initiated by R. I. Salganik at the Novosibirsk Institute of Cytology and Genetics [45]. A few months after birth, such age-related diseases as cataract and retinal dystrophy appeared in these animals [46-48]. N. G. Kolosova and colleagues in the same institute found that the development of these signs of aging was strongly decelerated by treatment with SkQ1 (50-250 nmol/kg per day with food). Concerning the developed cataract and retinal dystrophy, daily instillation to eye of single drop of 2.5·10-7 M SkQ1 cured the disease within 1.5 months. This was confirmed by both photographs of the eye fundus (data by N. G. Kolosova and A. Zh. Fursova) and electron microscopy (data by L. E. Bakeeva and V. B. Saprunova). A similar treatment of cataract was also favorable in Wistar rats [38]. Researchers of the Skryabin Veterinary Academy E. P. Kopenkin and L. F. Sotnikova jointly with I. I. Senin with his colleagues from our institute obtained a therapeutic effect of SkQ1 drops in retinopathies in dogs, cats, and horses. In total, 122 animals who failed to react to routine treatments were treated with SkQ1. The most clear positive effect was recorded in congenital retinal dysplasia (a radical improvement in 67% of cases) and in secondary degeneration of the retina (54%). In progressing retinal degeneration, SkQ1 was less effective (29%). In 66 cases, our patients were completely blind animals, and 50 of them began to see again upon treatment with SkQ1 drops (2.5·10-7 M) during two-to-six weeks.
These positive results are in contrast with the failure of researchers of Murphy's group who tried to treat congenital retinopathies of mice with MitoQ [49]. This may be explained by the closeness of the antioxidant and prooxidant concentrations of MitoQ [38]. However, the authors assumed [49] that the studied retinopathies cannot be cured by antioxidants at all.
To conclude this series of studies, we have turned to artificially induced eye diseases of animals, in particular, to experimental uveitis (I. I. Senin's group in our institute) and glaucoma (Helmholtz Institute, V. P. Erichev and coworkers). Both these diseases are known to be associated with strong oxidative stress [50, 51], and in both cases SkQ1 drops gave favorable effect [38, 43]. Experiments with autoimmune uveitis induced by immunization of rabbits with a photoreceptor cell protein arestin and resulting in blindness were especially demonstrative. Into one eye of the blind animals, 2.5·10-7 M SkQ1 was dropped four times a day, and several days later the vision of the treated eye recovered whereas the non-treated eye was still completely blind. The same procedure prevented the development of uveitis if the drops were given shortly after the immunization. The treatment was successful in 100% of cases--it prevented uveitis and cured the developed disease [38]. P. P. Philippov from our institute together with E. N. Grigoryan (Institute of Developmental Biology) succeeded in modeling an aspect of the SkQ1 effect in the treatment of retinopathies: in roller cultures of eye fundus, SkQ1 decreased eightfold the transformation of the pigmented epithelium cells of the retina into phagocytes that could destroy this tissue [38].
A major work was performed in the Institute of Oncology (St. Petersburg) by the group of the Russian Gerontological Society President V. N. Anisimov. The effect of SkQ1 was studied on aging of normal mice without any form of progeria. SkQ1 (0.5, 5, or 50 nmol/kg) noticeably decreased the age-related mortality of the animals, and the effect was especially demonstrative on the first 20% of the deceased animals (Fig. 4). The lifespan of these animals was increased 2.5-fold. For 50% of the deceased animals the effect was 30%, and for the long-lived mice (the last 20% of the cohort studied) it was only 15%. In other words, the so-called rectangularization of the mortality curve took place. Similar effect was revealed for invertebrates--Drosophila (data of E. G. Pasyukova et al., Institute of Molecular Genetics) and Daphnia (data of O. F. Filenko, Faculty of Biology, Moscow State University).
The effect of SkQ1 on lifespan was associated with improvement of the life quality. In particular, regular estrous cycles lost in 70% of mice by the 22nd month of life were similar to those in young mice if the old mice received SkQ1 with water. The antioxidant dramatically changed the pattern of causes of death. Thus, SkQ1 strongly decreased mortality from infections, which increased in untreated mice because of the aging-associated decline of immune response. Mortality from some types of tumors was also decreased. On the other hand, SkQ1 was impotent against mammary gland cancer, which became the main cause of death of the animals treated with SkQ1 (the experiments were conducted on females). SkQ1 had virtually no influence on mortality of the HER-2 mice predisposed to mammary gland cancer, which did not live to one year (as compared with the lifespan of normal mice which was more than two years under the same conditions) [43].Fig. 4. Rectangularization of survival curves of yeast cells (a) [52], human fibroblasts (b) [38], and mice (c) [43] upon switching off the programs of suicide of cells or aging of the organism. The lower curves present control, the upper curves show switching off of the programs by cycloheximide (a) or SkQ1 (b and c).
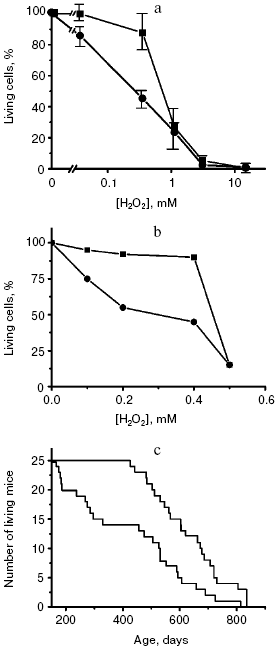
In experiments of B. P. Kopnin, M. P. Lichinitser et al. (Russian Oncology Center, Moscow), very low doses of SkQ1 (5 nmol/kg) significantly increased lifespan of mice deprived of p53 protein and as a result suffering from oxidative stress. These mice die mainly because of lymphomas by the 250th day of life. The survival curve of such mice given SkQ1 with water was noticeably shifted to the right, along with a pronounced decrease in the ROS level in their spleen cells [43]. It was shown in the same laboratory that the human intestinal carcinoma cells lacking p53 produced tumors much faster on grafting to athymic mice. Such an increased aggressiveness of the carcinoma cells was completely abolished if the mice were given SkQ1 (50 nmol/kg) [43].
Our data on the effect of SkQ1 and SkQR1 on animals in vivo are summarized in Table 2. One can see that 20 different signs of aging are mitigated by SkQs, a fact suggesting that SkQs are potent geroprotectors. It is remarkable that the geroprotective effect of SkQ1 consists not only in an increase in the lifespan but also in improvement of the quality of life during the second half of the life. In other words, it prolongs youth.
Table 2. SkQs decelerate aging of
animals

It is interesting to compare (i) the effect of SkQ1 on fibroblast death caused by H2O2, (ii) the effect of cycloheximide on the H2O2-induced death of yeast and (iii) the SkQ1 effect on the mouse lifespan (Fig. 4). No doubt the former two cases represent a blockade of the cell suicide program: in the fibroblasts it is blocked due to prevention by SkQs of the effect of ROS on the mitochondrial membrane, and in the yeast this blockade is caused by switching off synthesis of proteins necessary for phenoptosis. Note that in all the three cases the mortality curve is similarly changed, namely it is rectangularized. At high H2O2 (about 0.5 mM), the cells seem to die independently of the apoptotic or phenoptosis programs but simply because of a direct toxic action of this ROS. Therefore, inhibition of the suicide programs fails to save the cells. It is possible that in the case of treatment of mice with SkQs we are dealing with a complete inhibition of the aging program, and the animals die because the organism has accumulated too many injuries during such a long life.
Another possibility consists in that at very old age an aging program sensitive to low doses of SkQ is replaced by another program which is insensitive to such doses. Just this possibility seems to be realized in Drosophila. Here the geroprotective effect of SkQs was observed only during the very early stages of aging and then disappeared. If the hypothesis about two aging programs is also true for mammals, the lifespan in the late ages can be prolonged either by increasing the dose of SkQs or by combination of SkQ with, e.g. biguanide type geroprotectors, which act mainly in the end of life [53]. Corresponding experiments are now commenced in the group of V. N. Anisimov. Concurrently the effect of SkQ1 on the lifespan of mice mutant in eEF2 kinase is under study in the laboratory of A. G. Ryazanov. The lifespan of this mutant is longer due to decreased late mortality [20]. We are also going to test a p66shc-/- mutant [18] whose mortality curve is similar to that of the above-mentioned mutant.
The above findings indicate that SkQ1 itself is very promising for creating a new generation of drugs acting on aging-related processes as an antioxidant specifically addressed to mitochondria. On the other hand, the action of SkQs is not limited to their geroprotective effect. Experiments with heart arrhythmia, myocardial and kidney infarctions, stroke, wound healing, tumor inoculation, etc. were performed on young animals. It is certainly reasonable to suggest that SkQs simply “purify the dirtiest place in the cell” that results in a nonspecific favorable effect on very different aspects of the vital activity of cells. However, this explanation looks rather unlikely. In this case, it remains unclear why some plants synthesizing separately plastoquinone and such a lipophilic membrane-penetrating cation as berberin did not create during evolution their combination capable of removing ROS from the interior of plant mitochondria which are subjected to even stronger “oxygen danger” than mitochondria of animals because in plants the cytosol is saturated with oxygen generated by chloroplasts.
There is an alternative: certain constituents of the aging program as a slow phenoptosis can be also used in other fatal programs inducing a fast, or acute, phenoptosis. Here, as well as in the case of aging program, a basic question of biology arises, how genetic programs can exist which are absolutely counterproductive for an individual and kill it.
In the nineteenth century Ch. Darwin [7], A. P. Wallace, and A. Weissmann [8] supposed that death of an individual can be altruistic, being useful for family or community. In 1964, W. D. Hamilton published a series of two articles entitled “Genetic Evolution of Social Behavior” [54]. In 1976, the book “The Selfish Gene” [55] by R. Dowkins was published, where the author developed and popularized the idea of Hamilton that not a species, group, or even an individual was the main unit of selection, but that the gene was such a unit. In essence, here we are dealing not with the well-being of a community but with dictatorship of the genome, which is the only self-reproducing biological structure, and, hence, its retention, development, and expansion has priority compared to the well-being of an individual or a group of individuals. According to this concept, an organism is only a construction, a machine, serving interests of the genome.
Some years ago I formulated the so-called “Samurai Law of Biology--It is better to die than to be wrong”, or in more extended form, “Complex biological systems (from organelles and higher) are equipped with programs of self-elimination which are actuated when this system becomes dangerous for any other system of higher position in the biological hierarchy” [2]. Combined with concept about the genome dictatorship, this principle means that any critical state of the organism when it is unable to ensure the safety of its genome and therefore can produce offspring with the significantly altered genome has to be a signal for the organism's self-elimination, i.e. phenoptosis. As spoke Monsieur Bahys, a good-for-nothing physician in Molière's comedy “L'Amour médecin” (“Love as a Healer”)--“Il vaut mieux mourir selon les règles, que de réchapper contre les règles” (“It is better to die according to rules than to recover against the rules”).
It is likely that mechanisms of rapid phenoptosis and slow phenoptosis (aging) are mediated by intramitochondrial ROS at an early stage of the process. If this hypothesis is true, the favorable effect of SkQ not only in aging but in very different acute diseases of young organisms may be explained by quenching of these ROS.
It is likely that SkQs can be used as a tool in “rebellion of machines”--in the attempt of Homo sapiens to put an end to genome tyranny and to cancel those of the genome-dictated programs that are useful for the genome but counterproductive for the individual. Acute phenoptosis of a human actuated in a critical state and terminating his existence by biochemical suicide according to the above-mentioned Bahys' principle seems to be as harmful an atavism as aging. Possibly, there are other genetic programs counterproductive for the organism. Cancellation of all of them would symbolize a conversion of humans to Homo sapiens discatenatus (from Latin catena - shackles) that would be a highest achievement of twenty-fist century medicine.
I am grateful to leaders of the groups participating in the project for excellent work: V. N. Anisimov, Yu. N. Antonenko, L. E. Bakeeva, B. Cannon, B. V. Chernyak, O. F. Filenko, E. N. Grigorian, V. I. Kapel'ko, N. G. Kolosova, E. P. Kopenkin, B. P. Kopnin, G. A. Korshunova, M. P. Lichinitser, I. V. Manukhov, E. N. Mokhova, M. S. Muntyan, E. G. Pasyukova, P. P. Philippov, E. I. Rogaev, E. K. Ruuge, A. G. Ryazanov, I. I. Senin, I. I. Severina, M. V. Skulachev, L. F. Sotnikova, V. N. Tashlitsky, Yu. M. Vassiliev, M. Yu. Vyssokikh, L. S. Yaguzhinsky, D. B. Zorov--and to their coworkers. I am grateful to MSU Rector and Chairman of the Advisory Board V. A. Sadovnichii for his invaluable support, advice, and attention, and also to the Board members: O. S. Vikhansky who has played a decisive role at the start of the project and to Deans of the MSU Faculties M. P. Kirpichnikov and V. A. Tkachuk. I would like to express my gratitude to leaders of “RAInKo” and “Mitotechnology” companies S. V. Karabut and A. A. Grigorenko for the fruitful co-operation. I am particularly grateful to the “Ikontri” Consulting company Chief V. V. Perekhvatov and his coworkers I. V. Skulachev and K. V. Skulachev who have promoted not only organization of the whole entertainment but also to develop a computer program for its management. I am very grateful to Deputy Director of Belozersky Institute V. A. Drachev, to my Secretary O. O. Malakhovskaya, and to all services of the Institute because without their everyday help this venture would have inevitably failed. In conclusion, I would like to emphasize that the work could be realized due to the generous financial support by O. V. Deripaska, to whom I am very sincerely thankful.
REFERENCES
1.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
2.Skulachev, V. P. (2003) in Topics in Current
Genetics. Model Systems in Ageing (Nystrom, T., and Osiewacz, H.
D., eds.) Vol. 3, Springer-Verlag, Berlin-Heidelberg, pp. 191-238.
3.Lewis, K. (2000) Microbiol. Mol. Biol. Rev.,
64, 503-514.
4.Longo, V. D., Mitteldorf, J., and Skulachev, V. P.
(2005) Nature Rev. Genet., 6, 866-872.
5.Skulachev, V. P. (2005) Vestn. Ros. Akad.
Nauk, 75, 831-843.
6.Skulachev, V. P., and Longo, V. D. (2005) Ann.
N. Y. Acad. Sci., 1057, 145-164.
7.Darwin, Ch. (1871) The Descent of Man,
Murray, London.
8.Weissmann, A. (1889) Essays upon Heredity and
Kindred Biological Problems, Claderon Press, Oxford.
9.Harman, D. (1956) J. Gerontol., 11,
298-300.
10.Skulachev, V. P. (1999) Mol. Asp. Med.,
20, 139-184.
11.Grivennikova, V. G., and Vinogradov, A. D. (2006)
Biochim. Biophys. Acta, 1757, 553-561.
12.Lambert, A. J., Boysen, H. M., Buckingham, J. A.,
Yang, T., Podlutsky, A., Austad, S. N., Kunz, T. H., Buffenstein, R.,
and Brand, M. D. (2007) Aging Cell, 6, 607-618.
13.Buffenstein, R. (2005) J. Gerontol. Biol.
Sci., 60, 1369-1377.
14.Andziak, B., and Buffenstein, R. (2006) Aging
Cell, 5, 525-532.
15.Andziak, B., O'Connor, T. P., Qi, W., DeWaal, E.
M., Pierce, A., Chaudhuri, A. R., van Remmen, H., and Buffenstein, R.
(2006) Aging Cell, 5, 463-471.
16.Andziak, B., O'Connor, T. P., and Buffenstein, R.
(2005) Mech. Ageing Dev., 126, 1206-1212.
17.Labinsky, N., Csiszar, A., Orosz, Z., Smith, K.,
Rivera, A., Buffenstein, R., and Ungvari, Z. (2006) Am. J. Physiol.
Heart Circ. Physiol., 291, H2698-H2704.
18.Migliaccio, E., Giorgio, M., Mele, S., Pelicci,
G., Revoldi, P., Pandolfi, P. P., Lanfrancone, L., and Pelicci, P. G.
(1999) Nature, 402, 309-313.
19.Liu, X., Jiang, N., Hughes, B., Bigras, E.,
Shoubridge, E., and Hekimi, S. (2006) Gen. Dev., 19,
2424-2434.
20.Chu, H.-P., Grigorian, I. A., Dorovkov, M. V.,
Nagele, R. G., Komarova, E. A., Gudkov, A. V., Harrison, D. E., and
Ryazanov, A. G. (2007) Nature, in press.
21.Hagen, T. M., Liu, J., Lykkesfeldt, J., Wehr, C.
M., Ingersoll, R. T., Vinarsky, V., Bartholomew, J. C., and Ames, B. N.
(2002) Proc. Natl. Acad. Sci. USA, 99, 1870-1875.
22.Atamna, H., Robinson, C., Ingersoll, R., Elliott,
H., and Ames, B. N. (2001) FASEB J., 15, 196-204.
23.Howes, R. M. (2006) Ann. N. Y. Acad. Sci.,
1067, 22-26.
24.Goldstein, N. (2002) Biochemistry
(Moscow), 67, 161-170.
25.Liberman, E. A., Topali, V. P., Tsofina, L. M.,
Jasaitis, A. A., and Skulachev, V. P. (1969) Nature, 222,
1076-1078.
26.Grinius, L. L., Jasaitis, A. A., Kadziauskas, Yu.
L., Liberman, E. A., Skulachev, V. P., Topali, V. P., Tsofina, L. M.,
and Vladimirova, M. A. (1970) Biochim. Biophys. Acta,
216, 1-12.
27.Bakeeva, L. E., Grinius, L. L., Jasaitis, A. A.,
Kuliene, V. V., Levitsky, D. O., Liberman, E. A., Severina, I. I., and
Skulachev, V. P. (1970) Biochim. Biophys. Acta, 216,
12-21.
28.Liberman, E. A., and Skulachev, V. P. (1970)
Biochim. Biophys. Acta, 216, 30-42.
29.Skulachev, V. P. (1989) Membrane
Bioenergetics, Springer Verlag, Berlin.
30.Severin, S. E., Skulachev, V. P., and Yaguzinsky,
L. S. (1970) Biokhimiya, 35, 1250-1257.
31.Smith, R. A., Porteous, C. M., Coulter, C. V.,
and Murphy, M. P. (1999) Eur. J. Biochem., 263,
709-716.
32.Kelso, G. F., Porteous, C. M., Coulter, C. V.,
Hughes, G., Porteous, W. K., Ledgerwood, E. C., Smith, R. A., and
Murphy, M. P. (2001) J. Biol. Chem., 276, 4588-4596.
33.Murphy, M. P., and Smith, R. A. (2007) Annu.
Rev. Pharmacol. Toxicol., 47, 629-656.
34.James, A. M., Cocheme, H. M., Smith, R. A., and
Murphy, M. P. (2005) J. Biol. Chem., 280,
21295-21312.
35.Kelso, G. F., Porteous, C. M., Hughes, G.,
Ledgerwood, E. C., Gane, A. M., Smith, R. A., and Murphy, M. P. (2002)
Ann. N. Y. Acad. Sci., 959, 263-274.
36.Saretzki, G., Murphy, M. P., and von Zglinicki,
T. (2003) Aging Cell, 2, 141-143.
37.Jauslin, M. L., Meier, T., Smith, R. A., and
Murphy, M. P. (2003) FASEB J., 17, 1972-1974.
38.Antonenko, Yu. N., Archipova, L. T., Archipova,
M. M., Bakeeva, L. E., Chernyak, B. F., Domnina, L. V., Fursova, A.
Zh., Grigorian, E. N., Ivanova, O. Yu., Izyumov, D. S., Khailova, L.
S., Klishin, S. S., Kolosova, N. G., Kopenkin, E. P., Korshunov, S. S.,
Korshunova, G. A., Kovaleva, N. A., Lyamzaev, K. G., Muntyan, M. S.,
Nepryakhina, O. K., Pashkovskaya, A. A., Philippov, P. P.,
Pletjushkina, O. Yu., Pustovidko, A. V., Rokitskaya, T. I., Ruuge, E.
K., Saprunova, V. B., Senin, I. I., Severina, I. I., Simonyan, R. A.,
Skulachev, I. V., Skulachev, M. V., Sotnikova, L. F., Sumbatyan, N. V.,
Tashlitsky, V. N., Trofimova, N. A., Vassiliev, Yu. M., Vyssokikh, M.
Yu., Yaguzhinsky, L. S., and Skulachev, V. P. (2007), in press.
39.Lakowski, B., and Hekimi, S. (1996)
Science, 272, 1010-1013.
40.Kruk, J., Jemiola-Rzeminska, M., and Strzalka, K.
(1997) Chem. Phys. Lipids, 87, 73-80.
41.Roginsky, V., Barsukova, T., Loshadkin, D., and
Pliss, E. (2003) Chem. Phys. Lipids, 125, 49-58.
42.Skulachev, V. P., Bakeeva, L. E., Chernyak, B.
V., Domnina, L. V., Minin, A. A., Pletjushkina, O. Yu., Saprunova, V.
B., Skulachev, I. V., Tsyplenkova, V. G., Vasiliev, J. M., Yaguzhinsky,
L. S., and Zorov, D. B. (2004) Mol. Cell. Biochem.,
256/257, 341-358.
43.Skulachev, V. P., et al., in preparation.
44.Trifunovic, A., Wreeenberg, A., Falkenberg, M.,
Spelbrink, J. N., Rovio, A. T., Bruder, C. E., Bohlooly, Y. M., Gidlof,
S., Oldfors, A., Wilbom, R., Tornell, J., Jacobs, H. T., and Larsson,
N.-G. (2004) Nature, 429, 417-423.
45.Solov'eva, N. A., Morozkova, T. S., and Salganik,
R. I. (1975) Genetika, 11, 63-71.
46.Kolosova, N. G., Lebedev, P. A., Aidagulova, S.
V., and Morozkova, T. S. (2003) Bull. Exp. Biol. Med.,
136, 415-419.
47.Sergeeva, S., Bagryanskaya, E., Korbolina, E.,
and Kolosova, N. (2006) Exp. Gerontol., 41, 141-150.
48.Kolosova, N. G., Shcheglova, T. V., Sergeeva, S.
V., and Loskutova, L. V. (2006) Neurobiol. Aging, 27,
1289-1297.
49.Vlachantoni, D., Tulloch, B., Taylor, R. W.,
Turnbull, D. M., Murphy, M. P., and Wright, A. F. (2006) Invest.
Ophthalmol. Vis. Sci., E-5773.
50.Rajendram, R., Saraswathy, S., and Rao, N. A.
(2007) Br. J. Ophthalmol., 91, 531-537.
51.Moreno, M. C., Campanelli, J., Sande, P., Sanez,
D. A., Keller Sarmiento, M. I., and Rosenstein, R. E. (2004)
Free Rad. Biol. Med., 37, 803-812.
52.Madeo, F., Frohlich, E., Ligr, M., Grey, M.,
Sigrist, S. J., Wolf, D. H., and Frohlich, K.-U. (1999) J. Cell
Biol., 145, 757-767.
53.Anisimov, V. N., Semenchenko, A. V., and Yashin,
A. I. (2003) Biogerontology, 4, 297-307.
54.Hamilton, W. D. (1964) J. Theor. Biol.,
7, 1-16, 17-52.
55.Dowkins, R. (1976) The Selfish Gene,
Oxford University Publishers, Oxford.