REVIEW: Versatile Functions of p53 Protein in Multicellular Organisms
P. M. Chumakov1,2
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; fax: (499) 135-1405; E-mail: peter@chumakov.com2Lerner Research Institute, Cleveland Clinic Foundation, 9500 Euclid Ave., Cleveland, OH 44195, USA; E-mail: chumakp@ccf.org
Received June 28, 2007
The p53 tumor suppressor plays a pivotal role in multicellular organism by enforcing benefits of the organism over those of an individual cell. The task of p53 is to control the integrity and correctness of all processes in each individual cell and in the organism as a whole. Information about the state of ongoing events in the cell is gathered through multiple signaling pathways that convey signals modifying activities of p53. Changes in the activities depend on the character of damages or deviations from optimum in processes, and the activity of p53 changes depending on the degree of the aberration, which results in either stimulation of repair processes and protective mechanisms, or the cessation of further cell divisions and the induction of programmed cell death. The strategy of p53 ensures genetic identity of cells and prevents the selection of abnormal cells. By accomplishing these strategic tasks, p53 may use a wide spectrum of activities, such as its ability to function as a transcription factor, by inducing or repressing different genes, or as an enzyme, by acting as an exonuclease during DNA reparation, or as an adaptor or a regulatory protein, intervening into functions of numerous signaling pathways. Loss of function of the p53 gene occurs in virtually every case of cancer, and deficiency in p53 is an unavoidable prerequisite to the development of malignancies. The functions of p53 play substantial roles in many other pathologies as well as in the aging process. This review is focused on strategies of the p53 gene, demonstrating individual mechanisms underlying its functions.
KEY WORDS: genetic stability, tumor suppressors, apoptosis, carcinogenesis, aging, cell growth regulationDOI: 10.1134/S0006297907130019
Abbreviations: CTD) C-terminal domain; DBD) DNA-binding domain; TA-domain) transcription activation domain.
Perhaps no other protein has been studied so thoroughly during the last
few years as p53. More than 40,000 papers have been published on p53
during the quarter of the century since its discovery, and the number
of studies continues to grow steadily. Such interest in p53 is due
primarily to the key role that this protein plays in the prevention of
cancer, the disease most feared by humankind. The elucidation of the
mechanisms related to functions of p53 has proceeded in parallel with
the uncovering of basic mechanisms underlying intracellular signal
transduction, genome reparation, regulation of cell divisions and cell
death, coordinate balancing of metabolic processes, and regulation of
cell-to-cell communications. Quite surprisingly, p53 functions were
found to be tightly related to each of the listed processes, and the
connections occurred at many levels, forming an integral regulatory
network. p53 was found to be not only capable of recognizing certain
deviations and errors that occur in the cell, but of responding very
discriminately depending on the degree of the deviation, which either
assists the correction of the affected processes, or defines further
fate of the cell. Some functions of p53 in the organism are similar to
those of a conductor of an orchestra, although the other functions
resemble those of a judge: p53 supervises proper execution of
evolutionary programs and controls proper pace and modes; it can also
define fate of cells in the organism.
While playing its control function, p53 is not required for the execution of processes: it intervenes only in cases of dangerous deviations from optimum. Likewise, p53 is not required for normal differentiation and development of the organism: the phenotype of p53-knockout mice is almost indistinguishable from that of the wild-type mice. The biological role of p53 is to ensure genome stability and genetic identity of cells in the multicellular organism. Acknowledging its important role in the organism, p53 is often described metaphorically, either as the guardian of the genome [1], or the guardian angel [2], or the gene of cell's conscience [3], and mutant p53 was compared to a fallen angel [4]. The definitions reflect the important role of p53 in preventing diseases and the dramatic events that follow the loss of p53 functions. Functions of p53 exclude extremes in physiological processes and thus prevent the development of pathologies. Functional insufficiency in p53 unavoidably leads to the development of serious diseases. On the other hand, many pathological processes are accompanied by hyperactive p53, which by itself can complicate the outcome of diseases. In addition to cancer, the deregulated activities of p53 are involved not only in malignant diseases: they also accompany cardiovascular, neurodegenerative, infectious and metabolic diseases, as well as participate in the processes of aging.
The task of this review is to discuss the significance of multiple functions of p53 and their influence on different pathways and processes in the cell. We would like to discuss apparently opposing strategies p53 takes in ensuring genetic stability and maintaining overall homeostasis, which play decisive roles in protecting health. We do not touch a rather diverse array of processes that lead to deficiency of p53-dependent mechanisms in cancer, as well as the issues related to potential significance of p53-dependent mechanisms for cancer therapy. These topics have been discussed in detail in our recent review [5].
A HISTORICAL OVERVIEW
The view considering a breakdown in the genes controlling cell divisions as a major cause of cancer became widely accepted in the second half of the 20th century [6]. Studies on viral carcinogenesis formed a concept describing oncogenes as mutant regulatory genes that induce malignant transformation [7]. By the early 1970s the technologies for studying molecular functions of genes had been developed, which boosted studies of the mechanisms of cell transformation by viral oncogenes. The small DNA tumor virus SV40 became a popular object for studying transformation in cultured rodent cells. Particularly promising were the studies of the SV40-encoded large-T antigen, as it was found to be the single gene that is necessary and sufficient for cell transformation [8].
In 1979, several reports described a cellular protein with apparent molecular weight of 53 kD that was able to form a tight complex with SV40 large-T antigen in transformed mouse cells [9-12]. Initially the protein was named a cellular T-antigen. The protein was considered as the potential critical target for SV40 large-T antigen, which is capable of mediating its transforming effects. In independent studies, the protein was found to accumulate in a variety of cell types transformed by other agents, as well as in cancer cells derived from human tumors [13]. In sera from cancer patients, antibodies reacting with cellular T-antigen (which soon was named p53) were frequently found, suggesting the involvement of the protein in human carcinogenesis [14]. The data suggested that, being a tumor antigen, p53 represents a frequent marker of tumor cells. The first hint on a possible function of p53 was obtained when microinjection into cultured cells of antibodies to p53 was found to cause a delay in the entrance to the S-phase of the cell cycle [15]. The finding suggested a role of p53 in regulation of cell divisions, which further boosted interest in this protein.
Successful molecular cloning of cDNA to p53 [16, 17], and then of the gene itself [18, 19], laid the foundation for modern studies at the molecular genetics level. By introduction into cells of recombinant expressor constructs, it was found that p53 can cooperate with oncogenic Ras in malignant transformation, and when introduced into primary rodent cells it can increase the number of cell divisions and even immortalize cells in culture [20-22]. It was found that the oncogenic properties of p53 can be significantly activated by the introduction of artificial mutations into different positions of the protein-coding region of p53 [23]. These findings added p53 to the list of potential oncogenes, while the frequent accumulation of p53 in human tumors suggested an involvement of altered (oncogenic?) p53 in cancer.
In 1989, however, the results were reconsidered with the realization that most of the initially cloned p53 cDNAs contained point mutations. Apparently, the observed oncogenic properties of p53 corresponded to mutant forms of p53 [24]. In fact, when p53 cDNA isolated from normal cells was expressed the oncogenic effects were not observed, and quite opposite, the expression resulted in the suppression of cell divisions in cell culture [25-27]. In human tumors frequently observed point mutations in one allele of the p53 gene coincided with deletions of the region containing the second allele [28]. This indicated that in human tumors the p53 gene is a frequent target for inactivation and suggested that the p53 gene is in fact a tumor suppressor gene, rather than an oncogene [29].
Confirming the anti-oncogene nature of p53 were the phenotypes of mice carrying experimental deletions of the p53 gene. Quite surprisingly, it was found that the newborn mice carrying homozygous deletion of the entire p53 gene display no apparent phenotypic differences compared to the wild-type mice. The p53 knockout mice develop normally with no apparent abnormalities until the age of 2-3 months, when an increased rate of malignant diseases becomes evident. By the age of 9 months, almost all homozygous p53 knockout mice die of malignant diseases, which are mostly thymic lymphomas [30]. Cultured embryonic fibroblasts obtained from the p53-knockout mice display tremendous chromosome instability, leading to preponderance of the polyploid and aneuploid cells after only few divisions in cell culture [31, 32]. The hasty accumulation of aneuploid cells continues through the lifetime of mice, being one of the causes of frequent lymphomas.
THE FUNCTIONS OF p53 ENSURE ALTRUISTIC BEHAVIOR OF CELLS IN THE
MULTICELLULAR ORGANISM
Since the early 1990s, a number of mechanisms by which p53 lowers probability of malignant diseases have been discovered. The p53 protein is capable of several different functions, which collectively enforce the benefits of the whole organism through making sacrifice of individual cells. The p53 functions develop in evolution only in multicellular organisms. Owing to activities of p53 the cells in a multicellular organism display altruistic behavior: damaged or weak cells choose to commit suicide rather than to enter competition with their healthy counterparts. The prohibited intercellular competition would result in restricted selection of genetic variants that demonstrate certain growth or viability advantages under particular conditions. Thus, the function of p53 ensures genetic stability and genetic uniformity of somatic cells.
To succeed in this major task p53 uses several alternative strategies. It is monitoring the integrity of individual structures as well as supervising the proper execution of various processes within the cell. Apparently, to prevent the formation of genetically altered progeny the cell needs to either intensify repair processes, or it must perish. The p53 dependent control mechanisms define the legitimate limits for deviation from optimum and the legitimate tolerable levels of exposure to external and internal stresses. Thus, p53 carries an important prophylactic function by preventing damages inflicted by harmful influences. Any cell that came under exposure to unfavorable conditions or received damages either needs to intensify the repair processes, or it needs to stop the divisions or even die. The cell's fate depends on the level and activity of p53, which in turn depends on the character and the intensity of the influence, as well as on the tissue-specific tuning of mechanisms controlling the activity of p53. In addition, the activity of p53 controls fine tuning of the balance in glycolytic and aerobic metabolism, the activity of antioxidant systems, and overall economy of energy expense under conditions of scarcity. Owing to activity of p53 there is constant scanning, monitoring, and assessing of virtually all processes within the cells, and the p53 itself is acting similar to an “Executor of God's Will” within the cell, that enforces the priority of interests of the organism over needs of an individual cell.
How does p53 manage to play such complex functions? Apparently, p53 does its job through interacting with multiple components of a common regulatory system, while playing the role of its master element. The system includes a complex and branching sensor component, which gathers information about the state of different structures, conditions, and processes within the cell. The integrated information is then adapted into retuning of the systems that modify functional state of p53. In concordance with the changed activity, p53 influences the effector components of the system, which determine the resulting effects and responses. However, the p53-mediated effects depend substantially on the type of a cell, or on the tissue-specific context.
While being subject to complex regulation, the activities of p53 are quite diverse. The most studied one is the transcriptional activity of p53. The structure of the p53 protein molecule has domain composition typical for transcription factors. The domains are responsible for the recognition and binding to specific DNA elements and interaction with components of the transcriptional machinery and transcriptional activation. By binding to regulatory regions, p53 can either activate some genes, or repress transcription of others. The majority of the effects produced by p53 result from the combined action of the genes that p53 is controlling. However, there is a distinctive tissue-specific signature in responses to activated p53, which is the result of the unique combination of initial levels and magnitudes of changes in individual p53-regulated genes in different cell types.
The transcriptional function is not the only activity of p53. Several transcription-independent functions of p53 have been described, such as its proapoptotic effect produced through direct translocation into mitochondria, its direct involvement in DNA damage recognition and repair, etc. The functional consequences of all these diverse activities of p53 fit into the common strategy of p53, which is the enforcement of genetic stability. Therefore, p53 achieves its strategic goals by all available means. It not only plays a dispatching and supervising role, but in certain instances it acts as a direct performer of rather particular and local tasks.
REGULATION OF ACTIVITY OF p53
Until quite recently the dominant view was that in normal cells, which are not subjected to various stresses, there is no or minimal activity of p53. The p53 gene is transcribed constitutively, and the mRNA is translated to p53 protein. However, the p53 protein is notoriously unstable, which is due to changeable degradation process that occurs in ubiquitin-dependent and ubiquitin-independent systems of 20S and 26S proteasomes [33, 34] (Fig. 1).
Degradation of p53 in 26S proteasomes is initiated by a set of E3-type ubiquitin ligases. The most studied of the E3 ligases is Mdm2, which is the product of a p53-activated gene [35-37]. The resulting increase of p53 activity leads to upregulation of Mdm2, and respectively, to accelerated degradation of p53 in 26S proteasomes. Therefore, the Mdm2 and p53 proteins form an autoregulatory feedback loop, which controls the activities of p53. Some other E3 ubiquitin ligases (Cop1, Pirh2, ARF-BP/MULE, CHIP) have been recently found to be connected to regulation the of p53 levels [38-41]. Similarly to Mdm2, the ubiquitin ligases Cop1 and Pirh2 are transcriptional targets of p53, thus being subject to mutual feedback effects. Obviously, the degradation of p53 is subject to complex regulation that is not yet studied in detail.Fig. 1. Regulation of degradation of p53 protein in proteasomes.
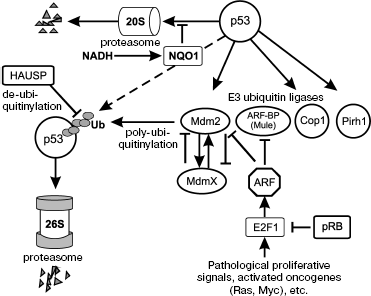
p53 can be also degraded by a ubiquitin-independent mechanism in 20S proteasomes [42]. This pathway for rapid protein turnover is characteristic for proteins that possess distinct unstructured regions; it is also used for rapid degradation of denatured proteins [43]. Masking of unstructured regions during the formation of certain protein-protein complexes can assist in stabilizing otherwise unstable proteins [34]. The protein molecule of p53 has unstructured regions in the N- and C-terminal parts, which drives its degradation in the 20S proteasomes. The entry of unstructured proteins into the 20S proteasomes is regulated by the NAD(P)H-dependent quinine oxidoreductase NQO1. In the presence of NADH the NQO1 binds to such proteins and prevents their degradation in the 20S proteasomes [34]. Various stresses that lead to DNA damage are able to increase the binding of NQO1 to p53, leading to its accumulation [42].
Degradation of p53 through the interaction with the ubiquitin ligase Mdm2 remains the most studied and, perhaps, the most important mechanism for regulation of its activity. The interaction of p53 with Mdm2 is subject to a fine regulation by a variety of mechanisms [44]. While some of the mechanisms are connected to regulation of Mdm2, others aim modifications in its target--the p53 itself [45].
An important player that regulates the activity of Mdm2 is the closely related protein MdmX. This protein has very similar composition [46], although unlike Mdm2 it does not have the E3 ubiquitin ligase activity [47]. The MdmX protein binds to the N-terminal region of p53, suppresses its transcription activity [48], but does not induce its degradation. In addition, MdmX can oligomerize with Mdm2 [49, 50], which leads to stabilization of Mdm2 [51] and accelerated degradation of MdmX [52]. Therefore, changes in the proportion of these two proteins can lead to fine regulation of levels and activity of p53.
The complex formation of p53 with Mdm2 and MdmX proteins is also a regulated process. Certain ribosomal proteins (L5, L11, and L23) bind to Mdm2, inhibit its activity toward p53 [53-55], and stimulate degradation of MdmX [56]. This mechanism is responsible for the activation of p53 under certain conditions that are characterized as ribosomal stresses [57, 58].
The activity of Mdm2 can be modified as a result of its binding to the transcription activator protein p300/CBP, leading to a switch in the ability of Mdm2 to conduct mono-ubiquitinylation of p53 toward preferential poly-ubiquitinylation [59], which is required for recognition and degradation of p53 in the 26S proteasomes [60]. Transcription factor YY1 [61, 62] and transcription repressor KAP1 [63] can also assist in degradation of p53, as their binding to Mdm2 stabilizes the complex of the latter with p53. Degradation of p53 can also be stimulated by binding of Mdm2 to gankyrin, which is capable of interacting with ATPase S6 of proteasomes [64].
Similar to p53, the MDM2 protein can also be degraded in the 26S proteasomes, although this process can be regulated by specific de-ubiquitinylating enzymes. One of such enzymes, the HAUSP protein, is able to remove the ubiquitin moieties from Mdm2 [65]. Another protein Daxx can form complexes with Mdm2 and HAUSP and prevent self-ubiquitinylation of Mdm2 [66], leading to its stabilization [67] and accelerated degradation of p53. In addition, HAUSP can remove ubiquitin from p53 itself leading to its stabilization [68]. By inducing opposite effects within the p53 degrading system, the HAUSP protein can mediate fine and discriminative regulation of p53 activity.
An important regulator of the Mdm2-dependent degradation of p53 is p14ARF (or ARF [69]), the product of an alternative reading frame of the CDKN2A gene that in addition encodes the CDKs inhibitor p16. ARF is a very basic protein that contains 20% arginine and no lysine residues. In the unbound state ARF is poorly structured, although it tends to form complexes with other proteins that neutralize the positive charge. ARF has tumor suppressor activity, and its absence leads to a phenotype that resembles deficiency of p53 [70]. One of the binding partners of ARF is the Mdm2 protein. By binding to Mdm2, ARF inhibits its ubiquitin ligase activity, leading to p53 stabilization and the induction of apoptosis [71-73].
Transcription of the ARF gene is subject to positive and negative regulation by complexes that contain transcription factor E2F1 [74, 75], which in turn is regulated by pRB. In normal tissues, the transcription level of ARF is low. However, upon oncogenic activation or sustained stimulation of proliferation, the ARF gene is activated at the transcription level. The accumulated ARF protein blocks Mdm2 and induces p53, which increases sensitivity of cells to apoptosis [76].
ARF can also block the other E3 ligase ARF-BP (or MULE), which also participates in degradation of p53. However, in addition to p53 the E3 ligase ARF-BP is involved in degradation of some other proteins, including a proapoptotic protein Mcl1 [77]. Therefore, the ARF protein serves as regulator and activator of several different systems that potentially prevent genetic lesions and protect an organism from the development of pathologies [41].
ARF is not the only factor that mediates upregulation of p53 in response to oncogene activation. Recently a quinine oxidoreductase Seladin-1, which is known as one of the key enzymes in cholesterol biosynthesis [78], was found to be able to displace p53 from its complex with Mdm2 in response to oxidative stress and oncogene activation [79]. Therefore, there exists a large network of factors that interact with each other and determine the rate of ubiquitinylation and degradation of p53.
In addition to changes in the systems that regulate levels of p53, important roles are played by various modifications of the p53 molecule, which not only change quantity, but also the quality and character of p53. The modifications include phosphorylation, acetylation, methylation, and covalent attachment of ubiquitin and ubiquitin-like proteins SUMO and NEDD8.
An additional level of complexity relates to a recently discovered array of different alternatively-spliced products of the p53 gene [80], and an additional N-terminally-truncated protein isoforms that originate from the internal initiation of translation [81, 82]. The significance of multiple isoforms of p53 is yet to be determined.
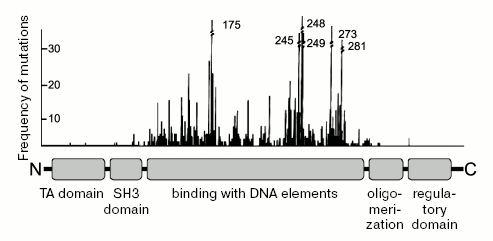
The active molecule of the p53 protein (at least the one that functions as a transcription factor) exists as a homo-tetramer [83]. The monomeric subunit of p53 has characteristic domain organization (Fig. 2). The N-terminal region (amino acids 1-73) contains a multipartite transcription activation (TA) domain and an adjacent (amino acids 63-97) proline-rich SH3 domain. The region responsible for the recognition and binding to the specific elements in the DNA is localized in the central third of the protein molecule (amino acids 94-312). This region is the target for the majority of oncogenic point mutations within the p53 gene. The oligomerization domain is localized closer to the C-terminus (amino acids 325-355), proximal to the basic and unstructured C-terminal domain (amino acids 360-393) that plays an important role in the regulation of p53 activity [84].
Covalent modifications in the p53 protein are introduced by a variety of enzyme systems in response to different influences. The modifications change the functional portrait of p53, which mediates adequate responses to changing conditions. More than 20 different amino acid residues in the p53 molecule are known to be acceptors for covalent modification [85]. The sites of modification cluster within the N- and C-terminal regions of p53. Modifications within the N-terminal region of p53 (such as phosphorylation of Ser15, Ser20, and Thr18) can interfere with the binding of p53 to Mdm2 and other E3-ligases, which leads to protein stabilization [3]. Depending on the type of modifications within the p53 molecule, there can be modulation of its ability to interact with co-activators and co-repressors of transcription and changes in the preferences for the p53 binding to responsive elements in different genes. Various modifications at the C-terminal region of p53, such as phosphorylation, acetylation, methylation, and covalent attachment of ubiquitin-like proteins SUMO [86] and NEDD-8 [87] neutralize the inhibiting effect of the C-terminal regulatory segment, leading to further stabilization of p53, either positive or negative modulation of its activity, and changes in intracellular localization.Fig. 2. Main functional domains of p53. The upper panel shows the distribution by p53 molecules of frequencies of oncogenic mutations revealed in human tumors.
Different conditions change the ability of p53 to interact and cooperate functionally with a variety of other proteins, although it is difficult to predict what particular impact a particular interaction would have on p53 functions, as the result depends substantially on the particular cell type.
Many different proteins are able to bind to p53 and introduce covalent modifications in its molecule. These proteins include more than thirty different protein kinases, a few protein phosphatases, several ubiquitin ligases, and factors that regulate interaction of p53 with the protein degradation machinery. There are also several methylases, deubiquitinylating enzymes, acetyltransferases, deacetylating enzymes, and proteins that participate in coupling to p53 of SUMO and NEDD8. In addition to the p53-modifying enzymes, p53 interacts with a number of proteins and forms functionally significant complexes. For example, a protein belonging to the peptidyl-proline isomerase family, Pin1, binds to the N-terminal region of p53 following its modification by phosphorylation and introduces conformational changes that modify the activity of p53 [88, 89]. Various components of the transcriptional machinery interact with p53 during its function as a transcription factor. Some proteins can bind to p53 depending on its redox state (HIF1alpha, Ref-1, thioredoxin, Wox1, COX2, NQO1) and affect its function [90-95]. p53 can bind to a large group of proteins that participate in DNA repair (RAD51, 53BP1, BRCA1/2, BARD1, MDC1, HMG1, BLM, WRN, MRE1, RPA1, ERCC6, SNF5, DNApolalpha, mtDNApolgamma), which either affects the efficiency of repair systems, or serves as part of a mechanism of signal transduction to p53. Particularly important is the interaction of p53 with the ASPP1 and ASPP2 proteins [96, 97] and with their antagonist iASPP [98]. The interaction changes relative affinity of p53 binding to different DNA elements located within the p53-modulated genes and thus modulates the output of the p53 induction--either toward increasing survival or toward the induction of cell death [99]. By acting through a transcription-independent mechanism, p53 can induce apoptosis by directly interacting with certain apoptosis regulator proteins, such as Bax [100], Bcl-XL [101], Bnip3L [102], and Scotin [103]. Finally, it is known that p53 can participate in interactions with dozens of other proteins, although the functional significance of some of the interactions is yet to be determined.
TRANSDUCTION OF SIGNALS TO p53
In early studies it was observed that the activity of p53 is stimulated in response to DNA damage, as well as in response to transfection into cells of partially degraded DNA [104-106]. In was suggested that p53 is capable of blocking divisions of cells that acquired damaged DNA. However, later it became clear that there are several alternative mechanisms of p53 induction, not only in response to different types of direct DNA damage, but also in response to various genotoxic influences, or the conditions that do not necessarily affect DNA directly, but that can eventually compromise the genome integrity [44].
Various faults and errors in a wide variety of physiological processes create conditions that can increase the risk of generation of genetically altered cells. There is a growing list of conditions that apparently affect p53 functions, suggesting that p53 monitors and responds to a variety of potential hazards. We already mentioned the induction of p53 in response to deregulated balance of proliferative signals, which particularly occurs during sustained activation of oncogenes. In this case it was found that the alarming signals are generated and transduced through the transcription factor E2F1, which controls the expression of products required for DNA synthesis and simultaneously stimulates transcription of the ARF gene [107]. The overexpressed ARF protein inhibits the activity of Mdm2, leading to accumulation of p53. Although, as oncogene activation induces dramatic changes in many signaling pathways, the induction of p53 can be initiated though several alternative mechanisms. Indeed, severe changes in morphology, unusual interactions with extracellular matrix, altered cell-to-cell contacts, changes in metabolism, elevated levels of reactive oxygen species, unusual prolonged activation of some signal pathways that leads to a misbalanced processes, accelerated consuming of energy resources and local depletion of nutrients, exhausted pools of nucleotides--these and many other features acquired in response to oncogene activation can send multiple alarming signals to p53 [44, 108].
Several characteristic conditions have been described that lead to activation of p53, which include the exhaustion of nucleotide pools [109], faults in cytoskeleton (altered polymerization of actin fibers and microtubules) [110-112], altered ribosome biogenesis [113], the condition of hypoxia and ischemia [114], hyperoxia [115], lack or deficit of certain growth factors and cytokines [116, 117], altered adhesion and focal contacts [118], defective integrins [119], abnormal attachment of cells to a substrate (leading to the p53-dependent anoikis, or death of unattached cells) [120], accumulation of polyploid cells [32, 121], formation of micronuclei [122], destruction or malformation of mitotic spindle [112], hypo- and hyperthermia [123, 124], and exposure to nitric oxide (NO) [125]. These conditions induce characteristic modifications, either within the protein molecule of p53, or in the systems that control levels and activity of p53. In turn, changes in the activity of p53 determine the fate of an altered cell. The fate depends significantly on the tissue origin or the cell type, as different cell types display a rather individual range of parameters that is recognized as normal or optimal.
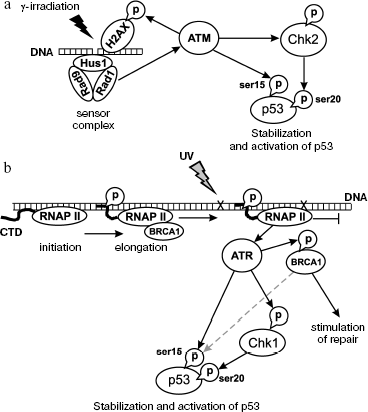
To date the DNA damage-induced mechanisms of p53 activation are studied in most detail. As an example, let us consider processes that take place after exposure to two different DNA damage-inducing influences - ionizing gamma radiation and UV (Fig. 3). The ionizing radiation produces mostly breaks in DNA, while the UV causes cross-links in DNA due to formation of thymine dimers. The DNA breaks are recognized through the action of several sensor proteins, such as heterotrimers of RAD9, RAD1, and Hus1 proteins that act together with the RAD17 protein. The signal is converted to activation of the ATM kinase, which phosphorylate several targets [126, 127]. One of the targets is histone H2AX, and its phosphorylation in the damaged regions of chromatin serves as a signal for the recruitment of repair systems. In addition, the ATM kinase phosphorylates several proteins that participate in activation of p53. The ATM kinase can participate in direct activation of p53 by phosphorylation of its Ser15 [128]. Other targets of the ATM kinase include the checkpoint kinase Chk2 [129], which becomes active and in turn phosphorylates p53 at Ser20 [130].
Recognition of bulky DNA damage that blocks the process of elongation by RNA polymerase II is achieved through different mechanisms. The bulky DNA damage is produced, in addition to UV, by certain other influences, such as the anticancer drug cisplatin. This type of damage is characterized by the introduction of road blocks on DNA that prevent further movement of RNA polymerase II. The RNA polymerase II transcription complex is equipped with an associated system that is entitled to remove DNA damage, which is called the transcription-coupled repair (TCR) system [131]. In addition, the RNA polymerase II complex plays a role of a sensor of bulky DNA damage that blocks transcription. During RNA synthesis, the polymerase is able to scan rather long portions of the genome. It is quite significant that transcription inhibitors that block RNA polymerase II movement during the elongation phase can also activate p53, which is found modified at Ser15 and Lys382 [132]. Very similar processes are initiated during blocking the elongation by the bulky DNA damage. In either case, there is activation of ATR kinase, which is similar to the ATM kinase [133]. Like the ATM kinase, the ATR kinase can phosphorylate p53 directly at Ser15, but in addition it activates a checkpoint kinase Chk1, which similar to the Chk2 kinase can phosphorylate p53 at Ser20 [134]. The other mechanisms that recognize stalked polymerase include the BRCA1 protein, which associates with the RNA polymerase II during the elongation phase when its C-terminal domain is fully phosphorylated [135]. After transcription elongation block, the BRCA1 protein is modified by phosphorylation leading to its release from the transcription complex [136]. As BRCA1 can interact with many different proteins, its release from the RNA polymerase II complex can, perhaps, facilitate the recruitment of repair enzymes; it can additionally signal to systems that activate p53 [137].Fig. 3. Scheme of basic mechanisms of p53 induction by penetrating radiation (a) and ultraviolet irradiation (b).
There are many other pathways that can potentially transmit signal to p53 either from the systems that recognize DNA damage or from the DNA repair systems. The p53 itself can serve as one of the sensors, as it can bind preferentially to the damaged regions on DNA [138-141]. Owing to its C-terminus, p53 can bind to the damaged regions, and it can even serve as a repair enzyme itself. The p53 protein molecule possesses 3´-5´-exonuclease activity, which is able to excise damaged DNA regions, and the gap can then be filled in by other components of the repair systems [142]. It has been suggested that the direct involvement in DNA damage repair represents the most ancient function of p53, which is supported by the fact that the exonuclease activity is present even in the ancestral p53 of invertebrates [143]. Functions of p53 have probably evolved from its direct participation in DNA repair toward the acquisition of the ability to supervise over the whole system controlling genetic stability. The latter could become possible as p53 acquired the activity of a transcription factor.
By binding to the regions of DNA damage, p53 can become exposed to phosphorylation by the DNA-dependent protein kinase (DNA-PK), or by the ATM and ATR kinases, which lead to its nuclear accumulation and activation of its transcription function. There are other collateral connections linking p53 to repair systems as p53 can interact with several proteins that are either repair enzymes or components of the DNA replication machinery. These include RPA [144], BLM [145], WRN [146], RAD51 [147], ERCC2/3/6, and XRCC9 proteins [148]. Although functional consequences of these complexes are still not quite clear, it looks like the interactions can affect both the functions of p53 and the efficiency of the repair processes. One of the examples is the ability of p53 to interact with Ref-1 protein [94], which acts as an endonuclease that removes apurinic regions in DNA [149, 150]. In turn, the Ref-1 protein can functionally modify p53 by reducing one of its oxidized cysteines [151]. Therefore, the processes couple the systems of replication and DNA repair with p53. The coupling allows p53 to gather information regarding the state of DNA well before the onset of DNA synthesis.
TRANSCRIPTION ACTIVITY OF p53
Although p53 has several very important activities, being an enzyme, or an adapter protein, or a co-factor in a set of active protein complexes, the most functionally significant activity of p53 is certainly the ability to regulate transcription of genes and to serve as a transcription factor [152, 153]. The role of p53 in transcription is diverse and controversial. When p53 binds to its specific responsive DNA elements it can either activate or repress transcription of appropriate genes. It can also repress genes without binding to DNA owing to its ability to inhibit the general transcription machinery through blocking transcription factors CBP, TBP, and Sp1 [154-156].
The specificity of transcription activation depends on the ability of p53 to interact with the regulatory regions of certain genes. Active tetrameric p53 can recognize and bind the DNA sequence elements that are composed of two adjacent ten-nucleotide segments, the half-sites, each separated by few nucleotides. The consensus structure of the p53-binding element includes two half-sites, each composed of two tandem pentamers with tail-to-tail orientation: PuPuPuCA/TA/TGPyPyPy-(Nn)-PuPuPuCA/TA/TGPyPyPy, where Pu is purine and Py is pyrimidine [157-159]. There are rather large variations from the consensus structure as single or multiple changes can be found in one or in both half-sites [160]. The degeneracy of the p53-binding element specifies the heterogeneity in p53 binding, which partially explains the apparent lack of synchrony in the induction of individual p53-regulated genes [161, 162]. The p53-binding elements can be located not only in immediate upstream regions of the gene, but often at large distances and even in the intronic regions [163]. Some of the p53-regulated genes may have two or even more p53-binding elements, which are often separated by large distances. p53 can also recognize elements that have substantial deviations from the consensus structure, and still the elements can be rather efficient in mediating the transcription activation [164, 165]. The rules for composition of such elements are still not clear. Along with the sequences that bind p53 and mediate transcription activation, there is a kind of DNA elements that conducts p53-dependent transcription repression. Such elements have a similar structure, but the pentamers in their half-sites are oriented in tail-to-head order. By binding to such elements p53 undergoes conformational changes that provide affinity toward components of the Sin3A complex containing histone deacetylases, leading to transcriptional repression of the corresponding gene [166, 167].
The p53 protein recognizes and binds to specific DNA elements through its central DNA-binding domain (DBD). The active p53 is organized as a homotetramer in which each monomeric p53 is bound through its oligomerization domain located near the C-terminus, and the DBD of each monomer is bound to the appropriate quarter-site of the DNA element [168]. Amino acid changes in virtually any residue of the DBD (this domain occupies approximately 100 amino acids) make the DNA binding looser. When a mutation hits certain critical regions, which are important for holding several loops of the protein chain, the whole interface between p53 and DNA collapses and the mutated protein looses completely the ability to bind the DNA elements.
Virtually every amino acid change within the DBD leads to loss of activity, which explains very high frequency of missense mutations in the p53 gene in nearly half of all cancer cases. To date approximately 20,000 types of missense mutants of the p53 gene are described. Apparently, each of the mutants provides some extent of inhibition of p53 functions leading to positive selection for mutant p53 in tumors [29, 169].
In early studies it has been established that mutant forms of p53 can cause transdominant (or dominant-negative) effect on the endogenous wild-type p53 [170, 171]. Meanwhile, in tumors the mutations in one allele of the p53 gene are usually accompanied by deletions of the second allele owing to the partial or complete loss of the second chromosome 17 [28]. In later experiments when ratios between mutant and wild-type p53 were controlled, it was found that the transcription activity is completely inhibited only in cases when there are more than three mutant p53 subunits per tetrameric complex [172]. In the mixed tetrameric complex containing both mutant and wild-type p53 the affinity of binding to DNA in the region that contact the mutant p53 subunit is substantially reduced, which leads to the reduced binding affinity of the whole complex. The lower affinity leads to functional consequences. While higher levels of p53 are required for the activation of strongly p53-responsive genes, the genes that initially contain low-affinity p53-binding elements can loose the p53 responsiveness completely. Certainly, mutations in one copy of the p53 gene result in certain mitigation of genetic stability control, which increases probability of the second allele loss through a genomic deletion. In the end, the cell losses completely the control over genetic integrity and it enters the process of evolution that is characteristic for unicellular organisms. The process proceeds toward maximal autonomy of the cell, leading to the formation of malignant tumor.
Similar mechanisms may underlie the functional heterogeneity of the p53-binding elements. The affinity of interaction between each monomer of the tetrameric p53 would be lower depending on the number of exceptions from the consensus structure in each of the pentameric quarter-sites of the p53-binding element. The mismatches would result in substantial conformational distortions in the tetrameric complex bound to DNA, thus affecting the overall affinity of the interaction. As a result, the induction kinetics for genes that have p53 elements with certain deviation from the high-affinity consensus structure may vary substantially, and therefore each of the p53-regulated genes responds individually to a range of p53 levels that is unique to a particular gene. The individual tuning of the p53-mediated effects for each of the responsive genes allows p53 to play a role of a cunning conductor for the whole orchestra of the p53-regulated genes. The effects of p53 represent the sum of activities produced by individual levels of hundreds of the p53 regulated genes, and the tuning of the whole system allows p53 to produce effects that are adequate to any particular condition of the cell.
The C-terminal domain (CTD) of p53 is also involved in interactions with DNA. This unstructured domain is rich in serine and arginine residues and serves as a target for various regulatory modifications (phosphorylation, acetylation, methylation, ubiquitinylation, binding of SUMO and NEDD8), which affect both the stability of p53 and its transcription activity. The CTD of p53 is capable of nonspecific sequence-independent DNA binding, with particular binding preference toward nonlinear DNA segments, including damaged regions of DNA [138-141].
Many earlier studies focused on the ability of the CTD to inhibit the specific DNA binding activity of p53, which had been testing in an in vitro reaction measuring shifts in electrophoretic mobility of short oligonucleotide duplexes representing consensus sequence of the p53-binding element [173]. According to the early model the modifications within the CTD remove structural obstacles that prevent interaction of p53 with specific DNA elements, which converts latent non-active p53 to the DNA-bound transcriptionally active form [174]. This model was rejected after it was found that the CTD does not affect interaction of the p53 consensus elements contained within longer segments of DNA. By a chromatin immunoprecipitation method, it has been established that the CTD modifications do not affect the presence of p53 in the region of the p53-binding element on chromatin [175, 176]. It seems likely that prior to binding to a specific DNA element p53 interacts with a nonspecific DNA stretch through its CTD, which leads to conformational changes and to activation of the central DNA-binding domain. Such activation does not occur in case of binding to short consensus oligonucleotide duplexes that do not contain flanking stretches of DNA. Therefore, binding to genomic DNA includes certain steric modifications within the CTD that expose the central DNA-binding domain. It is likely that the regulatory function of CTD plays a role in the modulation of the transcription activity of p53, which is already bound to a DNA element, or it can affect the stability of p53. One can also speculate that modifications within the CTD act as a switch in p53 activities, from the recognition and excision of damaged regions of DNA toward the initiation of its transcription functions.
The DNA binding activity of p53 can be also regulated by changes in the redox state of the protein, particularly by reducing the cysteines residues. The redox-active protein Ref-1 can play such a role depending on the redox state in the cell. It was found that treatment with selenomethionine, which affects redox conditions in the cell, can enhance the activity of Ref-1; this affects the spectrum of the p53-modulated genes in favor of the DNA repair genes [177-181]. The DNA-binding activity of p53 can be also modulated by certain metabolites. High levels of ADP or dADP increase DNA binding, while ATP and GTP act in the opposite direction [182]. The inhibition of DNA binding occurs also in case of increased levels of TDP and NAD+ [183], which establishes a direct link between the rate of major metabolic processes in the cell and the activity of p53.
The products of the p53 family--the p63 and p73 proteins--can also affect the efficiency of interaction of p53 with its DNA elements, as all members of the p53 family bind to very similar (if not identical) DNA elements. An additional level of complexity arises from the fact that each member of the p53 family has several isoforms. The isoforms have distinct functions, and by binding to each other in heterooligomeric complexes they can produce multiple functional combinations [184]. Apparently, the interactions among different isoforms might modify substantially both the activities of p53 and the activities of its family members. Binding of p53 to a DNA element does not necessarily induce transcription activation, because the activation may require some additional influences that eliminate the repressor effect from certain p53-interacting proteins, such as Mdm2 and MdmX. Therefore, the DNA element that is already occupied by a particular type of complex would be somewhat protected from the influences of other isoforms. In light of this competition, the tissue-specific differences in the p53-mediated responses may be explained by the fact that there is wide variation in the expression levels of p63 and p73 isoforms in different cell types.
The presence of mutant p53 in tumor cells results in certain modifications in the activities of p63 and p73, which is reflected by the acquisition of so-called gain of function activities of mutant p53 [185-191]. The gain of function activities promote further increase in malignancy and increased resistance to anticancer therapy [192, 193].
The transcriptional activation is determined by the N-terminal segment of p53, which contains several regions interacting with the transcription machinery and recruiting factors that modify local chromatin structure. The N-terminal segment of p53 contains frequent combination of aspartate and glutamate residues characteristic to the acid activator domains of proteins that interact with components of the transcription machinery [194]. The transcription activation (TA) domain of p53 is weakly structured [195, 196], forming short alpha-helical regions [197], which collectively form a scaffold for binding of several factors. Introduction of single amino acid substitutions within the TA domain does not usually lead to substantial changes in the transcription activity, which explains the observed low frequency of tumor derived mutations falling in this region. However, introduction of paired mutations has allowed identifying two critical regions: the substitution of two hydrophobic amino acids Leu22 and Trp23 dramatically inhibits the transcription activation capacity [198], while mutations of the Trp53 and Phe54 lead to selective inhibition of induction of some proapoptotic p53 target genes [199]. Later the TA domain was divided into relatively independent subdomains located between amino acids 1-42 and 43-73, respectively [200], though the interaction of p53 with components of the transcriptional machinery involves some additional segments as well. The region between amino acids 63 and 97 is responsible for the interaction with peptidyl prolylisomerase Pin1, which mediates conformational changes within the TA domain following activating phosphorylation at the N-terminus of p53 [88, 89]; the same region binds to the transcriptional co-activator protein CBP/p300 [201]. The TA domain is flanked by a recently identified repressor domain (located between amino acids 100 and 116), although the mechanism of its function has not yet been determined [202].
The details of transcription activation by p53 are still not quite understood, although there are numerous evidences suggesting that the binding of p53 to a specific DNA element in a gene assists the recruitment of chromatin remodeling factors. This leads to promoter opening for its further interaction with the components of the transcription machinery [203-205]. Some of the components include the p53-interacting histone acetyltransferases and histone metyltransferases, thus modifying not only the opened region of chromatin but also the p53 itself, which changes its activities [206-210]. In addition to its effects on chromatin, p53 participates in transcription activation by direct recruitment of certain basal transcription factors, such as TFIIA and TFIID [194, 211]. Finally, an additional model for the p53-mediated transcription regulation cannot be excluded that involves stimulation of re-initiation, or elongation of preformed short attenuator transcript, which has been frozen shortly after its initiation [212].
The control regions of genes that determine transcription of the downstream sequences have composite organization and bind to many different factors. Transcription factors can interact with each other both functionally and physically, which complicates predictions concerning the final effect. The ability of p53 to activate or repress a gene is determined not only by the presence of appropriate DNA binding element, but also by additional transcription factors that control expression of the gene.
Bioinformatic calculations suggest that more than 4000 genes in the human genome contain p53 binding elements [213], although the experimental estimations based on chromatin immunoprecipitation reduce the number of p53 regulated genes to 500-1600 [214, 215]. Remarkably, only half of the genes that actually bind p53 to the DNA elements demonstrate increased transcription upon p53 activation; the other half of the genes respond to p53 by reduced transcription [215].
The ambiguity of the responses to p53 may be explained by cross talk between different transcription factors. Various physiological or pathological influences usually induce several different signaling pathways that involve a number of transcription factors. Therefore, response to the influences that induce p53 may differ substantially from that observed in the experiments involving simple overexpression of recombinant p53. Particularly, in response to limited supply of nutrients, the combined effect of interacting p53 and transcription factor Foxo3a results in a switch from the p53-dependent repression of SIRT1 gene to its activation [216]. Another significant example is the interaction of p53 with the transcription factor NFkappaB. It is known that NFkappaB affects regulation of apoptosis through several different pathways, and usually the resulting effect is antiapoptotic. In certain systems, NFkappaB can even block the p53-induced apoptosis. However, quite surprisingly, activities of NFkappaB were found to be required for the efficient induction of cell death mediated by activated p53 [217, 218]. Apparently, NFkappaB is able to cooperate with p53 in the regulation of certain genes, such as KILLER/DR5, which are required for the induction of apoptosis [219]. Cooperation of p53 with the transcription factor Miz is required for the efficient induction of CDKN1A (p21) gene [220]. Likewise, the p53-dependent activation of the BBC3 (PUMA) gene is inhibited by combined effect of p53 and SLUG, which protects hematopoietic precursor cells from p53-dependent apoptosis [221]. In cooperation with KLF5 there is abrogation of the p53-dependent repression of the gene encoding apoptosis inhibitor Survivin [222]. A functionally opposite effect is produced by the cooperation with transcription factors YB1 and MUC1, resulting in selective inhibition of the p53-mediated induction of certain proapoptotic genes [223, 224].
The balance between the proapoptotic and pro-survival p53-regulated genes can be regulated by products of the ASPP1/ASPP2/iASPP gene family. The ASPP1 and ASPP2 proteins can bind to p53 in the region overlapping the TA and DNA-binding domains leading to modified binding preference of p53 toward different DNA elements [97]. As result, the expression of p53-dependent proapoptotic genes is increased [96]. The iASPP protein serves as an antagonist of ASPP1 and ASPP2, leading to inhibition of the apoptotic activity of p53.
It is likely that very similar structural changes accompany phosphorylation of p53 at Ser46, which is located within the second segment of the TA domain. The Ser46 phosphorylation is mediated by protein kinase C (PKCdelta) [225], and the process of PKCdelta binding to p53 is assisted by the p53-regulated gene product p53DINP1 [226]. Meanwhile, another p53-regulated gene encoding protein phosphatase PPM1D/Wip1 acts in the opposite manner by interfering with phosphorylation at Ser46 [227]. The Ser46 phosphorylation facilitates the interaction of the TA domain of p53 with p62/Tfb1 [228], a subunit of the transcription factor TFIIH, leading to changes in the spectrum of the p53-induced genes in favor of proapoptotic genes [229] with simultaneous release of the p53-dependent repression of the genes encoding antiapoptotic factors, such as the galektin-3 protein [230]. Therefore, regulation of the transcription activity of p53 includes numerous components and mechanisms allowing the cells to respond discriminately to various situations in order to take optimal decisions.
THE p53-REGULATED GENES
The transcriptional activity of p53 specifies the majority of its functions. This is why quite significant efforts have been invested in the identification of p53-regulated genes. Tracing of the p53-regulated genes was conducted both by identification of potential p53-binding elements in the DNA, and by monitoring the changes in expression of genes that take place in response to activation or inhibition of p53. An important role in the identification of p53-regulated genes has been played the SAGE (serial analysis of gene expression) method [231, 232], as well as the use of hybridization with microarrays specific for human transcripts [233].
Two genes specifying negative regulators of cell divisions, the Gadd45 protein [163] and the cyclin-dependent kinase inhibitor p21 [234], were among the very first identified p53-activated genes. Then the list was enhanced by a set of genes that participate in the induction of apoptosis [235, 236]. The character of the identified p53-regulated genes was in good agreement with the observations that following activation of p53 there was either inhibition of the cell cycle progression at certain checkpoints, or even the induction of cell death. Further studies have revealed additional p53-regulated genes, which contributed to the understanding of a much wider role for p53 in the control of important processes in the cell. Below we will describe several groups of p53-regulated genes, each targeting characteristic functional output.
Genes participating in cell cycle control. The ability of cells to stop in late G1 phase of the cell cycle is important for the maintenance of genome stability, as it prevents the replication of damaged DNA. An important role in the p53-dependent G1 phase block is played by the p53-regulated p21, which inhibits CDK2 and CDK4, thus preventing the phosphorylation of pRB, the initiation of transcription of genes encoding cyclin K, hCDC4, p53RFP, and a set of genes responsible for the DNA biosynthesis [233]. Some other p53-regulated genes, such as BTG2 (inhibits cyclin E1) [237], and MCG10 [238] contribute to the p53-dependent G1 block. The p53 protein can also block the cell cycle progression during the DNA synthesis S-phase, and it is likely that the activity is mediated by alternative isoforms of the p53 protein [239], which participate in the induction of the 14-3-3sigma and p21 proteins [240]. Various damages and faults during the S-phase induce the ATR-dependent activation of the Chk1 kinase, which modifies p53 at Ser20. However, most likely the major function of the predominant full-length isoform of p53 during the S-phase is in the activation of DNA repair, rather than in the inhibition of the cell cycle [241]. The ability to block the G2/M transition is important to prevent segregation of damaged and incompletely replicated chromosomes. The block at this checkpoint occurs through the inhibition of Cdc2-cyclin B complex, while several p53-inducible genes, such as GADD45, BTG2, B99 (GTSE-1), REPRIMO, HZF, and MCG10 may assist in this process [241, 242].
Genes that participate in DNA repair. The genes encoding components of the global genome repair (GGR)--the DDB2 (XPE) [243] and XPC [244]--are among the targets of p53. This is why the p53-deficient cells tend to switch the excision repair from GGR to the transcription couples repair (TCR) [241]. Certain factors of the DNA mismatch repair, such as MSH2, PCNA, MLH1, and PMS2 [245-248] are also encoded by the p53-inducible genes. It is noteworthy that after severe DNA damage that induces substantial increase in the p53 level, the accumulating p53-dependent PMS2 is able to enhance the proapoptotic function of p73, which suggests a role for PMS2 as a damage dosimeter [241]. The p53 protein induces DNA polymerase eta_[249], which assists DNA repair at the replication forks. It can also inhibit processes that involve homologous recombination--not only through activity modulation owing to the direct binding to such factors as RPA, RAD51, WRN, and BLM, but also through the transcription of RAD51, WRN, and RecQ4 genes [250-252]. Finally, p53 induces the p53R2 gene encoding a structural homolog of ribonucleotide reductases, which is important for maintenance of the nucleotide pools during the DNA repair [253].
Genes that regulate angiogenesis and other tissue reactions. These genes play important roles by limiting tumor growth and by preventing events that aid survival and spreading of abnormal cells. Such p53-regulated genes include an angiogenesis inhibitor thrombospondin (TSP-1) [254], GD-AIF [255], BAI1 [256], inhibitors of tumor invasion and metastasis KAI1 [257], collagenase MMP2 [258], MASPIN [259], an inhibitor of the plasminogen activator protein PAI-1 [260], and several secreting protein factors which inhibit proliferation of exposed cells [261].
Genes participating in induction of cell death. This is, perhaps, the most abundant group of the p53-induced genes. There are several alternative pathways leading to programmed cell death, and virtually all of them can be controlled by a p53-regulated gene. Apoptosis is a type of programmed cell death that is mediated through the action of cysteinic proteases (caspases). Effector caspases 2, 3, and 7 execute the major dismantling of cellular structures. They are activated by initiator caspases 8 and 9. The activation of the initiator caspases includes two major mechanisms. One of the mechanisms involves activation of death receptors that bind to certain extracellular ligands, leading to the assembly of active protease complexes that activate the initiator caspase 8. This is the extrinsic apoptotic pathway. The second is the mitochondrial pathway. It is initiated by cytochrome c released from mitochondria and serves as an activating subunit for the latent protease Apaf1, while the latter activates the initiator caspase 9 [262]. Regulation of permeability of the outer membrane of mitochondria is maintained by balanced interaction of the Bcl2-like protein family members. One of the proteins acts by reducing the permeability and preventing the cytochrome c release (the antiapoptotic proteins), while the proapoptotic family members act in opposite manner, by assisting the proapoptotic mitochondrial pore opening. The p53 protein affects both the extrinsic and the mitochondrial apoptotic pathways [263-265]. By acting in the mitochondrial pathway, p53 represses transcription of the antiapoptotic Bcl2 protein and activates transcription of the proapoptotic proteins Bax [266], Noxa [267], p53AIP1 [229], and Puma [236, 268]. Also, p53 activates transcription of the APAF1 gene [269-271] and increases the sensitivity to extrinsic proapoptotic ligands by stimulating transcription of FAS (APO1) [272] and KILLER/DR5 genes [273]. In addition, p53 induces a number of other proteins that somehow participate in the apoptosis induction process. These proteins include Perp [274], Pidd [275], Wip1 [276], Scotin [103], GML, STAG1, p53CABC1, p53RDL1 [233], and others. A large group of p53-induced proteins is involved in changing the redox balance of the cell. This group includes PIG3, PIG8 [277], FDXR [278], and some other genes. According to one model [277], a significant elevation of intracellular reactive oxygen species (ROS) taking place in response to the combined action of the mentioned genes is able to assist in acceleration of apoptosis. Recently a novel strategy used by p53 in the process of cell death induction has been described, which includes transcriptional activation of a gene encoding a micro RNA (miRNA). The miRNA are short hairpin-like noncoding RNA that inhibit translation of certain mRNAs by direct binding through the short complementary segments [279]. The p53-activated miRNA miR34 can also affect the expression of a set of genes, although its particular mRNA targets have not yet been identified. Introduction of recombinant constructs expressing miR34 results in block in cell divisions and apoptosis. Apparently, by inducing the expression of miR34 p53 affects a whole spectrum of the genes that mediate the suppressor effect [280-283].
Antioxidant genes. Along with the p53-induced genes that elevate intracellular levels of ROS, another group of the genes upregulated by p53 encode a set of antioxidant proteins, such as glutathione peroxidases Gpx1 [284] and Gpx2 [285], superoxide dismutase Sod2 [284], aldehyde dehydrogenase Aldh4A1 [286], and two members of the sestrin family, PA26 (Sesn1) [287] and Hi95 (Sesn2) [288]. The sestrins participate in the regeneration of overoxidized peroxiredoxins (Prxs) [289]. The peroxiredoxins are cysteinic peroxidases that remove excess H2O2. In addition to being a harmful byproduct of mitochondrial respiration, the hydrogen peroxide serves as an important signaling molecule. The H2O2 can be produced from the superoxide molecule synthesized by the NADPH-dependent oxidases in response to interaction of the membrane receptors with their ligands. The produced H2O2 is able to modify certain redox-sensitive components of signal transduction pathways [290]. Therefore, the formation of H2O2 represents an important physiological process that is needed for the transduction of intracellular signals. The sestrins control the activity of peroxiredoxins, thus contributing to the protection of the genome from excessive oxidation and acquisition of mutations. Inhibition of p53 by the RNA interference method, as well as selective inhibition of sestrins, results in a substantial increase in DNA oxidation and accelerated mutagenesis [291]. Apparently, even without stresses p53 carries an important antioxidant function that maintains minimal levels of intracellular ROS. Remarkably, dietary supplementation of p53 knockout mice with an antioxidant results in normalization of the intracellular ROS levels and prevention of the early development of lymphomas [291]. Accumulating evidences suggest an involvement of the p53-regulated sestrins in the carcinogenesis. Oncogenic Ras can induce a substantial upregulation of the ROS levels, and one of the mechanisms involves transcriptional inhibition of sestrins. The increased ROS induces p53, which leads to irreversible growth arrest and/or apoptosis. In case of breakdowns of the p53-dependent mechanisms, the restriction of the cells expressing oncogenic Ras and overproducing ROS is abolished, and this leads to rapid proliferation of abnormal cells that have accelerated rate of mutagenesis due to the increased oxidation of DNA [292]. Therefore, the antioxidant function of p53 is one of the most important components of its suppressor activity that prevents tumor formation.
Genes affecting metabolism. In the early 1930s Otto Warburg described the property of cancer cells to use glycolysis as a major source of energy even under aerobic conditions [293]. The Warburg effect was not explained until recently, when it was found that perhaps the major contribution to the effect is played by the ability of p53 to induce transcription of the SCO2 gene, which encodes a protein factor required for the assembly of mitochondrial respiratory complex COXII [294]. The p53-knockout mice display a substantially lower physical endurance in swimming test, which is explained by the insufficiency of mitochondrial respiration. Another p53-regulated gene TIGAR was found to be important for the regulation of the glycolytic pathway. The gene encodes a protein that is homologous to phosphofructokinase, the glycolytic enzyme converting glucose-6-phosphate to fructose-2,6-biphosphate [295]. The reaction is reversible, as the phosphofructokinase has a biphosphatase domain responsible for the conversion of the fructose-2,6-biphosphate to fructose-6-phosphate, which is then isomerized to glucose-6-phosphate. The product of the TIGAR gene resembles the biphosphatase domain of phosphofructokinase. It can therefore block glycolysis at the glucose-6-phosphate stage, which activates the pentose phosphate shunt pathway. Deficiency in p53 results in accelerated glycolysis and downregulated pentose phosphate pathway, which is an important source of NAD(P)H. Therefore, relative deficit of NAD(P)H and the increased ROS, along with the enhanced glycolysis and reduced respiration represent common consequences of the p53 deficiency [296].
THE MITOCHONDRIAL FUNCTION OF p53
In addition to being capable of transcriptional upregulation of the proapoptotic gene, p53 can affect the mitochondrial cell death pathway by direct interaction with the proteins that regulate the permeability of mitochondrial membrane [297-301]. Following activation in response to various stresses, a fraction of p53 is translocated directly into mitochondria where it interacts with the anti- and proapoptotic proteins of the Bcl2 family. The interactions lead to the increased permeability of the outer mitochondrial membrane, cytochrome c release, and apoptosis induction. The process of translocation of p53 into mitochondria is regulated by the ubiquitinylation process: The poly-ubiquitinylation process takes place in the nuclei, and the poly-ubiquitinylated p53 is then degraded in the 26S proteasomes. Meanwhile, the mono-ubiquitinylated p53 targets mitochondria [302], to where it enters directly from the cytoplasm, without entering the nuclei. Therefore, the stress-stabilized p53 either enters the nuclei where it participates in transcription regulation and in other nuclear functions (such as DNA repair), or it goes directly to the mitochondria to sensitize the cells to apoptosis. The Mdm2 protein itself does not participate in the translocation of p53 into mitochondria, although its background level may be important for the mono-ubiquitinylation. However, the participation of some additional E3 ligases in the mono-ubiquitinylation of p53 cannot be ruled out.
After entering mitochondria, p53 is rapidly de-ubiquitinylated by the mitochondrial HAUSP protein, which converts it to an active form. Then p53 interacts with BH4 domains of the antiapoptotic proteins BclXL and Bcl2, while the interacting domain on p53 corresponds to its DNA-binding domain [101]. The latter explains the paradoxical fact that oncogenic mutations in p53 disrupt simultaneously both the transcriptional and the mitochondrial activities [300]. The binding to antiapoptotic proteins displaces the associated Bax and Bid proteins boosting their proapoptotic activity. During its activation by the cytoplasmic p53, the Bax protein undergoes conformational changes and an oligomerization, which assists in its translocation from the cytosol into mitochondria [100, 303]. In addition, in mitochondria p53 binds to Bak and assists in its oligomerization and functional activation [297, 298, 300]; it also displaces Bak from its complex with antiapoptotic Mcl-1 [298]. The described interactions induce cytochrome c release and the induction of apoptosis even in the absence of transcriptional activation of the p53-regulated proapoptotic genes. It is noteworthy that a polymorphic variant of p53 (Arg72) is much more active in the mitochondrial translocation process as compared to the Pro72 variant of p53, and this property is in good agreement with the much weaker proapoptotic activity of the latter [304].
The direct induction of apoptosis by p53 perhaps represents an immediate response to very massive damages. For example, following irradiation of radiosensitive tissues (thymus or spleen) the translocation of p53 into mitochondria and activation of effector caspase 3 are observed during the first 30 min, which is long before the accumulation of the proapoptotic products owing to the induction of the p53-regulated genes. The latter products specify the second wave of apoptosis induction, which is detected after 6-7 h following the stress exposure [297]. The presence of p53 in the cytoplasm further sensitizes the cell to the products of the p53-regulated genes PUMA and NOXA. The Puma protein can efficiently displace p53 from its complex with BclXL, while the released p53 can activate the proapoptotic function of Bax. In addition, Puma is able to displace the proapoptotic proteins Bim and Bid from the complex with BclXL, which further increases the efficiency of apoptosis [303, 305, 306]. Thus, through its ability to act simultaneously at different levels and to use distinct mechanisms, p53 is capable of responding quickly and strongly to severe stresses, and to guide a delayed, but still very efficient program for the elimination of damaged cells.
REGULATORY LOOPS THAT CONTROL ACTIVITIES OF p53
Activities of p53 are triggered by numerous and very diverse signals. Certainly, in settling the cell's fate p53 plays a role of a supreme judge that evaluates thoroughly the entire upcoming information about the cell's condition, its compliance with the rules of behavior for the particular cell type under the particular circumstances. Based on this information, p53 makes decisions regarding the measures that would be optimal for the whole organism. p53 is able to play such a complex role due to the existing tight functional connections with virtually all signaling pathways in the cell, which occur through interactions and feed-back regulations involving numerous individual components of the pathways. To date at least ten distinct regulatory circuits have been identified that form negative or positive feedback loops [307]. However, it should be kept in mind that the dissection of individual circuits is rather artificial, as the circuits are actually embedded into the global regulatory network that interconnects all processes within the cell, allowing p53 to respond to any deviation from the optimum.
As an example, let us consider a fragment of the regulatory circuit involving p53, Mdm2, and ARF proteins. The ARF protein binds Mdm2 and prevents ubiquitinylation and degradation of p53, leading to accumulation of p53 [308]. The ARF gene transcription is activated by the transcription factors E2F1 [309] and beta-catenin [310] and is repressed by p53 itself. In turn, the ARF expression is increased in response to oncogenic Ras and Myc [76]. ARF can also regulate the activity of Myc [311]. Being localized to the nucleoli, the ARF protein affects the processing of ribosomal RNA and the maturation of ribosome subunits [312]. In turn, in the nucleoli Mdm2 binds to ribosomal proteins L5, L11, and L23, which results in down-regulation of its ubiquitin ligase activity [55]. The pRB protein can form complexes with p53 and with Mdm2, which leads to p53 activation and elevated apoptosis [313]. Increased level of free (not bound to pRB) transcription factor E2F1 switches the activity of p53 toward apoptosis. Similarly to Mdm2, the pRB protein can be inhibited through phosphorylation by the Cdc2/cyclin E kinase complex. p53 activates the synthesis of p21, which inhibits Cdc2/cyclin E, thus affecting the pRB/Mdm2 complex and tuning up the p53-dependent apoptosis. DNA damage induces the ATM-dependent phosphorylation of p53 and Mdm2; like in the case of p53-Mdm2-pRB complex formation, this leads to the enhanced activity of p53 and to apoptosis [314].
Listing of various components of positive and negative regulatory feed-back loops involving p53 can be continued with no end, because each of the interacting components is linked to numerous additional elements of signal transduction pathways. It should also be kept in mind that any given regulatory interaction has quite individual value in different cell types, as the overall tuning of signaling pathways as well as the levels and activities of individual regulatory proteins may have strong cell type specificity.
p53 ACTS UNDER ANY CIRCUMSTANCES
Until recently, it was considered that the activity of p53 is characteristic of cells that either were exposed to stresses or had acquired certain damages. It was considered that the function of p53 is mediated mainly by restrictive measures against cells that represent risk for acquiring inherited mutations. The discovery of additional functions of p53 that are directed toward stimulation of DNA repair, regulation of glycolytic and respiratory metabolism, regulation of the levels of basic metabolites, as well as of the levels of ROS suggests that the roles of p53 in the control of processes in cells and in the organism as a whole is virtually limitless. The function of p53 is to ensure optimal balance in each and every process in every cell type and under any circumstances. The p53 protein is entitled to intervene into processes in case of any deviation from the optimum. One can assume that activities of p53 are absent under the condition of ideal optimum. However, it is unlikely that such a condition ever exists, which is why even subtle and transient misbalancing caused by exercise or any other physiological load could induce certain functional changes in p53.
One can formally suggest two major modes of p53 function. Under mild, physiologically acceptable levels of stress that include exercising of all sorts, dietary abuses, mild inflammatory processes, etc., the activity of p53 is directed to restore homeostasis, to mobilize adequate repair, to choose the optimal balance of energy sources, to switch biosynthesis processes, and to protect the genome from mutagenic influences of ROS. Upon reaching certain physiologically tolerable threshold of damages the activity of p53 is switched toward the elimination of genetically hazardous damaged cells, which is achieved either by the induction of apoptosis or by other kinds of genetic death, such as the terminal exit of the cells from further cell divisions.
The outcomes from the absence or deficiency in the background activity of p53 are evident from the studies observing behavior of cells or animals after artificial inhibition of p53 and maintaining under non-stressful conditions. Even without stresses, the p53-knockout cells demonstrate substantial increase in intracellular levels of ROS, leading to increased DNA oxidation, elevated mutagenesis, and chromosome instability [291]. The effects are due to the deficiency in the p53-mediated tuning of the antioxidant defense mechanisms toward expedited and efficient neutralization of the peroxide compounds that form during normal functioning of signal transduction pathways. When subjected to physical exercises the p53-deficient animals demonstrate insufficiency in normal switching of metabolic processes, weak aerobic processes, and prevalence of glycolysis, which is particularly manifested by the reduced endurance of the p53-knockout mice in the swimming test [294].
One of the background activities of p53 affects the IGF-1-Akt-mTOR regulatory circuit, which controls the adaptive switches in glucose metabolism [315, 316]. The function of the circuit is affected by the availability of glucose and ensures the most favorable mode for energy metabolism. An AMP-dependent protein kinase AMPK serves as a sensor component of the circuit, being activated in response to reduced ATP/AMP ratio. The AMPK is able to activate by phosphorylation the GTPase TSC2 [317], which acting in a complex with TSC1 inhibits the activity of the G-protein RHEB, an activator of the mTOR kinase [318]. Low availability of glucose induces inhibition of mTOR kinase that promotes autophagy, the digestion of certain volumes of cytoplasm inside the cell. The autophagy is manifested by the formation inside the cell of the extended vacuoles that represent large regions of the cytoplasm surrounded by a membrane. The vacuoles are fused with lysosomes, and the content is digested by the lysosomal enzymes, thus supplying the cell with recycled nutrients [319]. The mTOR kinase phosphorylates the 4EBP1 protein that inhibits translation initiation factor 4E [320]; it can also phosphorylate the S6 kinase that participates in ribosome biogenesis and in the delivery and consumption of oxygen [321]. Therefore, active mTOR kinase promotes efficient translation and energy consumption under conditions of glucose excess. Inhibition of mTOR results in the prevalence of catabolic processes and energy saving. The processes in the circuit are regulated by the insulin-like growth factor IGF-1 and by the Akt kinase. IGF-1 activates the Akt-1 kinase, which inhibits FOXO, the transcription factors of the fork-head like family. The latter induce changes in the expression of many genes that participate in metabolism and antioxidant defense [316, 322]. Akt-1 can also inhibit activity of the TSC2 GTPase, leading to the activation of mTOR kinase [323].
The p53 protein controls transcription of several key components of the IGF-1-Akt-mTOR regulatory circuit: it can activate the synthesis of both subunits of AMPK, protein phosphatase PTEN, GTPase TSC2, and IGF-1 inhibitor IGF-BP3 [315]. Activation of p53 leads to the inhibition of mTOR and to the prevalence of catabolic processes, energy saving, increased activity of FOXO, enhanced autophagy, and reduced level of intracellular ROS. Along with the inhibition of the IGF1-Akt-mTOR circuit, the p53 protein boosts the more economical processes of aerobic respiration. Thus, tolerable and physiologically relevant conditions introduce mild p53-dependent corrections to the metabolism and switch to a more economical energy mode, with simultaneous reduction in the ROS levels.
The stress-induced activation of p53 occurs in response to severe stresses, substantial damages, or constant oncogene activation, and the outcomes of the deficiency in such responses are much more evident. The deficiency leads to the accumulation of genetically altered cells, which start to compete with each other resulting in the selection of the cells that are most rapidly growing are increasingly insensitive to local conditions. The process leads unavoidably to the development of tumors. However, the relative significance of two alternative mechanisms of stress-induced activation of p53 for the prevention of cancer is not quite clear. In some experimental models the antitumor activity of p53 was mainly ARF mediated, while the DNA-damage induced activation was quite dispensable [324, 325], whereas in other models it was found that genotoxic stresses are still very important for p53 activation and tumor suppression, even in the absence of functional ARF pathway [326, 327]. The contradiction is perhaps due to the cell-specific differences, which emphasizes the importance of redundancy of alternative p53-dependent mechanisms.
THE p53 PROTEIN AND AGING
Being in the center of mechanisms that supervise correct execution of genetic programs, p53 should certainly be related to the processes of organism aging. There is a clear distinction between the aging process and the diseases that frequently accompany old age. It is still not quite clear whether aging of the organism represents a programmed process, or it simply proceeds at the accelerated rate after certain mechanisms that regenerate and repair tissues start to fail at older ages. The quite substantial differences in the life span of organisms belonging to different species suggest the existence of certain genetic predetermination. On the other hand, commonly there are quite substantial differences in the lifetime of individual organisms within the same species, and there are powerful evidences in favor of genetic inheritance of the longevity trait. Good heredity, perhaps, can be considered as the first and the most important among the factors that favor longevity [328]. Lifestyle can be considered as the second important factor, as there are numerous arguments in favor of the substantial influence of habits, diet, and living conditions on the span of an individual's life [329].
Calories restriction in the diet represents the most clear and reliable factor affecting longevity [330]. The evolutionary significance of the correlation is easily understood: under limitation of nutrients, an organism needs to extend its existence to reach better times and to save the seed of life. In contrast, under vast excess of nutrients life is usually intense, energy consuming, fertile, and full of stresses. Such lifestyle leads to more substantial wear and leads to a shorter lifespan [331, 332].
There are also good arguments in favor of the importance of oxidative processes in the development of the aging processes. The organism is well equipped with powerful antioxidant mechanisms that efficiently protect its different components from oxidation and ensure timely repair of damages. However, the mechanisms are depleted with age and tend to fail.
The existing views on the role of p53 in the aging process are quite controversial. Various genetic manipulations with the p53 gene in mice have yielded rather ambiguous results. First, it is practically impossible to study aging process in p53 knockout mice because the mice do not live until the age of one year, succumbing to malignancies [30]. The introduction to the mouse genome of constantly activated p53 results in a significant reduction in the frequency of sporadic tumors, though the mice develop premature aging phenotype [333]. On the other hand, the introduction of an additional copy of unmodified p53 gene [334], or certain down-regulation of the Mdm2 gene expression [335], results in a protection against cancer formation without any reduction in the lifespan. In humans, there is a polymorphism in the codon 72 of p53 [336]. The Arg72 allele of p53 differs from the Pro72 variant in much higher resistance to malignancies, which is connected with its efficient mitochondrial proapoptotic function. The individuals homozygous in the Pro72 allele, while displaying more frequent development of malignant diseases, generally live longer and have increased mean lifespan compared to the individuals possessing the Arg72 allele [337]. The results suggest that the downside of better anticancer defense could be the acceleration of the aging processes [338].
How one could explain this paradox and are there means for overcoming the negative sides of p53? To find an answer to the question we need to consider critical determinants that specify expression of either of the opposing effects of p53. Low levels of p53 contribute to optimal balancing of various processes leading to the lower risk of acquiring mutations and to the enhanced rate of repair. Obviously, these functions of p53 are favorable both for cancer prevention and for delaying the aging processes. Lifespan extension due to calorie restriction probably does not occur without critical influences from p53, as the physiologically relevant levels of p53 are able to inhibit the IGF-mTOR circuit, and glucose deficiency induces adaptive upregulation of p53 activity through AMPK activation. Autophagy may also be beneficial for delaying the aging process, and it was found to be stimulated by the p53-dependent gene DRUM [339]. The autophagy leads to the efficient regeneration and re-juvenescence of the cytoplasm, as it assists in removing damaged proteins and organelles, especially worn-out mitochondria that are known to overproduce ROS [340]. Low levels of p53 can also reduce risk of mutations and damages to other components of the cell due to its antioxidant function. The antioxidant function of p53 delays the process of depletion of cell division capacity, as under the influence of ROS there is accelerated erosion of telomeres leading to the removal of several telomeric repeats in one cell division [341]. Therefore, under moderate lifestyle the activity of p53 is directed both against the risk of malignant diseases and against premature aging.
The other side of p53 function is connected with the radical measures that are taken in case of severe stresses, intoxications, irradiations, inflammations and infections, dietary excesses with the increased consumption of calories, metabolic diseases (such as diabetes), drug abuses, etc. These conditions induce strong activation of p53, leading to a qualitatively different responses, which include the induction of apoptosis of gravely damaged cells. Apoptosis is accompanied by the massive outbreak or oxygen radicals, affecting not only the cells that are committed to die, but also the whole microenvironment [342]. The latter can induce increased oxidation of DNA, excessive mutagenesis, changes in the extracellular matrix, the accumulation of damaged proteins, the development of pathologic changes in tissues, such as preferential death of parenchyma and the development of fibrosis [343], etc. Likewise, chronic inflammation processes are accompanied by constant oxidative stress in the affected tissues, which results in accelerated telomere erosion and local exhaustion of the tissue regeneration capacity. The condition leads to the development of “local progerias”, which is local aging of tissues that leads to pathologies, such as various neurodegenerative diseases, pulmonary emphysema and fibrosis, renal pathologies, etc. [332]. Apparently, p53 plays a substantial role in the development of these pathological conditions. Thus, functions of p53 not only restrict abnormal cells, but also define the fate of individual people in cases of unhealthy life habits. Proper understanding of the opposing functions of p53 confirms the undeniable advantages of preventive approaches in the fight with diseases. Wise and moderate way of life is certainly the basis of good health and longevity, as is this case p53 indeed becomes our Guardian Angel.
As the breakdown of the p53 gene was found in the majority of cancer cases, unprecedented efforts have been invested in the study of its functions. It was established that the protein molecule of p53 has several quite independent activities. The majority of functions of p53 are attributed to its ability to act as a transcription factor and to activate the expression of numerous genes. Other activities of p53 specify transcription repression of another set of genes, or enable its involvement in DNA repair as a DNA exonuclease, or to affect many different processes in the cell by binding to numerous proteins (enzymes, transcription factors, adapter proteins, etc.). In spite of the vast diversity of different activities of the p53 molecule, each of them contributes to a common macrofunction, which is maintenance of genetic identity of somatic cells in a multicellular organism. The biological role of p53 is related to its important “social” function in the organism, which ensures benefits of the organism over those of an individual cell. The function of p53 specifies altruistic behavior of cells in a multicellular organism, which is manifested by separate decisions of individual cells to commit suicide. Activities of p53 are changing depending on the condition and balance of virtually each and every process within the cell. Numerous connections converge on p53 and convey upcoming signals that report about all sorts of structural damages, deviations from optimum in processes, and the activity of p53 changes depending on the degree of the aberration, which results in either stimulation of repair processes and protective mechanisms, or the cessation of further cell divisions and the induction of programmed cell death. The strategy of p53 ensures genetic uniformity of cells and excludes competition among cells and selection of cells with certain advantages to local conditions. Loss of functions of the p53 gene is observed in virtually every cancer case, and conversely, the loss of p53 unavoidably leads to the development of cancer. The understanding of mechanisms underlying functions of p53 not only allows the development of new approaches to cancer treatment, but also defines important strategies for the prevention of numerous diseases and for delaying the process of aging.
This work was supported by the Russian Foundation for Basic Research grant 05-04-48979, grant No. 55005603 from the Howard Hughes Medical Institute, and the grants from the National Institutes of Health R01 CA104903 and R01 AG025278.
REFERENCES
1.Lane, D. P. (1992) Nature, 358,
15-16.
2.Royds, J. A., and Iacopetta, B. (2006) Cell
Death Differ., 13, 1017-1026.
3.Chumakov, P. M. (2000) Biochemistry
(Moscow), 65, 28-40.
4.Deppert, W. (2007) Oncogene, 26,
2142-2144.
5.Almazov, V. P., Kochetkov, D. A., and Chumakov, P.
M. (2007) Mol. Biol. (Moscow), 41, 459-466.
6.Kufe, D. W., Holland, J. F., and Frei, E. (2003)
Cancer Medicine, 6th Edn., BC Decker Publisher, NY.
7.Huebner, R. J., and Todaro, G. J. (1969) Proc.
Natl. Acad. Sci. USA, 64, 1087-1094.
8.Tooze, J., and Acheson, N. H. (1980) DNA Tumor
Viruses; Molecular Biology of Tumor Viruses, Cold Spring Harbor
Laboratory Publisher, Cold Spring Harbor, N. Y.
9.Chang, C., Simmons, D. T., Martin, M. A., and Mora,
P. T. (1979) J. Virol., 31, 463-471.
10.Kress, M., May, E., Cassingena, R., and May, P.
(1979) J. Virol., 31, 472-483.
11.Lane, D. P., and Crawford, L. V. (1979)
Nature, 278, 261-263.
12.Linzer, D. I., and Levine, A. J. (1979)
Cell, 17, 43-52.
13.DeLeo, A. B., Jay, G., Appella, E., Dubois, G.
C., Law, L. W., and Old, L. J. (1979) Proc. Natl. Acad. Sci.
USA, 76, 2420-2424.
14.Barque, J. P., Danon, F., Peraudeau, L., Yeni,
P., and Larsen, C. J. (1983) Embo J., 2, 743-749.
15.Mercer, W. E., Nelson, D., DeLeo, A. B., Old, L.
J., and Baserga, R. (1982) Proc. Natl. Acad. Sci. USA,
79, 6309-6312.
16.Oren, M., and Levine, A. J. (1983) Proc. Natl.
Acad. Sci. USA, 80, 56-59.
17.Chumakov, P. M., Iotseva, V. S., and Georgiev, G.
P. (1982) Dokl. AN SSSR, 267, 1272-1275.
18.Wolf, D., Laver-Rudich, Z., and Rotter, V. (1985)
Mol. Cell Biol., 5, 1887-1893.
19.Bukhman, V. L., Ninkina, N. N., Chumakov, P. M.,
Khilenkova, M. A., and Samarina, O. P. (1987) Genetika,
23, 1547-1554.
20.Eliyahu, D., Raz, A., Gruss, P., Givol, D., and
Oren, M. (1984) Nature, 312, 646-649.
21.Jenkins, J. R., Rudge, K., and Currie, G. A.
(1984) Nature, 312, 651-654.
22.Parada, L. F., Land, H., Weinberg, R. A., Wolf,
D., and Rotter, V. (1984) Nature, 312, 649-651.
23.Jenkins, J. R., Rudge, K., Chumakov, P., and
Currie, G. A. (1985) Nature, 317, 816-818.
24.Hinds, P., Finlay, C., and Levine, A. J. (1989)
J. Virol., 63, 739-746.
25.Baker, S. J., Markowitz, S., Fearon, E. R.,
Willson, J. K., and Vogelstein, B. (1990) Science, 249,
912-915.
26.Eliyahu, D., Michalovitz, D., Eliyahu, S.,
Pinhasi-Kimhi, O., and Oren, M. (1989) Proc. Natl. Acad. Sci.
USA, 86, 8763-8767.
27.Finlay, C. A., Hinds, P. W., and Levine, A. J.
(1989) Cell, 57, 1083-1093.
28.Baker, S. J., Fearon, E. R., Nigro, J. M.,
Hamilton, S. R., Preisinger, A. C., Jessup, J. M., vanTuinen, P.,
Ledbetter, D. H., Barker, D. F., Nakamura, Y., White, R., and
Vogelstein, B. (1989) Science, 244, 217-221.
29.Levine, A. J., Momand, J., and Finlay, C. A.
(1991) Nature, 351, 453-456.
30.Donehower, L. A., Harvey, M., Slagle, B. L.,
McArthur, M. J., Montgomery, C. A., Jr., Butel, J. S., and Bradley, A.
(1992) Nature, 356, 215-221.
31.Tsukada, T., Tomooka, Y., Takai, S., Ueda, Y.,
Nishikawa, S., Yagi, T., Tokunaga, T., Takeda, N., Suda, Y., Abe, S.,
et al. (1993) Oncogene, 8, 3313-3322.
32.Agapova, L. S., Ilyinskaya, G. V., Turovets, N.
A., Ivanov, A. V., Chumakov, P. M., and Kopnin, B. P. (1996) Mutat.
Res., 354, 129-138.
33.Brooks, C. L., and Gu, W. (2006) Mol.
Cell., 21, 307-315.
34.Asher, G., Reuven, N., and Shaul, Y. (2006)
Bioessays, 28, 844-849.
35.Haupt, Y., Maya, R., Kazaz, A., and Oren, M.
(1997) Nature, 387, 296-299.
36.Momand, J., Zambetti, G. P., Olson, D. C.,
George, D., and Levine, A. J. (1992) Cell, 69,
1237-1245.
37.Oliner, J. D., Kinzler, K. W., Meltzer, P. S.,
George, D. L., and Vogelstein, B. (1992) Nature, 358,
80-83.
38.Dornan, D., Wertz, I., Shimizu, H., Arnott, D.,
Frantz, G. D., Dowd, P., O'Rourke, K., Koeppen, H., and Dixit, V. M.
(2004) Nature, 429, 86-92.
39.Leng, R. P., Lin, Y., Ma, W., Wu, H., Lemmers,
B., Chung, S., Parant, J. M., Lozano, G., Hakem, R., and Benchimol, S.
(2003) Cell, 112, 779-791.
40.Chen, D., Kon, N., Li, M., Zhang, W., Qin, J.,
and Gu, W. (2005) Cell, 121, 1071-1083.
41.Shmueli, A., and Oren, M. (2005) Cell,
121, 963-965.
42.Asher, G., and Shaul, Y. (2005) Cell
Cycle, 4, 1015-1018.
43.Yano, M., Koumoto, Y., Kanesaki, Y., Wu, X., and
Kido, H. (2004) Biomacromolecules, 5, 1465-1469.
44.Horn, H. F., and Vousden, K. H. (2007)
Oncogene, 26, 1306-1316.
45.Lavin, M. F., and Gueven, N. (2006) Cell Death
Differ., 13, 941-950.
46.Shvarts, A., Steegenga, W. T., Riteco, N., van
Laar, T., Dekker, P., Bazuine, M., van Ham, R. C., van der Houven van
Oordt, W., Hateboer, G., van der Eb, A. J., and Jochemsen, A. G. (1996)
Embo J., 15, 5349-5357.
47.Jackson, M. W., and Berberich, S. J. (2000)
Mol. Cell Biol., 20, 1001-1007.
48.Francoz, S., Froment, P., Bogaerts, S., de
Clercq, S., Maetens, M., Doumont, G., Bellefroid, E., and Marine, J. C.
(2006) Proc. Natl. Acad. Sci. USA, 103, 3232-3237.
49.Sharp, D. A., Kratowicz, S. A., Sank, M. J., and
George, D. L. (1999) J. Biol. Chem., 274,
38189-38196.
50.Tanimura, S., Ohtsuka, S., Mitsui, K., Shirouzu,
K., Yoshimura, A., and Ohtsubo, M. (1999) FEBS Lett.,
447, 5-9.
51.Gu, J., Kawai, H., Nie, L., Kitao, H.,
Wiederschain, D., Jochemsen, A. G., Parant, J., Lozano, G., and Yuan,
Z. M. (2002) J. Biol. Chem., 277, 19251-19254.
52.De Graaf, P., Little, N. A., Ramos, Y. F.,
Meulmeester, E., Letteboer, S. J., and Jochemsen, A. G. (2003) J.
Biol. Chem., 278, 38315-38324.
53.Dai, M. S., and Lu, H. (2004) J. Biol.
Chem., 279, 44475-44482.
54.Dai, M. S., Zeng, S. X., Jin, Y., Sun, X. X.,
David, L., and Lu, H. (2004) Mol. Cell Biol., 24,
7654-7668.
55.Zhang, Y., Wolf, G. W., Bhat, K., Jin, A., Allio,
T., Burkhart, W. A., and Xiong, Y. (2003) Mol. Cell Biol.,
23, 8902-8912.
56.Gilkes, D. M., Chen, L., and Chen, J. (2006)
Embo J., 25, 5614-5625.
57.Gilkes, D. M., and Chen, J. (2007) Cell
Cycle, 6, 151-155.
58.Lindstrom, M. S., Deisenroth, C., and Zhang, Y.
(2007) Cell Cycle, 6, 434-437.
59.Grossman, S. R., Deato, M. E., Brignone, C.,
Chan, H. M., Kung, A. L., Tagami, H., Nakatani, Y., and Livingston, D.
M. (2003) Science, 300, 342-344.
60.Weissman, A. M. (2001) Nat. Rev. Mol. Cell
Biol., 2, 169-178.
61.Gronroos, E., Terentiev, A. A., Punga, T., and
Ericsson, J. (2004) Proc. Natl. Acad. Sci. USA, 101,
12165-12170.
62.Sui, G., el Affar, B., Shi, Y., Brignone, C.,
Wall, N. R., Yin, P., Donohoe, M., Luke, M. P., Calvo, D., Grossman, S.
R., and Shi, Y. (2004) Cell, 117, 859-872.
63.Wang, C., Ivanov, A., Chen, L., Fredericks, W.
J., Seto, E., Rauscher, F. J., 3rd, and Chen, J. (2005) Embo J.,
24, 3279-3290.
64.Higashitsuji, H., Higashitsuji, H., Itoh, K.,
Sakurai, T., Nagao, T., Sumitomo, Y., Masuda, T., Dawson, S., Shimada,
Y., Mayer, R. J., and Fujita, J. (2005) Cancer Cell, 8,
75-87.
65.Li, M., Brooks, C. L., Kon, N., and Gu, W. (2004)
Mol. Cell, 13, 879-886.
66.Tang, J., Qu, L. K., Zhang, J., Wang, W.,
Michaelson, J. S., Degenhardt, Y. Y., El-Deiry, W. S., and Yang, X.
(2006) Nat. Cell Biol., 8, 855-862.
67.Cummins, J. M., and Vogelstein, B. (2004) Cell
Cycle, 3, 689-692.
68.Li, M., Chen, D., Shiloh, A., Luo, J., Nikolaev,
A. Y., Qin, J., and Gu, W. (2002) Nature, 416,
648-653.
69.Sherr, C. J. (2001) Nat. Rev. Mol. Cell
Biol., 2, 731-737.
70.Kamijo, T., Zindy, F., Roussel, M. F., Quelle, D.
E., Downing, J. R., Ashmun, R. A., Grosveld, G., and Sherr, C. J.
(1997) Cell, 91, 649-659.
71.Kamijo, T., Weber, J. D., Zambetti, G., Zindy,
F., Roussel, M. F., and Sherr, C. J. (1998) Proc. Natl. Acad. Sci.
USA, 95, 8292-8297.
72.Pomerantz, J., Schreiber-Agus, N., Liegeois, N.
J., Silverman, A., Alland, L., Chin, L., Potes, J., Chen, K., Orlow,
I., Lee, H. W., Cordon-Cardo, C., and DePinho, R. A. (1998)
Cell, 92, 713-723.
73.Zhang, Y., Xiong, Y., and Yarbrough, W. G. (1998)
Cell, 92, 725-734.
74.Rowland, B. D., Denissov, S. G., Douma, S.,
Stunnenberg, H. G., Bernards, R., and Peeper, D. S. (2002) Cancer
Cell, 2, 55-65.
75.Aslanian, A., Iaquinta, P. J., Verona, R., and
Lees, J. A. (2004) Genes Dev., 18, 1413-1422.
76.Lowe, S. W., and Sherr, C. J. (2003) Curr.
Opin. Genet. Dev., 13, 77-83.
77.Zhong, Q., Gao, W., Du, F., and Wang, X. (2005)
Cell, 121, 1085-1095.
78.Wechsler, A., Brafman, A., Shafir, M., Heverin,
M., Gottlieb, H., Damari, G., Gozlan-Kelner, S., Spivak, I., Moshkin,
O., Fridman, E., Becker, Y., Skaliter, R., Einat, P., Faerman, A.,
Bjorkhem, I., and Feinstein, E. (2003) Science, 302,
2087.
79.Wu, C., Miloslavskaya, I., Demontis, S., Maestro,
R., and Galaktionov, K. (2004) Nature, 432, 640-645.
80.Bourdon, J. C., Fernandes, K., Murray-Zmijewski,
F., Liu, G., Diot, A., Xirodimas, D. P., Saville, M. K., and Lane, D.
P. (2005) Genes Dev., 19, 2122-2137.
81.Ray, P. S., Grover, R., and Das, S. (2006)
EMBO Rep., 7, 404-410.
82.Yang, D. Q., Halaby, M. J., and Zhang, Y. (2006)
Oncogene, 25, 4613-4619.
83.Jeffrey, P. D., Gorina, S., and Pavletich, N. P.
(1995) Science, 267, 1498-1502.
84.Laptenko, O., and Prives, C. (2006) Cell Death
Differ., 13, 951-961.
85.Appella, E., and Anderson, C. W. (2001) Eur.
J. Biochem., 268, 2764-2772.
86.Rodriguez, M. S., Desterro, J. M., Lain, S.,
Midgley, C. A., Lane, D. P., and Hay, R. T. (1999) Embo J.,
18, 6455-6461.
87.Xirodimas, D. P., Saville, M. K., Bourdon, J. C.,
Hay, R. T., and Lane, D. P. (2004) Cell, 118, 83-97.
88.Zacchi, P., Gostissa, M., Uchida, T., Salvagno,
C., Avolio, F., Volinia, S., Ronai, Z., Blandino, G., Schneider, C.,
and Del Sal, G. (2002) Nature, 419, 853-857.
89.Zheng, H., You, H., Zhou, X. Z., Murray, S. A.,
Uchida, T., Wulf, G., Gu, L., Tang, X., Lu, K. P., and Xiao, Z. X.
(2002) Nature, 419, 849-853.
90.An, W. G., Kanekal, M., Simon, M. C., Maltepe,
E., Blagosklonny, M. V., and Neckers, L. M. (1998) Nature,
392, 405-408.
91.Anwar, A., Dehn, D., Siegel, D., Kepa, J. K.,
Tang, L. J., Pietenpol, J. A., and Ross, D. (2003) J. Biol.
Chem., 278, 10368-10373.
92.Chang, N. S. (2002) Int. J. Mol. Med.,
9, 19-24.
93.Corcoran, C. A., He, Q., Huang, Y., and Sheikh,
M. S. (2005) Oncogene, 24, 1634-1640.
94.Jayaraman, L., Murthy, K. G., Zhu, C., Curran,
T., Xanthoudakis, S., and Prives, C. (1997) Genes Dev.,
11, 558-570.
95.Ueno, M., Masutani, H., Arai, R. J., Yamauchi,
A., Hirota, K., Sakai, T., Inamoto, T., Yamaoka, Y., Yodoi, J., and
Nikaido, T. (1999) J. Biol. Chem., 274, 35809-35815.
96.Samuels-Lev, Y., O'Connor, D. J., Bergamaschi,
D., Trigiante, G., Hsieh, J. K., Zhong, S., Campargue, I., Naumovski,
L., Crook, T., and Lu, X. (2001) Mol. Cell, 8,
781-794.
97.Bergamaschi, D., Samuels, Y., Jin, B.,
Duraisingham, S., Crook, T., and Lu, X. (2004) Mol. Cell Biol.,
24, 1341-1350.
98.Bergamaschi, D., Samuels, Y., O'Neil, N. J.,
Trigiante, G., Crook, T., Hsieh, J. K., O'Connor, D. J., Zhong, S.,
Campargue, I., Tomlinson, M. L., Kuwabara, P. E., and Lu, X. (2003)
Nat. Genet., 33, 162-167.
99.Sullivan, A., and Lu, X. (2007) Br. J.
Cancer, 96, 196-200.
100.Chipuk, J. E., Kuwana, T., Bouchier-Hayes, L.,
Droin, N. M., Newmeyer, D. D., Schuler, M., and Green, D. R. (2004)
Science, 303, 1010-1014.
101.Petros, A. M., Gunasekera, A., Xu, N.,
Olejniczak, E. T., and Fesik, S. W. (2004) FEBS Lett.,
559, 171-174.
102.Fei, P., Wang, W., Kim, S. H., Wang, S., Burns,
T. F., Sax, J. K., Buzzai, M., Dicker, D. T., McKenna, W. G., Bernhard,
E. J., and El-Deiry, W. S. (2004) Cancer Cell, 6,
597-609.
103.Bourdon, J. C., Renzing, J., Robertson, P. L.,
Fernandes, K. N., and Lane, D. P. (2002) J. Cell Biol.,
158, 235-246.
104.Di Leonardo, A., Linke, S. P., Clarkin, K., and
Wahl, G. M. (1994) Genes Dev., 8, 2540-2551.
105.Kastan, M. B., Onyekwere, O., Sidransky, D.,
Vogelstein, B., and Craig, R. W. (1991) Cancer Res., 51,
6304-6311.
106.Maltzman, W., and Czyzyk, L. (1984) Mol.
Cell Biol., 4, 1689-1694.
107.Lin, A. W., and Lowe, S. W. (2001) Proc.
Natl. Acad. Sci. USA, 98, 5025-5030.
108.Vousden, K. H., and Lane, D. P. (2007) Nat.
Rev. Mol. Cell Biol., 8, 275-283.
109.Agarwal, M. L., Agarwal, A., Taylor, W. R.,
Chernova, O., Sharma, Y., and Stark, G. R. (1998) Proc. Natl. Acad.
Sci. USA, 95, 14775-14780.
110.Blagosklonny, M. V., Schulte, T. W., Nguyen,
P., Mimnaugh, E. G., Trepel, J., and Neckers, L. (1995) Cancer
Res., 55, 4623-4626.
111.Rubtsova, S. N., Kondratov, R. V., Kopnin, P.
B., Chumakov, P. M., Kopnin, B. P., and Vasiliev, J. M. (1998) FEBS
Lett., 430, 353-357.
112.Sablina, A. A., Agapova, L. S., Chumakov, P.
M., and Kopnin, B. P. (1999) Cell Biol. Int., 23,
323-334.
113.David-Pfeuty, T., Nouvian-Dooghe, Y., Sirri,
V., Roussel, P., and Hernandez-Verdun, D. (2001) Oncogene,
20, 5951-5963.
114.Graeber, T. G., Peterson, J. F., Tsai, M.,
Monica, K., Fornace, A. J., Jr., and Giaccia, A. J. (1994) Mol. Cell
Biol., 14, 6264-6277.
115.O'Reilly, M. A., Staversky, R. J., Stripp, B.
R., and Finkelstein, J. N. (1998) Am. J. Respir. Cell Mol.
Biol., 18, 43-50.
116.Agarwal, M. L., Taylor, W. R., Chernov, M. V.,
Chernova, O. B., and Stark, G. R. (1998) J. Biol. Chem.,
273, 1-4.
117.Blandino, G., Scardigli, R., Rizzo, M. G.,
Crescenzi, M., Soddu, S., and Sacchi, A. (1995) Oncogene,
10, 731-737.
118.Sablina, A. A., Chumakov, P. M., Levine, A. J.,
and Kopnin, B. P. (2001) Oncogene, 20, 899-909.
119.Bachelder, R. E., Marchetti, A., Falcioni, R.,
Soddu, S., and Mercurio, A. M. (1999) J. Biol. Chem.,
274, 20733-20737.
120.Nikiforov, M. A., Hagen, K., Ossovskaya, V. S.,
Connor, T. M., Lowe, S. W., Deichman, G. I., and Gudkov, A. V. (1996)
Oncogene, 13, 1709-1719.
121.Agapova, L. S., Turovets, N. A., Ivanov, A. V.,
Ilyinskaya, G. V., and Chumakov, P. M. (1996) Genetika,
32, 1080-1087.
122.Sablina, A. A., Ilyinskaya, G. V., Rubtsova, S.
N., Agapova, L. S., Chumakov, P. M., and Kopnin, B. P. (1998) J.
Cell Sci., 111 (Pt. 7), 977-984.
123.Seo, Y. R., Smith, M. L., Han, S. S.,
Fairbairn, D. W., O'Neill, K. L., and Ryu, J. C. (1999) Res. Commun.
Mol. Pathol. Pharmacol., 104, 285-292.
124.Matijasevic, Z., Snyder, J. E., and Ludlum, D.
B. (1998) Oncol. Res., 10, 605-610.
125.Forrester, K., Ambs, S., Lupold, S. E., Kapust,
R. B., Spillare, E. A., Weinberg, W. C., Felley-Bosco, E., Wang, X. W.,
Geller, D. A., Tzeng, E., Billiar, T. R., and Harris, C. C. (1996)
Proc. Natl. Acad. Sci. USA, 93, 2442-2447.
126.Iliakis, G., Wang, Y., Guan, J., and Wang, H.
(2003) Oncogene, 22, 5834-5847.
127.Niida, H., and Nakanishi, M. (2006)
Mutagenesis, 21, 3-9.
128.Sarkaria, J. N., Busby, E. C., Tibbetts, R. S.,
Roos, P., Taya, Y., Karnitz, L. M., and Abraham, R. T. (1999) Cancer
Res., 59, 4375-4382.
129.Matsuoka, S., Huang, M., and Elledge, S. J.
(1998) Science, 282, 1893-1897.
130.Chehab, N. H., Malikzay, A., Appel, M., and
Halazonetis, T. D. (2000) Genes Dev., 14, 278-288.
131.Hanawalt, P. C. (1989) Genome,
31, 605-611.
132.Ljungman, M., O'Hagan, H. M., and Paulsen, M.
T. (2001) Oncogene, 20, 5964-5971.
133.Tibbetts, R. S., Brumbaugh, K. M., Williams, J.
M., Sarkaria, J. N., Cliby, W. A., Shieh, S. Y., Taya, Y., Prives, C.,
and Abraham, R. T. (1999) Genes Dev., 13, 152-157.
134.Chehab, N. H., Malikzay, A., Stavridi, E. S.,
and Halazonetis, T. D. (1999) Proc. Natl. Acad. Sci. USA,
96, 13777-13782.
135.Scully, R., Anderson, S. F., Chao, D. M., Wei,
W., Ye, L., Young, R. A., Livingston, D. M., and Parvin, J. D. (1997)
Proc. Natl. Acad. Sci. USA, 94, 5605-5610.
136.Krum, S. A., Miranda, G. A., Lin, C., and Lane,
T. F. (2003) J. Biol. Chem., 278, 52012-52020.
137.Ljungman, M., and Lane, D. P. (2004) Nat.
Rev. Cancer, 4, 727-737.
138.Bakalkin, G., Selivanova, G., Yakovleva, T.,
Kiseleva, E., Kashuba, E., Magnusson, K. P., Szekely, L., Klein, G.,
Terenius, L., and Wiman, K. G. (1995) Nucleic Acids Res.,
23, 362-369.
139.Lee, S., Cavallo, L., and Griffith, J. (1997)
J. Biol. Chem., 272, 7532-7539.
140.Wetzel, C. C., and Berberich, S. J. (2001)
Biochim. Biophys. Acta, 1517, 392-397.
141.Zotchev, S. B., Protopopova, M., and
Selivanova, G. (2000) Nucleic Acids Res., 28,
4005-4012.
142.Mummenbrauer, T., Janus, F., Muller, B.,
Wiesmuller, L., Deppert, W., and Grosse, F. (1996) Cell,
85, 1089-1099.
143.Janus, F., Albrechtsen, N., Dornreiter, I.,
Wiesmuller, L., Grosse, F., and Deppert, W. (1999) Cell Mol. Life
Sci., 55, 12-27.
144.Dutta, A., Ruppert, J. M., Aster, J. C., and
Winchester, E. (1993) Nature, 365, 79-82.
145.Garkavtsev, I. V., Kley, N., Grigorian, I. A.,
and Gudkov, A. V. (2001) Oncogene, 20, 8276-8280.
146.Blander, G., Kipnis, J., Leal, J. F., Yu, C.
E., Schellenberg, G. D., and Oren, M. (1999) J. Biol. Chem.,
274, 29463-29469.
147.Sturzbecher, H. W., Donzelmann, B., Henning,
W., Knippschild, U., and Buchhop, S. (1996) Embo J., 15,
1992-2002.
148.Wang, X. W., Vermeulen, W., Coursen, J. D.,
Gibson, M., Lupold, S. E., Forrester, K., Xu, G., Elmore, L., Yeh, H.,
Hoeijmakers, J. H., and Harris, C. C. (1996) Genes Dev.,
10, 1219-1232.
149.Herring, C. J., West, C. M., Wilks, D. P.,
Davidson, S. E., Hunter, R. D., Berry, P., Forster, G., MacKinnon, J.,
Rafferty, J. A., Elder, R. H., Hendry, J. H., and Margison, G. P.
(1998) Br. J. Cancer, 78, 1128-1133.
150.Xanthoudakis, S., Miao, G., Wang, F., Pan, Y.
C., and Curran, T. (1992) Embo J., 11, 3323-3335.
151.Hanson, S., Kim, E., and Deppert, W. (2005)
Oncogene, 24, 1641-1647.
152.Raycroft, L., Wu, H. Y., and Lozano, G. (1990)
Science, 249, 1049-1051.
153.Liu, G., and Chen, X. (2006) J. Cell
Biochem., 97, 448-458.
154.Kubicka, S., Kuhnel, F., Zender, L., Rudolph,
K. L., Plumpe, J., Manns, M., and Trautwein, C. (1999) J. Biol.
Chem., 274, 32137-32144.
155.Mack, D. H., Vartikar, J., Pipas, J. M., and
Laimins, L. A. (1993) Nature, 363, 281-283.
156.Ohlsson, C., Kley, N., Werner, H., and LeRoith,
D. (1998) Endocrinology, 139, 1101-1107.
157.Funk, W. D., Pak, D. T., Karas, R. H., Wright,
W. E., and Shay, J. W. (1992) Mol. Cell Biol., 12,
2866-2871.
158.El-Deiry, W. S., Kern, S. E., Pietenpol, J. A.,
Kinzler, K. W., and Vogelstein, B. (1992) Nat. Genet., 1,
45-49.
159.Hoh, J., Jin, S., Parrado, T., Edington, J.,
Levine, A. J., and Ott, J. (2002) Proc. Natl. Acad. Sci. USA,
99, 8467-8472.
160.Tomso, D. J., Inga, A., Menendez, D., Pittman,
G. S., Campbell, M. R., Storici, F., Bell, D. A., and Resnick, M. A.
(2005) Proc. Natl. Acad. Sci. USA, 102, 6431-6436.
161.Inga, A., Storici, F., Darden, T. A., and
Resnick, M. A. (2002) Mol. Cell Biol., 22, 8612-8625.
162.Kondratov, R. V., Kuznetsov, N. V., Pugacheva,
E. N., Almazov, V. P., Prasolov, V. S., Kopnin, B. P., and Chumakov, P.
M. (1996) Mol. Biol. (Moscow), 30, 613-620.
163.Kastan, M. B., Zhan, Q., el-Deiry, W. S.,
Carrier, F., Jacks, T., Walsh, W. V., Plunkett, B. S., Vogelstein, B.,
and Fornace, A. J., Jr. (1992) Cell, 71, 587-597.
164.Zheng, X., and Chen, X. (2001) FEBS
Lett., 489, 4-7.
165.Contente, A., Dittmer, A., Koch, M. C., Roth,
J., and Dobbelstein, M. (2002) Nat. Genet., 30,
315-320.
166.Johnson, R. A., Ince, T. A., and Scotto, K. W.
(2001) J. Biol. Chem., 276, 27716-27720.
167.Dannenberg, J. H., David, G., Zhong, S., van
der Torre, J., Wong, W. H., and Depinho, R. A. (2005) Genes
Dev., 19, 1581-1595.
168.Cho, Y., Gorina, S., Jeffrey, P. D., and
Pavletich, N. P. (1994) Science, 265, 346-355.
169.Hamroun, D., Kato, S., Ishioka, C., Claustres,
M., Beroud, C., and Soussi, T. (2006) Hum. Mutat., 27,
14-20.
170.Gannon, J. V., Greaves, R., Iggo, R., and Lane,
D. P. (1990) Embo J., 9, 1595-1602.
171.Levine, A. J. (1989) Princess Takamatsu
Symp., 20, 221-230.
172.Chan, W. M., Siu, W. Y., Lau, A., and Poon, R.
Y. (2004) Mol. Cell Biol., 24, 3536-3551.
173.Hupp, T. R., Meek, D. W., Midgley, C. A., and
Lane, D. P. (1992) Cell, 71, 875-886.
174.Hupp, T. R., Meek, D. W., Midgley, C. A., and
Lane, D. P. (1993) Nucleic Acids Res., 21, 3167-3174.
175.Kaeser, M. D., and Iggo, R. D. (2002) Proc.
Natl. Acad. Sci. USA, 99, 95-100.
176.Kaneshiro, K., Tsutsumi, S., Tsuji, S.,
Shirahige, K., and Aburatani, H. (2007) Genomics, 89,
178-188.
177.Brash, D. E., and Havre, P. A. (2002) Proc.
Natl. Acad. Sci. USA, 99, 13969-13971.
178.Fischer, J. L., Lancia, J. K., Mathur, A., and
Smith, M. L. (2006) Anticancer Res., 26, 899-904.
179.Gudkov, A. V. (2002) Nat. Med.,
8, 1196-1198.
180.Seo, Y. R., Kelley, M. R., and Smith, M. L.
(2002) Proc. Natl. Acad. Sci. USA, 99, 14548-14553.
181.Smith, M. L., Lancia, J. K., Mercer, T. I., and
Ip, C. (2004) Anticancer Res., 24, 1401-1408.
182.Okorokov, A. L., and Milner, J. (1999) Mol.
Cell Biol., 19, 7501-7510.
183.McLure, K. G., Takagi, M., and Kastan, M. B.
(2004) Mol. Cell Biol., 24, 9958-9967.
184.Morgunkova, A. A. (2005) Biochemistry
(Moscow), 70, 955-971.
185.Weisz, L., Oren, M., and Rotter, V. (2007)
Oncogene, 26, 2202-2211.
186.Dittmer, D., Pati, S., Zambetti, G., Chu, S.,
Teresky, A. K., Moore, M., Finlay, C., and Levine, A. J. (1993) Nat.
Genet., 4, 42-46.
187.Di Como, C. J., Gaiddon, C., and Prives, C.
(1999) Mol. Cell Biol., 19, 1438-1449.
188.Bensaad, K., Le Bras, M., Unsal, K., Strano,
S., Blandino, G., Tominaga, O., Rouillard, D., and Soussi, T. (2003)
J. Biol. Chem., 278, 10546-10555.
189.Marin, M. C., Jost, C. A., Brooks, L. A.,
Irwin, M. S., O'Nions, J., Tidy, J. A., James, N., McGregor, J. M.,
Harwood, C. A., Yulug, I. G., Vousden, K. H., Allday, M. J., Gusterson,
B., Ikawa, S., Hinds, P. W., Crook, T., and Kaelin, W. G., Jr. (2000)
Nat. Genet., 25, 47-54.
190.Lang, G. A., Iwakuma, T., Suh, Y. A., Liu, G.,
Rao, V. A., Parant, J. M., Valentin-Vega, Y. A., Terzian, T., Caldwell,
L. C., Strong, L. C., El-Naggar, A. K., and Lozano, G. (2004)
Cell, 119, 861-872.
191.Olive, K. P., Tuveson, D. A., Ruhe, Z. C., Yin,
B., Willis, N. A., Bronson, R. T., Crowley, D., and Jacks, T. (2004)
Cell, 119, 847-860.
192.Gloushankova, N., Ossovskaya, V., Vasiliev, J.,
Chumakov, P., and Kopnin, B. (1997) Oncogene, 15,
2985-2989.
193.Pugacheva, E. N., Ivanov, A. V., Kravchenko, J.
E., Kopnin, B. P., Levine, A. J., and Chumakov, P. M. (2002)
Oncogene, 21, 4595-4600.
194.Ko, L. J., and Prives, C. (1996) Genes
Dev., 10, 1054-1072.
195.Bell, S., Klein, C., Muller, L., Hansen, S.,
and Buchner, J. (2002) J. Mol. Biol., 322, 917-927.
196.Dawson, R., Muller, L., Dehner, A., Klein, C.,
Kessler, H., and Buchner, J. (2003) J. Mol. Biol., 332,
1131-1141.
197.Lee, H., Mok, K. H., Muhandiram, R., Park, K.
H., Suk, J. E., Kim, D. H., Chang, J., Sung, Y. C., Choi, K. Y., and
Han, K. H. (2000) J. Biol. Chem., 275, 29426-29432.
198.Lin, J., Chen, J., Elenbaas, B., and Levine, A.
J. (1994) Genes Dev., 8, 1235-1246.
199.Candau, R., Scolnick, D. M., Darpino, P., Ying,
C. Y., Halazonetis, T. D., and Berger, S. L. (1997) Oncogene,
15, 807-816.
200.Chang, J., Kim, D. H., Lee, S. W., Choi, K. Y.,
and Sung, Y. C. (1995) J. Biol. Chem., 270,
25014-25019.
201.Dornan, D., Shimizu, H., Burch, L., Smith, A.
J., and Hupp, T. R. (2003) Mol. Cell Biol., 23,
8846-8861.
202.Curtin, J. C., and Spinella, M. J. (2005)
Oncogene, 24, 1481-1490.
203.Bochar, D. A., Wang, L., Beniya, H., Kinev, A.,
Xue, Y., Lane, W. S., Wang, W., Kashanchi, F., and Shiekhattar, R.
(2000) Cell, 102, 257-265.
204.Hill, D. A., de la Serna, I. L., Veal, T. M.,
and Imbalzano, A. N. (2004) J. Cell Biochem., 91,
987-998.
205.Lee, D., Kim, J. W., Seo, T., Hwang, S. G.,
Choi, E. J., and Choe, J. (2002) J. Biol. Chem., 277,
22330-22337.
206.An, W., Kim, J., and Roeder, R. G. (2004)
Cell, 117, 735-748.
207.Avantaggiati, M. L., Ogryzko, V., Gardner, K.,
Giordano, A., Levine, A. S., and Kelly, K. (1997) Cell,
89, 1175-1184.
208.Barlev, N. A., Liu, L., Chehab, N. H.,
Mansfield, K., Harris, K. G., Halazonetis, T. D., and Berger, S. L.
(2001) Mol. Cell, 8, 1243-1254.
209.Hsu, C. H., Chang, M. D., Tai, K. Y., Yang, Y.
T., Wang, P. S., Chen, C. J., Wang, Y. H., Lee, S. C., Wu, C. W., and
Juan, L. J. (2004) Embo J., 23, 2269-2280.
210.Lill, N. L., Grossman, S. R., Ginsberg, D.,
DeCaprio, J., and Livingston, D. M. (1997) Nature, 387,
823-827.
211.Xing, J., Sheppard, H. M., Corneillie, S. I.,
and Liu, X. (2001) Mol. Cell Biol., 21, 3652-3661.
212.Braastad, C. D., Han, Z., and Hendrickson, E.
A. (2003) J. Biol. Chem., 278, 8261-8268.
213.Lu, X. (2005) Curr. Opin. Genet. Dev.,
15, 27-33.
214.Cawley, S., Bekiranov, S., Ng, H. H., Kapranov,
P., Sekinger, E. A., Kampa, D., Piccolboni, A., Sementchenko, V.,
Cheng, J., Williams, A. J., Wheeler, R., Wong, B., Drenkow, J.,
Yamanaka, M., Patel, S., Brubaker, S., Tammana, H., Helt, G., Struhl,
K., and Gingeras, T. R. (2004) Cell, 116, 499-509.
215.Wei, C. L., Wu, Q., Vega, V. B., Chiu, K. P.,
Ng, P., Zhang, T., Shahab, A., Yong, H. C., Fu, Y., Weng, Z., Liu, J.,
Zhao, X. D., Chew, J. L., Lee, Y. L., Kuznetsov, V. A., Sung, W. K.,
Miller, L. D., Lim, B., Liu, E. T., Yu, Q., Ng, H. H., and Ruan, Y.
(2006) Cell, 124, 207-219.
216.Nemoto, S., Fergusson, M. M., and Finkel, T.
(2004) Science, 306, 2105-2108.
217.Tergaonkar, V., Pando, M., Vafa, O., Wahl, G.,
and Verma, I. (2002) Cancer Cell, 1, 493-503.
218.Ryan, K. M., Ernst, M. K., Rice, N. R., and
Vousden, K. H. (2000) Nature, 404, 892-897.
219.Shetty, S., Graham, B. A., Brown, J. G., Hu,
X., Vegh-Yarema, N., Harding, G., Paul, J. T., and Gibson, S. B. (2005)
Mol. Cell Biol., 25, 5404-5416.
220.Herold, S., Wanzel, M., Beuger, V., Frohme, C.,
Beul, D., Hillukkala, T., Syvaoja, J., Saluz, H. P., Haenel, F., and
Eilers, M. (2002) Mol. Cell, 10, 509-521.
221.Wu, W. S., Heinrichs, S., Xu, D., Garrison, S.
P., Zambetti, G. P., Adams, J. M., and Look, A. T. (2005) Cell,
123, 641-653.
222.Zhu, N., Gu, L., Findley, H. W., Chen, C.,
Dong, J. T., Yang, L., and Zhou, M. (2006) J. Biol. Chem.,
281, 14711-14718.
223.Homer, C., Knight, D. A., Hananeia, L., Sheard,
P., Risk, J., Lasham, A., Royds, J. A., and Braithwaite, A. W. (2005)
Oncogene, 24, 8314-8325.
224.Wei, X., Xu, H., and Kufe, D. (2005) Cancer
Cell, 7, 167-178.
225.Yoshida, K., Liu, H., and Miki, Y. (2006) J.
Biol. Chem., 281, 5734-5740.
226.Nakanishi, K., Kukita, F., Asai, K., and Kato,
T. (2001) Neurosci. Lett., 304, 85-88.
227.Bulavin, D. V., Demidov, O. N., Saito, S.,
Kauraniemi, P., Phillips, C., Amundson, S. A., Ambrosino, C., Sauter,
G., Nebreda, A. R., Anderson, C. W., Kallioniemi, A., Fornace, A. J.,
Jr., and Appella, E. (2002) Nat. Genet., 31, 210-215.
228.Mayo, L. D., Seo, Y. R., Jackson, M. W., Smith,
M. L., Rivera Guzman, J., Korgaonkar, C. K., and Donner, D. B. (2005)
J. Biol. Chem., 280, 25953-25959.
229.Oda, K., Arakawa, H., Tanaka, T., Matsuda, K.,
Tanikawa, C., Mori, T., Nishimori, H., Tamai, K., Tokino, T., Nakamura,
Y., and Taya, Y. (2000) Cell, 102, 849-862.
230.Cecchinelli, B., Lavra, L., Rinaldo, C.,
Iacovelli, S., Gurtner, A., Gasbarri, A., Ulivieri, A., Del Prete, F.,
Trovato, M., Piaggio, G., Bartolazzi, A., Soddu, S., and Sciacchitano,
S. (2006) Mol. Cell Biol., 26, 4746-4757.
231.Tokino, T., Thiagalingam, S., el-Deiry, W. S.,
Waldman, T., Kinzler, K. W., and Vogelstein, B. (1994) Hum. Mol.
Genet., 3, 1537-1542.
232.Yu, J., Zhang, L., Hwang, P. M., Rago, C.,
Kinzler, K. W., and Vogelstein, B. (1999) Proc. Natl. Acad. Sci.
USA, 96, 14517-14522.
233.Nakamura, Y. (2004) Cancer Sci.,
95, 7-11.
234.El-Deiry, W. S., Tokino, T., Velculescu, V. E.,
Levy, D. B., Parsons, R., Trent, J. M., Lin, D., Mercer, W. E.,
Kinzler, K. W., and Vogelstein, B. (1993) Cell, 75,
817-825.
235.Polyak, K., Waldman, T., He, T. C., Kinzler, K.
W., and Vogelstein, B. (1996) Genes Dev., 10,
1945-1952.
236.Nakano, K., and Vousden, K. H. (2001) Mol.
Cell, 7, 683-694.
237.Rouault, J. P., Falette, N., Guehenneux, F.,
Guillot, C., Rimokh, R., Wang, Q., Berthet, C., Moyret-Lalle, C.,
Savatier, P., Pain, B., Shaw, P., Berger, R., Samarut, J., Magaud, J.
P., Ozturk, M., Samarut, C., and Puisieux, A. (1996) Nat.
Genet., 14, 482-486.
238.Zhu, J., and Chen, X. (2000) Mol. Cell
Biol., 20, 5602-5618.
239.Rohaly, G., Chemnitz, J., Dehde, S., Nunez, A.
M., Heukeshoven, J., Deppert, W., and Dornreiter, I. (2005)
Cell, 122, 21-32.
240.Hermeking, H., and Benzinger, A. (2006)
Semin. Cancer Biol., 16, 183-192.
241.Helton, E. S., and Chen, X. (2007) J. Cell
Biochem., 100, 883-896.
242.Harms, K., Nozell, S., and Chen, X. (2004)
Cell Mol. Life Sci., 61, 822-842.
243.Hwang, B. J., Ford, J. M., Hanawalt, P. C., and
Chu, G. (1999) Proc. Natl. Acad. Sci. USA, 96,
424-428.
244.Adimoolam, S., and Ford, J. M. (2002) Proc.
Natl. Acad. Sci. USA, 99, 12985-12990.
245.Scherer, S. J., Maier, S. M., Seifert, M.,
Hanselmann, R. G., Zang, K. D., Muller-Hermelink, H. K., Angel, P.,
Welter, C., and Schartl, M. (2000) J. Biol. Chem., 275,
37469-37473.
246.Xu, J., and Morris, G. F. (1999) Mol. Cell
Biol., 19, 12-20.
247.Chen, J., and Sadowski, I. (2005) Proc.
Natl. Acad. Sci. USA, 102, 4813-4818.
248.Shimodaira, H., Yoshioka-Yamashita, A.,
Kolodner, R. D., and Wang, J. Y. (2003) Proc. Natl. Acad. Sci.
USA, 100, 2420-2425.
249.Liu, G., and Chen, X. (2006) Mol. Cell
Biol., 26, 1398-1413.
250.Arias-Lopez, C., Lazaro-Trueba, I., Kerr, P.,
Lord, C. J., Dexter, T., Iravani, M., Ashworth, A., and Silva, A.
(2006) EMBO Rep., 7, 219-224.
251.Sengupta, S., Shimamoto, A., Koshiji, M.,
Pedeux, R., Rusin, M., Spillare, E. A., Shen, J. C., Huang, L. E.,
Lindor, N. M., Furuichi, Y., and Harris, C. C. (2005) Oncogene,
24, 1738-1748.
252.Yamabe, Y., Shimamoto, A., Goto, M., Yokota,
J., Sugawara, M., and Furuichi, Y. (1998) Mol. Cell Biol.,
18, 6191-6200.
253.Kimura, T., Takeda, S., Sagiya, Y., Gotoh, M.,
Nakamura, Y., and Arakawa, H. (2003) Nat. Genet., 34,
440-445.
254.Dameron, K. M., Volpert, O. V., Tainsky, M. A.,
and Bouck, N. (1994) Science, 265, 1582-1584.
255.Van Meir, E. G., Polverini, P. J., Chazin, V.
R., Su Huang, H. J., de Tribolet, N., and Cavenee, W. K. (1994) Nat.
Genet., 8, 171-176.
256.Nishimori, H., Shiratsuchi, T., Urano, T.,
Kimura, Y., Kiyono, K., Tatsumi, K., Yoshida, S., Ono, M., Kuwano, M.,
Nakamura, Y., and Tokino, T. (1997) Oncogene, 15,
2145-2150.
257.Mashimo, T., Watabe, M., Hirota, S., Hosobe,
S., Miura, K., Tegtmeyer, P. J., Rinker-Shaeffer, C. W., and Watabe, K.
(1998) Proc. Natl. Acad. Sci. USA, 95, 11307-11311.
258.Bian, J., and Sun, Y. (1997) Mol. Cell
Biol., 17, 6330-6338.
259.Zou, Z., Gao, C., Nagaich, A. K., Connell, T.,
Saito, S., Moul, J. W., Seth, P., Appella, E., and Srivastava, S.
(2000) J. Biol. Chem., 275, 6051-6054.
260.Kunz, C., Pebler, S., Otte, J., and von der
Ahe, D. (1995) Nucleic Acids Res., 23, 3710-3717.
261.Komarova, E. A., Diatchenko, L., Rokhlin, O.
W., Hill, J. E., Wang, Z. J., Krivokrysenko, V. I., Feinstein, E., and
Gudkov, A. V. (1998) Oncogene, 17, 1089-1096.
262.Alberts, B. (ed.) (2008) Molecular Biology
of the Cell. Extended Version, Garland Science Publisher, New
York.
263.Gottlieb, T. M., and Oren, M. (1998) Semin.
Cancer Biol., 8, 359-368.
264.Vousden, K. H., and Lu, X. (2002) Nat. Rev.
Cancer, 2, 594-604.
265.Vousden, K. H. (2000) Cell, 103,
691-694.
266.Miyashita, T., and Reed, J. C. (1995)
Cell, 80, 293-299.
267.Oda, E., Ohki, R., Murasawa, H., Nemoto, J.,
Shibue, T., Yamashita, T., Tokino, T., Taniguchi, T., and Tanaka, N.
(2000) Science, 288, 1053-1058.
268.Yu, J., Zhang, L., Hwang, P. M., Kinzler, K.
W., and Vogelstein, B. (2001) Mol. Cell, 7, 673-682.
269.Fortin, A., Cregan, S. P., MacLaurin, J. G.,
Kushwaha, N., Hickman, E. S., Thompson, C. S., Hakim, A., Albert, P.
R., Cecconi, F., Helin, K., Park, D. S., and Slack, R. S. (2001) J.
Cell Biol., 155, 207-216.
270.Moroni, M. C., Hickman, E. S., Lazzerini
Denchi, E., Caprara, G., Colli, E., Cecconi, F., Muller, H., and Helin,
K. (2001) Nat. Cell Biol., 3, 552-558.
271.Robles, A. I., Bemmels, N. A., Foraker, A. B.,
and Harris, C. C. (2001) Cancer Res., 61, 6660-6664.
272.Owen-Schaub, L. B., Zhang, W., Cusack, J. C.,
Angelo, L. S., Santee, S. M., Fujiwara, T., Roth, J. A., Deisseroth, A.
B., Zhang, W. W., Kruzel, E., et al. (1995) Mol. Cell Biol.,
15, 3032-3040.
273.Wu, G. S., Burns, T. F., McDonald, E. R., 3rd,
Jiang, W., Meng, R., Krantz, I. D., Kao, G., Gan, D. D., Zhou, J. Y.,
Muschel, R., Hamilton, S. R., Spinner, N. B., Markowitz, S., Wu, G.,
and el-Deiry, W. S. (1997) Nat. Genet., 17, 141-143.
274.Attardi, L. D., Reczek, E. E., Cosmas, C.,
Demicco, E. G., McCurrach, M. E., Lowe, S. W., and Jacks, T. (2000)
Genes Dev., 14, 704-718.
275.Lin, Y., Ma, W., and Benchimol, S. (2000)
Nat. Genet., 26, 122-127.
276.Fiscella, M., Zhang, H., Fan, S., Sakaguchi,
K., Shen, S., Mercer, W. E., Vande Woude, G. F., O'Connor, P. M., and
Appella, E. (1997) Proc. Natl. Acad. Sci. USA, 94,
6048-6053.
277.Polyak, K., Xia, Y., Zweier, J. L., Kinzler, K.
W., and Vogelstein, B. (1997) Nature, 389, 300-305.
278.Hwang, P. M., Bunz, F., Yu, J., Rago, C., Chan,
T. A., Murphy, M. P., Kelso, G. F., Smith, R. A., Kinzler, K. W., and
Vogelstein, B. (2001) Nat. Med., 7, 1111-1117.
279.Valencia-Sanchez, M. A., Liu, J., Hannon, G.
J., and Parker, R. (2006) Genes Dev., 20, 515-524.
280.Chang, T. C., Wentzel, E. A., Kent, O. A.,
Ramachandran, K., Mullendore, M., Lee, K. H., Feldmann, G., Yamakuchi,
M., Ferlito, M., Lowenstein, C. J., Arking, D. E., Beer, M. A., Maitra,
A., and Mendell, J. T. (2007) Mol. Cell., 26,
745-752.
281.He, L., He, X., Lim, L. P., de Stanchina, E.,
Xuan, Z., Liang, Y., Xue, W., Zender, L., Magnus, J., Ridzon, D.,
Jackson, A. L., Linsley, P. S., Chen, C., Lowe, S. W., Cleary, M. A.,
and Hannon, G. J. (2007) Nature, 447, 1130-1134.
282.Raver-Shapira, N., Marciano, E., Meiri, E.,
Spector, Y., Rosenfeld, N., Moskovits, N., Bentwich, Z., and Oren, M.
(2007) Mol. Cell, 26, 731-743.
283.Tarasov, V., Jung, P., Verdoodt, B., Lodygin,
D., Epanchintsev, A., Menssen, A., Meister, G., and Hermeking, H.
(2007) Cell Cycle, 6, 1586-1593.
284.Hussain, S. P., Amstad, P., He, P., Robles, A.,
Lupold, S., Kaneko, I., Ichimiya, M., Sengupta, S., Mechanic, L.,
Okamura, S., Hofseth, L. J., Moake, M., Nagashima, M., Forrester, K.
S., and Harris, C. C. (2004) Cancer Res., 64,
2350-2356.
285.Yan, W., and Chen, X. (2006) J. Biol.
Chem., 281, 7856-7862.
286.Yoon, K. A., Nakamura, Y., and Arakawa, H.
(2004) J. Hum. Genet., 49, 134-140.
287.Velasco-Miguel, S., Buckbinder, L., Jean, P.,
Gelbert, L., Talbott, R., Laidlaw, J., Seizinger, B., and Kley, N.
(1999) Oncogene, 18, 127-137.
288.Budanov, A. V., Shoshani, T., Faerman, A.,
Zelin, E., Kamer, I., Kalinski, H., Gorodin, S., Fishman, A., Chajut,
A., Einat, P., Skaliter, R., Gudkov, A. V., Chumakov, P. M., and
Feinstein, E. (2002) Oncogene, 21, 6017-6031.
289.Budanov, A. V., Sablina, A. A., Feinstein, E.,
Koonin, E. V., and Chumakov, P. M. (2004) Science, 304,
596-600.
290.Rhee, S. G., Kang, S. W., Jeong, W., Chang, T.
S., Yang, K. S., and Woo, H. A. (2005) Curr. Opin. Cell Biol.,
17, 183-189.
291.Sablina, A. A., Budanov, A. V., Ilyinskaya, G.
V., Agapova, L. S., Kravchenko, J. E., and Chumakov, P. M. (2005)
Nat. Med., 11, 1306-1313.
292.Kopnin, P. B., Agapova, L. S., Kopnin, B. P.,
and Chumakov, P. M. (2007) Cancer Res., 67,
4671-4678.
293.Warburg, O. H. (1947) Abh. der Deutschen
Akad. der Wissenschaften zu Berlin.
294.Matoba, S., Kang, J. G., Patino, W. D., Wragg,
A., Boehm, M., Gavrilova, O., Hurley, P. J., Bunz, F., and Hwang, P. M.
(2006) Science, 312, 1650-1653.
295.Bensaad, K., Tsuruta, A., Selak, M. A., Vidal,
M. N., Nakano, K., Bartrons, R., Gottlieb, E., and Vousden, K. H.
(2006) Cell, 126, 107-120.
296.Bensaad, K., and Vousden, K. H. (2007)
Trends Cell Biol., 17, 286-291.
297.Erster, S., Mihara, M., Kim, R. H., Petrenko,
O., and Moll, U. M. (2004) Mol. Cell Biol., 24,
6728-6741.
298.Leu, J. I., Dumont, P., Hafey, M., Murphy, M.
E., and George, D. L. (2004) Nat. Cell Biol., 6,
443-450.
299.Marchenko, N. D., Zaika, A., and Moll, U. M.
(2000) J. Biol. Chem., 275, 16202-16212.
300.Mihara, M., Erster, S., Zaika, A., Petrenko,
O., Chittenden, T., Pancoska, P., and Moll, U. M. (2003) Mol.
Cell, 11, 577-590.
301.Moll, U. M., Wolff, S., Speidel, D., and
Deppert, W. (2005) Curr. Opin. Cell. Biol., 17,
631-636.
302.Marchenko, N. D., Wolff, S., Erster, S.,
Becker, K., and Moll, U. M. (2007) Embo J., 26,
923-934.
303.Schuler, M., and Green, D. R. (2005) Trends
Genet., 21, 182-187.
304.Dumont, P., Leu, J. I., Della Pietra, A. C.,
3rd, George, D. L., and Murphy, M. (2003) Nat. Genet.,
33, 357-365.
305.Chipuk, J. E., and Green, D. R. (2006) Cell
Death Differ., 13, 994-1002.
306.Moll, U. M., Marchenko, N., and Zhang, X. K.
(2006) Oncogene, 25, 4725-4743.
307.Harris, S. L., and Levine, A. J. (2005)
Oncogene, 24, 2899-2908.
308.Honda, R., and Yasuda, H. (1999) Embo
J., 18, 22-27.
309.Zhu, J. W., DeRyckere, D., Li, F. X., Wan, Y.
Y., and DeGregori, J. (1999) Cell Growth Differ., 10,
829-838.
310.Damalas, A., Kahan, S., Shtutman, M.,
Ben-Ze'ev, A., and Oren, M. (2001) Embo J., 20,
4912-4922.
311.Datta, A., Nag, A., Pan, W., Hay, N., Gartel,
A. L., Colamonici, O., Mori, Y., and Raychaudhuri, P. (2004) J.
Biol. Chem., 279, 36698-36707.
312.Sugimoto, M., Kuo, M. L., Roussel, M. F., and
Sherr, C. J. (2003) Mol. Cell., 11, 415-424.
313.Xiao, Z. X., Chen, J., Levine, A. J.,
Modjtahedi, N., Xing, J., Sellers, W. R., and Livingston, D. M. (1995)
Nature, 375, 694-698.
314.Yamasaki, L. (2003) Cancer Treat. Res.,
115, 209-239.
315.Feng, Z., Hu, W., de Stanchina, E., Teresky, A.
K., Jin, S., Lowe, S., and Levine, A. J. (2007) Cancer Res.,
67, 3043-3053.
316.Levine, A. J., Feng, Z., Mak, T. W., You, H.,
and Jin, S. (2006) Genes Dev., 20, 267-275.
317.Shaw, R. J., Kosmatka, M., Bardeesy, N.,
Hurley, R. L., Witters, L. A., DePinho, R. A., and Cantley, L. C.
(2004) Proc. Natl. Acad. Sci. USA, 101, 3329-3335.
318.Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan,
K. L. (2002) Nat. Cell Biol., 4, 648-657.
319.Lum, J. J., Bauer, D. E., Kong, M., Harris, M.
H., Li, C., Lindsten, T., and Thompson, C. B. (2005) Cell,
120, 237-248.
320.Hay, N., and Sonenberg, N. (2004) Genes
Dev., 18, 1926-1945.
321.Thomas, G. (2002) Biol. Res., 35,
305-313.
322.Sarbassov, D. D., Guertin, D. A., Ali, S. M.,
and Sabatini, D. M. (2005) Science, 307, 1098-1101.
323.Zhang, Y., Gao, X., Saucedo, L. J., Ru, B.,
Edgar, B. A., and Pan, D. (2003) Nat. Cell Biol., 5,
578-581.
324.Christophorou, M. A., Ringshausen, I., Finch,
A. J., Swigart, L. B., and Evan, G. I. (2006) Nature,
443, 214-217.
325.Efeyan, A., Garcia-Cao, I., Herranz, D.,
Velasco-Miguel, S., and Serrano, M. (2006) Nature, 443,
159.
326.Bartkova, J., Horejsi, Z., Koed, K., Kramer,
A., Tort, F., Zieger, K., Guldberg, P., Sehested, M., Nesland, J. M.,
Lukas, C., Orntoft, T., Lukas, J., and Bartek, J. (2005) Nature,
434, 864-870.
327.Gorgoulis, V. G., Vassiliou, L. V., Karakaidos,
P., Zacharatos, P., Kotsinas, A., Liloglou, T., Venere, M., Ditullio,
R. A., Jr., Kastrinakis, N. G., Levy, B., Kletsas, D., Yoneta, A.,
Herlyn, M., Kittas, C., and Halazonetis, T. D. (2005) Nature,
434, 907-913.
328.Capri, M., Salvioli, S., Sevini, F., Valensin,
S., Celani, L., Monti, D., Pawelec, G., de Benedictis, G., Gonos, E.
S., and Franceschi, C. (2006) Ann. N. Y. Acad. Sci.,
1067, 252-263.
329.Depp, C. A., Glatt, S. J., and Jeste, D. V.
(2007) Curr. Psychiatry Rep., 9, 7-13.
330.Guarente, L. (2005) Mech. Aging Dev.,
126, 923-928.
331.Beckman, K. B., and Ames, B. N. (1998)
Physiol. Rev., 78, 547-581.
332.Droge, W. (2002) Physiol. Rev.,
82, 47-95.
333.Tyner, S. D., Venkatachalam, S., Choi, J.,
Jones, S., Ghebranious, N., Igelmann, H., Lu, X., Soron, G., Cooper,
B., Brayton, C., Hee Park, S., Thompson, T., Karsenty, G., Bradley, A.,
and Donehower, L. A. (2002) Nature, 415, 45-53.
334.Garcia-Cao, I., Garcia-Cao, M.,
Martin-Caballero, J., Criado, L. M., Klatt, P., Flores, J. M., Weill,
J. C., Blasco, M. A., and Serrano, M. (2002) Embo J., 21,
6225-6235.
335.Mendrysa, S. M., O'Leary, K. A., McElwee, M.
K., Michalowski, J., Eisenman, R. N., Powell, D. A., and Perry, M. E.
(2006) Genes Dev., 20, 16-21.
336.Bukhman, V. L., Ninkina, N. N., Chumakov, P.
M., Khilenkova, M. A., and Samarina, O. P. (1987) Genetika,
23, 1547-1554.
337.Van Heemst, D., Mooijaart, S. P., Beekman, M.,
Schreuder, J., de Craen, A. J., Brandt, B. W., Slagboom, P. E., and
Westendorp, R. G. (2005) Exp. Gerontol., 40, 11-15.
338.Campisi, J. (2003) Nat. Rev. Cancer,
3, 339-349.
339.Crighton, D., Wilkinson, S., and Ryan, K. M.
(2007) Autophagy, 3, 72-74.
340.Terman, A., Gustafsson, B., and Brunk, U. T.
(2007) J. Pathol., 211, 134-143.
341.Saretzki, G., and von Zglinicki, T. (2002)
Ann. N. Y. Acad. Sci., 959, 24-29.
342.Pletjushkina, O. Y., Fetisova, E. K., Lyamzaev,
K. G., Ivanova, O. Y., Domnina, L. V., Vyssokikh, M. Y., Pustovidko, A.
V., Vasiliev, J. M., Murphy, M. P., Chernyak, B. V., and Skulachev, V.
P. (2005) Cell Death Differ., 12, 1442-1444.
343.Poli, G., and Parola, M. (1997) Free Rad.
Biol. Med., 22, 287-305.