REVIEW: Noncoding RNAs and Chromatin Structure
S. A. Lavrov* and M. V. Kibanov
Institute of Molecular Genetics, Russian Academy of Sciences, pl. Kurchatova 2, 123182 Moscow, Russia; fax: (499) 196-0221; E-mail: slavrov@img.ras.ru* To whom correspondence should be addressed.
Received April 5, 2007
A number of examples of noncoding RNA-connected chromatin modifications in eukaryotes has been recently revealed. Four cases are under detailed consideration in the present review, namely Xist RNA-dependent X-chromosome inactivation in mammals, roX RNA-dependent hyperactivation of X-chromosome in the fruit fly (in both cases the goal is dosage compensation, equalization of transcription level from two X chromosomes in females and one in males), and two examples of RNAi-connected down-regulation of transcription--siRNA-dependent heterochromatin formation in fission yeast and RdDM (RNA-dependent DNA methylation) in plants (FWA gene regulation in Arabidopsis). Although overall quite different, each phenomenon demonstrates some common features of RNA-driven chromatin modification process, including the role of RNA in aiming of chromatin-modifying protein complexes to their targets and subsequent formation of self-maintaining specific chromatin conformation (DNA methylation, changes in histone code, and binding of self-assembling protein complexes).
KEY WORDS: RNA interference, chromatin structure, transcription regulationDOI: 10.1134/S0006297907130020
Abbreviations: DCC) dosage compensation complex; DDM1) decrease in DNA methylation 1; DMT) DNA methyltransferase; HAT) histone acetyltransferase; HDAC) histone deacetylase; HMG) high mobility group; HMT) histone methyltransferase; HP1) heterochromatic protein 1; MBD) methylcytosine binding domain; PC) Polycomb; PcG) Polycomb group; RdDM) RNA-dependent DNA methylation; RDRC) RNA-directed RNA polymerase complex; RISC) RNA-induced silencing complex; RITS) RNA-induced transcriptional silencing; RNAi) RNA interference; siRNA) short interfering RNA; Xic) X-chromosome inactivation center; Xist) X-inactive specific transcript; Xite) X-inactivation intergenic transcription element.
Apart from protein-coding mRNAs, in eukaryotic cells there are a number
of other types of RNA that do not translate but bear other functions.
RNAs may be structural components of cell organelles, like ribosomal
RNA, participate in protein synthesis (tRNA), and may have enzymatic
activity or regulatory functions.
Recently, the role of noncoding RNAs in transcription regulation has been revealed [1]. The most famous and well-studied examples of this kind are the dosage compensation systems: X-chromosome inactivation in mammals and X-chromosome upregulation in drosophila. In addition, participation of short interference RNAs (siRNA, component of RNA interference system) in transcription regulation has been shown for a number of systems. Pathways of this type exist in yeast, the ciliate Tetrahymena thermophila (in this case transcriptional repression is the preliminary step before heterochromatin elimination [2, 3]), higher plants, mammals [4-6], and insects [7].
This review is mainly devoted to dosage compensation systems of drosophila and mammals and two RNAi-related phenomena--RNA-dependent DNA methylation (RdDM) in plants and RNA-dependent heterochromatin formation in yeast. These pathways have been studied in-depth for the last several years, but there are still many unknown and difficult-to-explain problems there.
CHROMATIN STRUCTURE. BRIEF DESCRIPTION
Chromatin is defined as a complex of DNA and attached proteins of different types. In eukaryotes, the basic unit of chromatin is the nucleosome. The nucleosome is composed of eight histone molecules (which form core) and DNA of ~145 bp in length making 1.7 turns around the core. Neighbor nucleosomes may be located at different distances from each other: the density and regularity of its package depend on the functional state of the chromatin region. Active transcription correlates with overall chromatin decompactization, while heterochromatinized regions (below) demonstrate a dense and regular (with ~40 bp spacers) nucleosome pattern.
Regulatory regions of genes either are free of nucleosomes or contain so-called “positioned nucleosomes”, those with fixed position relative to gene sequence [8]. This type of arrangement is important for proper access of transcription factors to regulatory regions and the promoter. A group of proteins, so-called chromatin remodeling factors, is able to perform ATP-dependent nucleosome displacement [9]; this type of proteins is often necessary for transcription initiation or, the opposite, for repression. An example of chromatin remodeling factors is the swi/snf family of proteins, named for it first member, SWI2/SNF2. SWI2/SNF2 is required for transcription activation in yeast, and its Drosophila ortholog, BRAHMA, plays the same role. BRAHMA is a component of a multiprotein complex which contains more than ten other members and demonstrates chromatin remodeling activity [10]. Proteins with the same properties can be found in the majority of eukaryotes [11-13].
Nucleosome core is composed of eight histones: in pairs of molecules of H2A, H2B, H3, and H4 type. Histone H1 participates in nucleosome-nucleosome interactions and is necessary for chromatin packaging into a high-order structure: a 30-nm fiber. Histones are small, positively charged proteins with highly conserved primary structure. The histone molecule consists of a globular C-end and amorphous (with no distinct secondary structure) N-end. The N-end region goes beyond the nucleosome core and can interact with other chromatin proteins.
Apart from four main types, there are some minor variants of histones in the eukaryotic genome. These minor histones may have some special functions in chromatin organization. There are also many non-histone chromatin components, such as high mobility group proteins, constituents of kinetochore, nuclear envelope, transcription, translation, and reparation systems, etc.
Histones are not just structural components of chromatin; they actively participate in transcription regulation. The above-mentioned N-end regions (tails) play a key role in this process. Amino acids of histone tails undergo covalent modifications of several types. It may be acetylation, methylation, phosphorylation, ADP-ribosylation, and ubiquitylation. The diversity of covalent modifications suggests a “histone code”, which determines the possibility of transcription and the expression level of any particular gene [14-16]. The number of variants of covalent modifications is enough to make any nucleosome in the genome unique. Nucleosome structure and the types of covalent modifications are shown on Fig. 1.
Although full description of each possible histone modification type is beyond the limits of this review, we will consider several most important ones because of their universal role in gene regulation in eukaryotes.Fig. 1. Nucleosome structure and the types of histone modifications. a) Nucleosome structure according to X-ray diffraction analysis. Two orthogonal projections are shown; H2A, H2B, H3, and H4 histones are marked by different colors. b) Covalent histone modifications. Modified amino acids marked by 1-letter code and number of position from N-end of protein molecule. Ac, acetylation; Met, methylation; P, phosphorylation; Ub, ubiquitylation; ADP, ADP-ribosylation. Lysine and arginine can be methylated, lysine can be acetylated, and serine and glutamic acid can be phosphorylated. Repression-associated modifications are in blue, active chromatin state is in red. Gray color marks modifications connected to chromosome condensation in mitosis or meiosis.
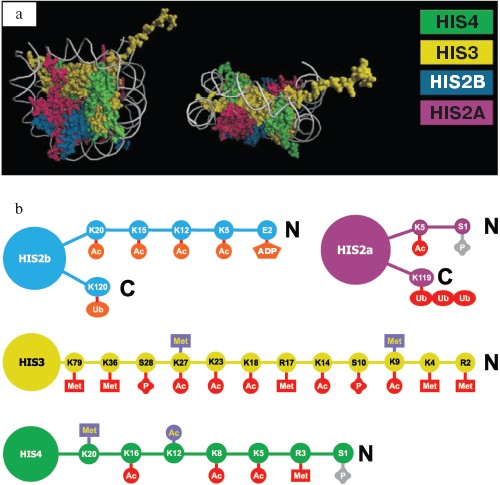
Acetylation. Acetylation of the N-end lysine residues of H3 and H4 histones is most well studied. This type of modification is performed by histone acetyltransferase enzymes (HATs). Acetylation typically correlates with active transcription, but there are some exceptions from this rule [17]. Acetylation of histone H4 at lysine-16 is particularly connected to transcription hyperactivation in case of fruit fly dosage compensation (below). Histone deacetylation correlates with gene repression and is provided by specific enzymes, histone deacetylases (HDACs).
Methylation. Lysine residues in histones can be mono-, di-, and tri-methylated. Methylation of lysine residues at positions 9 and 27 of H3 leads to transcription repression, while methylation of lysine-4 associates with active chromatin state. Methylation is performed by histone methyltransferase (HMT) enzymes; demethylation is not well studied [18].
Histone H3 methylated at lysine-9 is a heterochromatin mark in all eukaryotes studied up to now.
Originally, heterochromatin was defined as the part of chromatin remaining condensed during interphase of the cell cycle (in contrast to euchromatin). Centromeric and telomeric regions of chromosomes are usually heterochromatinized.
The bulk of heterochromatin is different types of repeats and transposable elements; it has a specific protein composition and specific pattern of histone modifications, including the above-mentioned methylated lysine-9 of H3. Euchromatic genes, being transferred into heterochromatin surroundings, typically undergo transcriptional repression (so-called heterochromatic position effect). The euchromatic region in heterochromatic environment can acquire heterochromatin molecular structure; the process is called heterochromatinization.
There are a number of proteins specifically associated with heterochromatin; they interact with each other and maintain compact chromatin structure. Heterochromatin proteins are well-studied in drosophila [19-21], but other eukaryotes have similar ones.
Typical heterochromatin proteins of fruit fly are:
- HDAC1, histone deacetylase. HDACs have been found in all studied eukaryotes (for example, yeast CLR3 and plant HDA6) [15, 22];
- SUV39, histone methyltransferase that methylates lysine-9 of histone H3. Orthologs have been found in yeast (CLR4), plants (KYP), and mammals and seem to exist in all eukaryotes [20, 23];
- HP1, heterochromatin protein 1. The yeast equivalent is SWI6, and orthologs have been found in plants and mammals. A specific feature of HP1 is the presence of chromodomain, a protein domain detected in a number of chromatin proteins. Chromodomains from different proteins differ in their sequence and are able to bind to differently methylated histones and, possibly, RNA [24-27]. The chromodomain of HP1 recognizes histone H3 methylated at lysine-9 [28].
There are a number of other heterochromatin proteins, e.g. the components of the DNA replication machinery [29], DNA-binding proteins (SUVAR 3-7) [30], and several types of histone acetyltransferases, deacetylases, and methyltransferases.
Apart from heterochromatin proteins, there is a second gene repression system in fruit fly: Polycomb group (PcG). The name comes from one of the members--chromodomain-containing POLYCOMB (PC) protein. The group includes more than 10 participants [31, 32].
Heterochromatin protein complexes cover genomic regions megabases in size. Polycomb group proteins, in contrast, perform “point” repression of euchromatic genes. One of the targets of Polycomb group is developmental loci, like Bithorax and Antennapedia clusters [33, 34], the total number of targets exceeding several hundred [35, 36]. Related systems have been found in plants, mammals, nematode Caenorhabditis elegans, and, obviously, exist in all higher eukaryotes [37-40]. Chromodomain of Polycomb, in contrast to HP1, recognizes histone H3 methylated at lysine-27 [24].
Not just histones, the DNA itself can also be covalently modified. This is DNA methylation--modification of cytosine residues resulting in methylcytosine formation. This type of DNA modification has been found in mammals and higher plants, where it is indispensable for proper transcription control. At the same time, in yeast, fruit fly, and C. elegans DNA methylation is either not found or negligible [41].
Usually, DNA sequences like CpG, CpNpG, and CpHpH (H is A, T, or C bases) are subjected to methylation. Clusters of CpG dinucleotides (so-called CpG-islands) exist in the regulatory regions of a number of genes. Enzymes called DNA-methyltransferases (DMT) are responsible for methylation in plants and mammals and are represented by a number of classes with different molecular properties [42, 43]. In most cases, methylation is associated with transcriptional repression. The mechanism of repression suggests recognition of methylated sequences by a complex of chromatin proteins, some of which contain a methylcytosine-binding domain (MBD) [44-46].
DOSAGE COMPENSATION SYSTEMS
In many organisms, sex chromosomes differ in their gene content. For instance, in mammals and the fruit fly Drosophila melanogaster, female X chromosome has gene content similar to that of autosomes. In contrast, the male Y chromosome is gene depleted and mainly heterochromatinized. In mammals and fruit fly, females have two X chromosomes while males have one X and one Y chromosome.
The unequal gene content of sex chromosomes results in dosage imbalance between the XX and XY sexes. This disbalance in gene dosage should be compensated to achieve parity of X-linked gene products in cells of both sexes. For that purpose, organisms employ different strategies of dosage compensation.
Dosage Compensation in Mammals
During evolution, some mammals have evolved a system of dosage compensation based on inactivation of one of two female X chromosomes early in development. According to recent data, inactivation takes place at the stage of the four-cell embryo [47, 48]. This process is regulated by a locus on the X chromosome called Xic (X-chromosome inactivation center) [49]. Xic is essential for the process. It determines the X chromosome to be inactivated and also participates in initiation of the phenomenon: transcription repression starts from the Xic locus. A mutant X chromosome with deleted Xic locus is not inactivated, and insertion of extra copies of Xic into one of the autosomes leads to inactivation of adjacent genes [50-52].
The Xic locus encodes two long transcripts: sense Xist (17 kb) and antisense Tsix, the latter being transcribed in the opposite direction to Xist and being much longer. Xist is active on the X chromosome that is going to be inactivated and is silent on the other, active X chromosome. The Tsix gene takes a vigorous part in the transcriptional repression of the Xist gene on the active X chromosome [1, 50, 53-56]. Processes of X chromosome inactivation by Xist and Xist repression by Tsix are closely interconnected, and misregulation of one of them leads to death of embryos at early stages of development if both X chromosomes either stay active or become silent.
Xist and X chromosome inactivation. Inactivation begins with the up-regulation of Xist gene transcription, and, as a result, the amount of RNA products per cell considerably increases [57]. At the beginning, nascent transcripts are accumulated near the Xic locus; then, they spread out in both directions along the X chromosome and coat it. In cases when the Xist gene does not work, the X chromosome stays active [58].
X chromosome inactivation is accompanied by covalent modifications of histone tails, such as deacetylation of histones H3 and H4, dimethylation of H3 lysine-9, trimethylation of H3 lysine-27, and demethylation of H3 lysine-4 [59-61]. Exchange of histone H2A for its heterochromatic counterpart macroH2A1 occurs at later stages of embryogenesis, together with DNA methylation and set-up of later timing of replication [62, 63]. Therefore, at the end of the process, the inactive X chromosome has all main features of heterochromatin.
It is still unclear by what mechanism the Xist gene leads to X chromosome inactivation. Up-regulation of nascent transcripts, their accumulation and, finally, X chromosome coating allow one to suppose that there are specific proteins which are recruited to the future inactive X chromosome by accumulating Xist transcripts. In particular, it has been discovered that Polycomb group (PcG) proteins--EED and ENX1 (also known as EZH2)--associate with the X chromosome in Xist-dependent mode at the very beginning of development [60, 64, 65]. This leads to methylation of lysine-27 of histone H3. It is possible that covalent modifications left by these proteins and enzymatic complexes associated with them may contribute to the maintaining of the inactive state of an X chromosome. The replacement of histone H2A for its minor heterochromatic variant macroH2A1 is also Xist-RNA dependent [66, 67].
It is also important that Xist expression only leads to X chromosome inactivation in differentiating cells; in cells, which have passed this stage, the mechanism does not work. In other words, there is some developmental window when inactivation can take place; outside this period inactivation is impossible [68]. This developmental window probably depends on the activity of EED and ENX1 proteins.
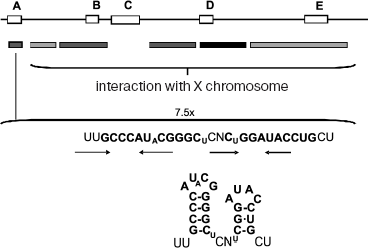
It has been shown that different sites along Xist RNA are responsible for the X chromosome localization of nascent transcripts, on one hand, and for silencing of X chromosome genes on the other [67] (Fig. 2). The first ones are distributed along the whole molecule of Xist RNA; they have no common sequences and are redundant in function. These sites probably have low affinity to some still unknown proteins, which are able to bind with them cooperatively. However, the region Repeat-A, which is crucial for silencing activity, has been found at the 5´-terminus of Xist RNA. Its deletion does not influence accumulation of nascent transcripts, but it completely abolishes X chromosome inactivation. This element contains several inverted repeats, which are able to form stem-loop structures, as a computer simulation shows. It is possible that some repressor proteins interact with these loops. Other evolutionarily conserved repeat elements, found earlier, do not influence dosage compensation [69, 70]. Proteins interacting with these functional elements of Xist RNA are now under intensive investigations.
Interestingly, free molecules of Xist RNA have not been found in the nucleus [67], probably due to instability and rapid degradation of Xist molecules out of complex with X chromosome or due to their cotranscriptional attachment to the X chromosome. In any case, this fact may be important for the restriction of the “action field” of Xist RNAs by X chromosome in cis and, therefore, for preventing autosome inactivation or second X chromosome mis-inactivation.Fig. 2. Mouse Xist-RNA structure. Schematic representation of Xist RNA. Evolutionarily conserved repeat elements found in other mammals (A-E) are depicted. Repeat-A on the 5´-terminus of Xist is important for silencing. This element contains 7.5 inverted repeats, which are able to form stem-loop structures according to computer simulation. Conserved nucleotides are highlighted. Small characters indicate less frequent base changes. GC base pairs are represented by a line, GU base pairs by a dot. The boxes below the map summarize the regions localizing Xist RNA to chromatin (black boxes represent the greatest activity).
Tsix is a negative regulator of Xist. There are at least two stages in X chromosome inactivation. First inactivation occurs in very early development, at the stage of the four-cell embryo [47, 48], and the paternal chromosome is always silenced. If dosage compensation is impaired at this stage, the embryo will die. The next stages are pre-implantation development and implantation. After implantation, the paternal X chromosome can be reactivated in cells of ICM (inner cell mass), and then silencing takes place again, but now in a “random” manner, that is on either of the X chromosomes [48, 71]. Preferential inactivation of paternal X chromosome is an example of genome imprinting, one of the variants of epigenetic inheritance that means a type of inheritance without changes in DNA sequence.
On the active X chromosome, the Xist gene is silent. There are several lines of evidences that antisense RNA Tsix plays a crucial role in the process of this silencing. Thus, when the Tsix gene does not work (as the result of deletion or mutation in the promoter region), Xist RNA is accumulated on the mutated X chromosome, which will be inactivated. However, continuous transcription of the Tsix gene prevents X chromosome inactivation [54, 55, 72, 73]. Embryos which inherit X chromosome with mutated Tsix gene from their father develop normally, and in this case only the paternal X chromosome is switched off in female embryos. Otherwise, most embryos of both sexes will die if they inherit mutated X chromosome from their mother, because in such situation inactivation affects both X chromosomes in females and the single X chromosome in males. Hence, it seems that X chromosome with mutated Tsix gene is inactivated, irrespective of its heritage. On the basis of these experiments, the hypothesis that Tsix gene transcription is responsible for the imprinted inactivation of paternal X chromosomes at the beginning of development has been proposed. But there are no signs of Tsix gene expression during early embryogenesis. Therefore, it is evident that the Tsix gene plays an important role in the phenomenon of dosage compensation, and its main function is the maintaining of the active state of an X chromosome, but it is unlikely that Tsix is involved in the choice between maternal and paternal X chromosomes, and there ought to be another mechanism responsible for preferential inactivation of paternal X chromosomes at the stage of early embryogenesis.
Based on recent investigations two new hypotheses concerning imprinted X chromosome inactivation have been proposed. According to one of them, paternal X chromosome is inherited already in pre-inactivated state [47]; from the other point of view, inactivation occurs very rapidly after the first zygote division [48], but at the same time, the Xist gene on the maternal X chromosome is epigenetically silent. In any case, the mechanism does not depend on the Tsix gene or its RNA product, at least during early development, but Tsix is necessary for its further maintaining because mutations in Tsix ultimately lead to Xist up-regulation and maternal X chromosome inactivation. The nature of this mechanism of imprinting is still unknown, but it has been revealed that its establishment occurs during oocytes maturation, meiosis prophase I [74]. There were some reports that methylation of the Xist gene promoter might be responsible for the imprinting [75-77], but further investigations did not confirm this idea [78, 79].
In short, the Xist and Tsix genes and their transcripts are all involved in the process of dosage compensation, and at the same time Tsix is a negative regulator of Xist. It seems that molecular mechanisms underlying this phenomenon are similar in the case of both RNA molecules, with the only difference that Xist affects the whole X chromosome, whereas Tsix affects the Xist gene only.
For instance, using differentiating embryonic stem (ES) cells as a model system, it has been shown that Tsix gene transcription through the Xist leads to Xist promoter methylation, probably through the recruitment of specific enzymes modifying histone N-terminal tails and DNA, and thus to Xist gene inactivation. Particularly, Tsix RNA has been co-immunoprecipitated with DNMT3a enzyme, which is able to methylate DNA de novo [80]. Earlier studies have provided information that Xist promoter is differently methylated in differentiated female cells, and this correlates with Xist gene activity: silent promoter is much more modified than its active counterpart [81].
At the same time, Xist promoters on both X chromosomes are partially methylated in equal manner in undifferentiated female ES cells [50, 82]. Therefore, it seems that functionally important methylation appears only during differentiation. If the Tsix gene is turned off a profound cell death will occur owing to mis-regulation of methylation and, as a result, activation of both Xist genes [57, 83]. It has been proposed that Tsix RNA modulates DNMT3a enzymatic activity that leads to Xist promoter methylation and gene inactivation on the future active X chromosome. This hypothesis is supported by the observations that DNMT3a enzyme is present on the X chromosome even before the beginning of inactivation and also in cells lacking Tsix transcripts [57].
There also might be another mechanism of Xist regulation by Tsix based on Xist-Tsix duplex formation as a result of their antiparallel nature, following by Xist RNA degradation in an RNA-interference pathway. But it is very unlikely that such full-length double-strand RNA really exists because of splicing peculiarities of nascent Tsix RNA [55, 84]. This process splices most of the Tsix RNA complementary with Xist RNA and leaves only minor overlap of about 1.9 kb with Xist 5´-terminus, the region that includes Repeat-A, which is important for the silencing activity of Xist RNA. So this may suggest that Tsix RNA works through its spliced forms by blocking this silencing domain of Xist RNA. However, it has been shown earlier that spliced forms of Tsix RNA by themselves are not sufficient for repression of Xist [84] and, moreover, most Tsix transcripts are truncated as the result of transcription stop within the gene. For instance, in humans Tsix RNA has been reported to truncate shortly after crossing of the 3´-terminus of Xist gene [85, 86].
In addition to Xist and Tsix genes, a new gene called Xite (X-inactivation intergenic transcription element) has been mapped recently on the Xic locus. This gene also encodes long noncoding RNA transcribed in the same direction as Tsix. There are several transcriptional start points clustered in two regions, with one located about 10 kb upstream of Tsix, and the second located about 15 kb upstream of Tsix [55, 87]. Xite is a lowly transcribed gene; the level of Xite RNA is about 10-60-fold less than Tsix RNA level in mouse ES cells. Deletion or mutation of Xite results in abnormal expression of Tsix and preferential silencing of X chromosome in cis [87]. At the same time, it seems that Xite action does not appear to depend on RNA itself. There is a suggestion that the nearest Xite promoter contains an enhancer element for Tsix [87, 88]. So, according to the current model, Xite works synergistically with Tsix to designate the active X chromosome. Owing to the presence of enhancer, Xite promotes continual expression of Tsix during cell differentiation; the constant expression of Tsix, in turn, prevents the up-regulation and spreading of Xist RNA and X chromosome inactivation.
Dosage Compensation in Drosophila
The fruit fly Drosophila melanogaster has also evolved a system of dosage compensation to achieve parity of products of X-linked genes between the sexes. But the strategy of the process differs from that used by mammals. Both female X chromosomes are transcribed with equal basal rate, whereas transcription from the single male X chromosome is two-fold increased.
Dosage compensation in Drosophila is mediated by a ribonucleoprotein complex known as Dosage Compensation Complex (DCC), consisting of six different proteins and two noncoding RNA molecules (see Fig. 3). This complex binds to numerous sites along the male X chromosome, so-called “entry points” [89]. Five proteins of the complex are generally known as MSL (male-specific lethal): MSL-1, -2, and -3, MLE (maleless), and MOF (males absent on the first). Mutations at these proteins prevent male X chromosome hypertranscription and lead to male lethality. The sixth protein member of the DCC, JIL-1, is also enriched on X chromosomes and interacts with MSL proteins [90, 91].
MOF is a histone acetyltransferase that modifies lysine-16 of histone H4 [92, 93]. A kinase activity has been shown for JIL-1 protein that is able to phosphorylate Ser10 of histone H3 in vitro [94-96]. HAT and kinase activities of MOF and JIL-1 proteins correspondingly suggest that histone code and epigenetic changes in male X chromosome chromatin structure are responsible for hypertranscription. So, histone H4 acetylated at lysine-16 is specific for dosage compensated X chromosome.Fig. 3. Dosage compensation in Drosophila melanogaster. Dosage compensation is mediated by a ribonucleoprotein complex known as the compensasome or DCC (a). Complex assembly occurs in a stepwise manner: MSL-1 and MSL-2 proteins appear to bind first with 30-35 high-affinity entry sites along the X chromosome, two of which are roX1 and roX2 genes (b). MLE, an RNA helicase, is required to integrate roX RNAs into the DCC. MSL-3 and MOF are also able to bind RNA by their chromodomains. Histone kinase JIL-1 has also been found on the X chromosome. Mature complexes can spread in cis and finally coat the entire X chromosome.
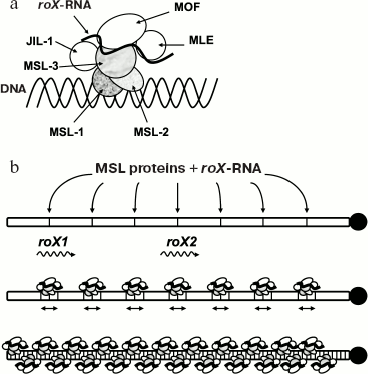
Together with protein factors, RNA molecules also take part in dosage compensation in Drosophila. There are at least two noncoding RNAs, roX1 (3.7 kb) and roX2 (0.6 kb), that are components of DCC [97]. As in the case with Xist RNA, roX RNAs (RNA on the X) are also synthesized on the X chromosome and transcription activation occurs as a result of their accumulation and spreading along the X chromosome, together with DCC proteins. This has been shown in experiments with extra copies of roX transgenes inserted into autosomes, when roX RNAs and DCC accumulation, gene hyperacetylation, and hypertranscription took place on the several thousands of kb from the insertions [98, 99]. Moreover, according to these results, roX RNAs can work regardless of sequence specificity and can also activate transcription of autosomal genes.
DCC assembly takes place exclusively in males, in spite of the fact that initial doses of MSL-1 and -3, MLE, and MOF proteins are maternally provided [100]. Such high specificity of DCC formation is attributed to expression of SXL protein, the product of Sex-lethal gene, in females but not in males. SXL inhibits translation of MSL-2 mRNA by blocking its interaction with the ribosome [101, 102]. Suppression of MSL-2 prevents DCC formation in females, as MSL-1 and -3 require MSL-2 protein for sustained expression and stability [103]. Moreover, MSL-2 lies at the base of DCC assembly [102]. For instance, expression of MSL-2 protein in females from transgenic mRNA lacking SXL-binding site leads to hypertranscription of both X chromosomes and, as a result, to female lethality [95, 104, 105]. It is important that continued expression and stability of roX RNAs also depend on MSL proteins [101, 103].
The exact role of roX RNAs in dosage compensation is still poorly understood. According to available data, roX RNAs are rather required for proper binding of DCC to and distribution over the X chromosome than for the DCC assembly. Approximately 30-35 high-affinity “entry sites” have been mapped on the X chromosome. They are proposed to serve as nucleation sites from which complexes can spread in cis to coat adjacent regions of the chromosome [97]. Interestingly, two of these entry sites coincide with roX1 and roX2 genes. Thus, roX RNAs, sequestering DCC proteins to the sites of their synthesis, can direct the mature complexes to the X chromosome. For instance, males are not viable in the absence of both roX genes because of the mis-localization of the complexes: they begin to bind with autosomes and heterochromatin sites [96]. Such males can be rescued by ectopic autosomal roX transcripts [96]. This indicates that roX RNAs can also work in trans to male X chromosome, suggesting that they also have a role in recognition of the X chromosome. The role of roX RNAs in targeting DCC complexes to the X chromosome has been also demonstrated in experiments when lethal females with MSL-2 protein expressing from the transgene survived in the absence of both roX genes [95, 106]. Both roX RNAs are interchangeable and full functional complexes can assemble even in the absence of one of the transcripts [105]. But there are some reports according to which roX2 RNA is more important [107]. The functional elements of roX RNAs, which may correspond to binding sites for MSL proteins or may be important for the recognition of X chromosome, are still mysterious.
An attempt to reveal some functional elements of roX1 RNA using deletion analysis has been made recently [106]. Overlapping deletions covering the entire roX1 gene were created and then tested for their ability to support dosage compensation in vivo. Each deletion lies between 260 and 400 nt in length. As a result, it was found that most roX1 transcripts carrying deletions retained nearly normal activity except for the deletion in the 3´-terminus, which, according to the results, is important for full activity and X localization.
These results are in good agreement with the absence of evident sequence homology between roX RNA molecules. Complex tertiary structure may be rather important for function of RNAs. The short 30 nt element shared between both roX RNAs could be deleted without obvious consequences [67]. Interestingly, evolutionarily conserved elements within mammalian Xist RNA are also dispensable for its function [96]. From another point of view, functional elements could be widely distributed along roX1 RNA and are redundant, so deletion of one or several of them does not alter dosage compensation. Whereas most of the deleted sequences did not considerably affect roX1 RNA function, deletion in the 3´-region led to male lethality. This region has been shown to contain inverted repeats, which, according to computer analysis, are able to form stem-loop structures. It is well known that stem-loop structures are frequent elements of secondary and tertiary structures of RNA molecules, and quite often they serve as binding sites for proteins. More thorough deletion analysis of the 3´-end revealed that inverted repeats are responsible, at least partially, for the roX1 RNA function, but there might also be other additional regulatory elements in this terminus.
On the other hand, several lines of evidence support the idea that MSL proteins by themselves even in the absence of roX RNAs are able to interact with X chromosome and hyperactivate transcription. For instance, in males double mutated on both roX RNAs, MSL proteins still can be detected on the X chromosome, though in a lesser extent. It is important that the distribution of the complexes is reminiscent of that in the case of wild type males [108]. It has been shown that a role of roX loci as “entry sites” for DCC complexes is independent of their transcription [96]. Moreover, only two sites from 30-35 identified overlap with loci of genes whose transcripts are known. It can be suggested that there are similar transcripts in the case of each “entry site”, but strong lethality of males mutated on both roX genes excludes this possibility.
Numerous studies indicate that roX genes play a double role in dosage compensation in Drosophila. On one hand, they are the source of transcripts directing MSL proteins preferentially to the X chromosome on which they reside; on the other hand, they belong to 30-35 “entry sites” at which DCCs assemble even in absences of the transcripts. So it seems that ability to bind MSL proteins is a feature of “entry sites” DNA sequences themselves and the roX RNAs serve rather as cofactors in this process sequestering or targeting MSL proteins to the X chromosome. In this light, one may suggest that there are other roX-like RNA molecules specific for a restricted number of genes [105], but till now roX RNAs are the sole example of regulatory system based on noncoding RNAs for the recruitment of transcription activation proteins.
RNA INTERFERENCE AND CHROMATIN STRUCTURE
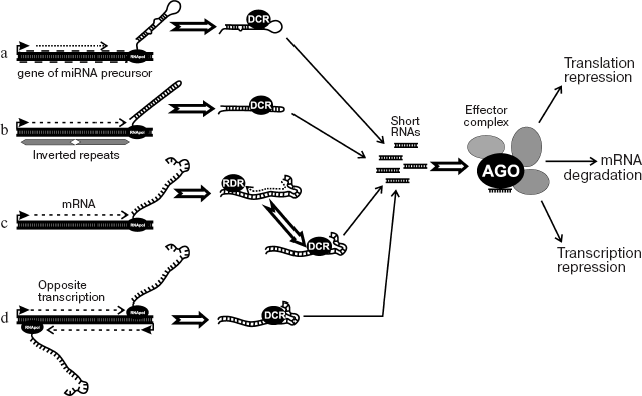
In the last decade, RNA-interference (RNAi) has been discovered and intensively studied [109-112]. The phenomenon is known in eukaryotic organisms from yeast to mammals and was originally described as a system of degradation of mRNA directed by short interfering RNAs. In eukaryotes, molecular mechanisms of this process are similar. The precursor, long double-stranded RNA, is generated in several ways (Fig. 4). These are transcription of inverted repeats, transcription of a DNA region in sense and antisense orientations (from two promoters), and activity of RNA-dependent RNA-polymerase. Double-stranded RNAs are split up to 22-26 bp fragments (siRNA) by ribonucleases of the Dicer family and then bind to protein complexes (RISC), where they cause degradation or distortion of translation of corresponding mRNAs. RISC protein complexes include proteins of the Argonaute family; in such complexes, siRNA discerns the target, the mRNA molecule that should be cleaved. In turn, degradation products can once more participate in the process, therefore effectively securing gene inactivation. Data have been recently obtained that the activity of siRNA may lead to repression at both post-transcription and chromatin structure levels [111]. Among such systems, the best understood are RNA-dependent heterochromatin formation with participation of RITS-complex in yeast and RNA-mediated DNA methylation (RdDM) in plants. The participation of siRNA in regulation of chromatin structure is also reported in other organisms. The table shows genes participating in heterochromatin formation and DNA methylation in yeast and plants, correspondingly.
Proteins involved in RdDM in plants and RNA-dependent heterochromatin formation in yeastFig. 4. RNAi and dsRNA production mechanisms. a) Transcription of miRNA (microRNA) precursor gene. The miRNA precursor genes have been found in a number of eukaryotes. They produce short non-translated RNAs with advanced secondary structure (hairpin loops). Dicer (DCR) cleaves these RNAs to miRNA molecules. In turn, miRNA binds to effector protein complex. b) Transcription of inverted repeats (palindromes). The result is RNA with the region of internal complementarity that gives hairpin structure. Double-strand stem of hairpin is cleaved by Dicer (DCR), resulting in siRNA. c) Transcription and subsequent synthesis of second strand with the involvement of RNA-dependent RNA polymerase (RDR). d) Transcription of the same sequence in both directions from two promoters. Double-strand RNA in this case is a result of annealing of two complementary RNAs. Effector protein complex binds siRNA. After that, transcriptional repression (RITS), mRNA slicing (RISC), or translational repression may occur. One of the components of effector complex is typically Argonaute family protein.
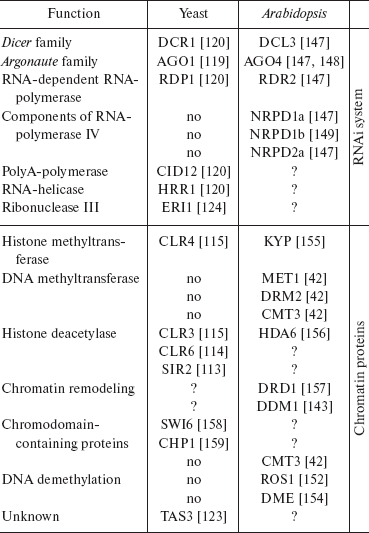
Note: Proteins are grouped according to function. There is no RNA-polymerase IV or DNA methylation-related proteins in yeast, and so the corresponding fields are empty. If functional equivalent of protein from one organism is unknown in other, the field is marked with “?”. Literature links are in brackets.
RITS complex in yeast. The budding yeast Schizosaccharomyces pombe is probably the simplest eukaryote possessing heterochromatin. Heterochromatin structure is intrinsic to pericentromeric areas, telomeres, and MAT-locus.
In yeast, centromeric areas are 35-110 kb in size and consist of a central part surrounded by innermost (imr) and outermost (otr) areas of repeats. The otr region contains tandem repeats of two types: dh and dg. MAT-locus includes an area of 3 kb that is 97% homological to dh/dg repeats. The area called cenH and surrounding area of about 10 kb are heterochromatinized. Fragments that are highly homologous to dh also occur in the area of telomere repeats.
The molecular structure of heterochromatin in plants, animals, and yeast show some common features. In contrast to plants and mammals, DNA methylation in yeast has not been found. During the formation of heterochromatin structure, histones are deacetylated by CLR3, CLR6, and SIR2 deacetylases. Later, the lysine of H3 histone is methylated at the ninth position by histone methyltransferase CLR4 [113-116]. Protein SWI6 (equivalent of HP1 in drosophila and mammals) recognizes the methylated histone and binds to chromatin. A complex that includes SWI6, CHP1, and CLR4 is able to oligomerize and spread along the chromatin, starting from the initiation point. This way, heterochromatinization occurs over a region several kb in size [117]. Genes are repressed in this area.
In pericentromeric regions and MAT-locus heterochromatin formation occurs with participation of components of the RNA interference system and siRNA produced from transcripts of pericentromeric repeats [118].
In yeast, there is just a single gene from each family Dicer (Dcr1), Argonaute (Ago1), and RNA-dependent RNA-polymerase (Rdp1). The deletion of any of these genes leads to distortion of the RNAi system and loss of pericentromeric heterochromatin. In the centromere area, no accumulation of lysine-methylated H3 histone or binding of SWI6 occurs; derepression of artificially introduced transgenes was observed. Accumulation of full-sized transcripts of dh/dg tandem repeats, which are normally split into siRNA, was also discovered [118]. Similar observations were made for MAT-locus, which contains the cenH region homologous to pericentromeric repeats.
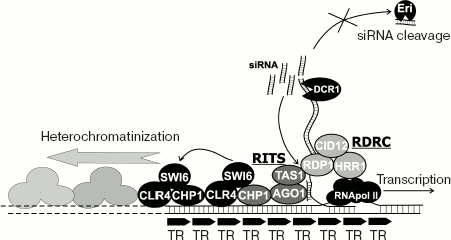
Using a biochemical approach, a protein complex was purified [119]. The RITS complex includes CHP (chromodomain-containing protein), AGO1, TAS1 (function is unclear), and siRNA itself.
In yeast, components of the RITS complex are attached to pericentromeric chromatin. Localization is dependent on DICER, and, therefore, on siRNA. Consequently, the simplest model suggests that siRNA resulting from the cleavage of transcripts of pericentromeric repeats by ribonuclease DICER become part of RITS complex and facilitate its binding to homologous regions of the genome, like the repeats themselves or cenH mating type locus. Later, SWI6 and histone methyltranferase CLR4 are attracted by protein-protein interactions, and heterochromatin packing is initiated.
However, the real picture is much more complicated. Apart from RITS, RDRC complex was identified, with the components RDP1 (RNA-dependent RNA polymerase), HRR1 (RNA-helicase), and CID12, polyA-polymerase. The complex shows RNA-dependent RNA-polymerase activity and, similarly to RITS, is associated with pericentromeric chromatin. The binding to heterochromatin depends upon DICER, RITS components, and CLR4/SWI6. Vice versa, mutations in RDRC components distort binding of RITS-complex and heterochromatin formation [120, 121]. In short, RNAi-mediated heterochromatin formation requires RNA synthesis on RNA.
It was found that binding of RITS-complex to chromatin depends on histone methyltranferase CLR4. In CLR4 mutants, purified RITS complex lacks siRNA, and the total amount of siRNA in pericentromeric regions diminishes [120]. The binding between RDP1 and chromatin was also distorted. Therefore, the statement that structural components of heterochromatin are necessary for maintaining intact assemblage RITS-RDRC and efficient processing of pericentrometic transcripts to siRNA is as true as it is true that siRNA associating with RITS initiates heterochromatin packing.
Results of two papers unequivocally indicate that RITS complex bind not to DNA, but newly produced RNA. RNAi-induced changes in chromatin were shown to occur only if target sequence homologous to siRNA is transcribed. In this case, protein AGO1 of RITS interacts with newly produced RNA.
Mutation in RPB2, one of subunits of RNA-polymerase II, leads to distortion of siRNA formation, methylation of H3 histone, and heterochromatin assembly. The mutation does not distort transcription itself; this indicates a physical link between the siRNA production and basic transcriptional machinery [122].
Addition of protein TAS3, one of the components of RITS, to newly synthesized transcript of gene ura4 by means of an RNA-binding domain inserted into TAS3 sequence initiates target gene repression, heterochromatin formation, and appearance of siRNA corresponding to the target gene [123]. The initiation of ura4 transcription is not distorted (amount of RNA-polymerase bound to ura4 sequence does not change).
Buhler et al. [123] showed that the appearance of ura4-derived siRNA after TAS3 binding does not result in repression of the second copy of ura4 in the genome. These data confirm the model that describes siRNA formation in yeast as a local process occurring cotranscriptionally. A gene was found, Eri1, mutation in which leads to repression of the second copy of ura4. Eri1 (enhancer of RNAi) is a highly conservative ribonuclease. It was also found in the nematode C. elegans. In yeast, mutation in Eri1 increases RNAi-induced repression [124], apparently distorting degradation of siRNA.
We believe that in yeast siRNA formation (at least in case of pericentromeric repeats) is linked to transcription and is performed by a compound protein complex with several functions: RNA splitting up to siRNA, RNA amplification by means of RNA-dependent RNA-polymerase, heterochromatin packing, and, maybe, siRNA degradation. The presence of DICER in the complex is not shown; there is a possibility that it is needed only to initiate the process. However, in yeast DICER is located in the nucleus. Figure 5 shows the proposed scheme of RNAi-mediated heterochromatin formation.
RdDM in plants. RNA-dependent DNA methylation. DNA methylation is a reversible covalent modification of some nucleotides (as a rule, cytosine in symmetrical CpG, CpNpG, or asymmetrical CpHpH sequences, where H is A, C, or T) that is common in plants and mammals. However, insects, yeasts, and the nematode C. elegans either completely lack DNA methylation or this process does not hold a specific role in gene regulation.Fig. 5. RNA-dependent heterochromatin formation in yeast. Possible mechanism of pericentromeric and MAT-locus heterochromatin formation. RNA produced from centromeric direct tandem repeats (TR) is in-place converted to double-strand RNA with the help of RDRC complex (including HRR1, SID12, and RNA-dependent RNA-polymerase RDP1). dsRNA is also in-place cleaved by DCR1 (DICER) ribonuclease giving siRNA. siRNA binds to RITS complex (including TAS1, CHP1, and AGO1, Argonaute-family protein). siRNA-containing RITS complex interacts with transcript from tandem repeats (via siRNA-based recognition of complementary sequence) and launches heterochromatin formation (maybe CHP1, the common component of RITS and heterochromatin complex SWI6-CLR4, is a mediator). Heterochromatin then spreads from the initiation point along the chromosome by self-assembling process (CLR4 methylates histone H3 at lysine-9, SWI6 binds to methylated histone, CLR4 in turn binds to SWI6). It seems that components of RITS, RDRC, and heterochromatin proteins interact with each other and form a common complex. siRNA interacts with effector complex in the site of production. Apparently, there is no free diffusion of siRNA from the production point because of activity of ribonuclease Eri1.
In plants and mammals, DNA methylation is believed to be among the main mechanisms of epigenetic inheritance. As a rule, methylation correlates with transcriptionally repressed state of chromatin: pericentromeric heterochromatin, inactive X-chromosome in mammals, various repeats, and mobile elements are methylated. Regulatory areas in genes are also methylated in the course of their inactivation; in several cases, active/inactive state is stably inherited in several generations of plants, producing so-called epialleles. Cases of gene imprinting (variations in expression level between maternally and paternally inherited alleles) are also methylation-related.
The efficiency of methylation as an epigenetic mark depends on the system of maintaining of methylation of symmetric sequences (CpG) through the cell cycle. The mechanism of the process is similar to semiconservative DNA replication. After DNA replication, such sites are in “half-methylated” state. Maintenance methyltransferase (MET1 in Arabidopsis, DMNT1 in mammals) recognize these sequences and reestablish full methylation; the process repeats each cell cycle. In plants, histone-modifying proteins (histone deacetylase HDA6) and chromatin modifier DDM1 participate in the process. Maintenance of asymmetric sites has a more complicated mechanism, which includes for CpNpG sites chromomethylase CMT3 (the protein is characterized by DNA methyltransferase activity and contains a chromodomain), KYP (histone methyltransferase, which methylates lysine-9 of H3 histone), chromatin modifier DRD1 (swi/snf family), and de novo DNA methyltransferase DRM2 [125, 126].
As far as the complex of DNA-methyltransferases and chromatin proteins is responsible for maintenance of methylated state, de novo methylation and repression, according to the latest ideas, is connected to the RNA-interference system. Of interest is that siRNA-induced epigenetic modifications of the genome were first revealed in plants.
Transgenic constructions that contain sequence of viroid RNA (PSTVd, potato spindle tuber viroid) were inserted into the genome of Nicotiana. It was revealed that viroid sequences became methylated in the genome if the plant is infected by viroid and the latter passes a cycle of autonomous RNA replication [127]. Therefore, DNA sequence in the genome was methylated if complementary double-stranded RNA was present in the cell.
Transcription of a transgene construction that contains NOS gene promoter (nopaline synthase) as a part of an inverted repeat leads to repression and methylation of the transgene. RNA resulting from the transcription of such a construction contains a hairpin (double-stranded region). This region was cleaved to siRNA about 23 nucleotides long, which indicates dsRNA degradation via RNA-interference mechanism [128].
RNA-dependent methylation of regulatory sequences of genes, mobile elements, direct and inverted repeats, and centromeric satellite DNA was shown in numerous examples [42, 111, 128-140].
In plants the RNA-interference system is very complicated and redundant. In Arabidopsis four genes of the Dicer family and ten members of the Argonaute family have been detected. In addition, RNA-dependent RNA-polymerases (six genes) and plant-specific DNA-dependent RNA-polymerase IV are parts of this system [141]. In plants, both RNAi-linked transcriptional repression and classic posttranscriptional gene silencing (PTGS) occur. Moreover, different types of genetic elements, such as genes, centromeric satellites, and direct and inverted repeats, have different mechanisms of transcription repression, in which different proteins participate.
To date, regulation of methylation and transcriptional repression is characterized in maximal detail in case of the FWA gene of Arabidopsis thaliana. The corresponding product is a transcription factor that is responsible for the time of flowering of the plant. The gene is expressed in endosperm and repressed in other tissues. The repressed state associates with methylation of upstream region of the gene [142]. This area is formed by retrotransposon SINE3 [143] and contains two pairs of direct repeats, which are necessary for repression. In the case of FWA, methylation of both symmetric CpG and asymmetric CpHpH sequences takes place. Demethylation of repeats in tissues of the adult plant leads to the gene expression and “phenotype of late flowering”. This state is stably inherited in several generations (so-called epimutation fwa) [142].
Epimutation fwa can occur incidentally or be induced by mutations in DDM1 and MET1 genes [142]. DDM1 is a protein of swi/snf family accomplishing ATP-dependent chromatin remodeling [144]. MET1 is a DNA methyltransferase responsible for maintaining methylation of symmetric CpG sequences. In DDM1 and MET1 mutants, gradual demethylation of some elements of the genome takes place in the course of several generations, resulting in distortions of gene regulation and various phenotypic effects [145].
In the Arabidopsis genome, bacterial transformation allows one to introduce a construction containing an additional copy of the FWA gene. The transgene integrates in the genome and, in the normal case, is methylated and repressed with a probability of 100%. Consequently, the transformation of wild-type plants by FWA gene does not lead to “late flowering phenotype”; endogenous and transgenic copies are both inactive.
If the genes of Arabidopsis that are necessary for the methylation and repression of FWA are mutated, no inactivation of the extra copy of FWA will happen after introduction of transgenic construction. Screening based on this approach revealed a range of genes necessary for de novo methylation. These are:
- DRM2, DNA methyltransferase, that is necessary for de novo methylation of symmetrical (CpG) and asymmetrical sequences;
- DRD1, a protein belonging to the swi/snf family (chromatin remodeling protein);
- AGO4, a member of the Argonaute family. Proteins of this family are present in all eukaryotes; they are able to bind short RNA (like siRNA) and are included in effector complexes (RITS, RISC);
- DCL3, a member of the Dicer family. Ribonuclease that cuts long dsRNA into siRNA;
- RDR2, RNA-dependent RNA-polymerase. The synthesis of the second strand of RNA on RNA-template results in formation of dsRNA, which is later cut up to siRNA;
- NRPD1a and NRPD1b, subunits of plant-specific DNA-dependent RNA-polymerase IV (RNApol IV). In plants, RNApol IV transcribes repeats and is necessary for repression of mobile elements and other types of repeated sequences. It exists in two variants: NRPD1a-NRPD2 (RNApol IVa) and NRPD1b-NRPD2 (RNApol IVb) complexes. NRPD2 is a common subunit for both types. The complexes have different functions: RNApol IVa is needed for siRNA synthesis, and RNApol IVb is needed for methylation and transcriptional repression.
In addition, it was shown that siRNA is formed on direct repeats of the 5´-area of gene FWA [143, 146].
So, besides chromatin modifier protein DRD1 and methyltransferase DRM2, the components of RNAi pathway in plants are necessary for de novo methylation and repression of FWA transgenes. This fact points to a link between RNAi and transcription-level repression [147]. The case of FWA is not unique: for example, the AGO4 protein participates in SUPERMAN gene inactivation [148].
Further studies uncover the order of events ultimately leading to FWA repression. It has been shown that DCL3, NRPD1a, and RDR2 are required for siRNA production from FWA repeats [149]. NRPD1b, AGO4, DRD1, and DRM2 act at a downstream stage and are responsible for DNA methylation and repression of the transgene.
In situ immunostaining demonstrates that DCL3, RDR2, NRPD1b, and AGO4 localize in Cajal bodies, subnuclear structure near the nucleolus. Cajal bodies are responsible for RNA processing. DRD1, DRM2, and NRPD1a have been found in chromatin and co-localize with repeats. NRPD1b and AGO4 have been detected in both Cajal bodies and chromatin.
Taking into account available data, we may suggest the following mechanism of RdDM for the FWA locus (Fig. 6). In the normal state, FWA is repressed and the upstream repeats are methylated in both symmetrical (CpG) and asymmetrical (CpHpH) sites. After the round of DNA replication in S-phase, the symmetrical sites reestablish their methylation state by means of a semi-conservative mechanism with participation of MET1-DDM1-HDA6 complex, where MET1 is maintenance methyltransferase, DDM1 is chromatin-modifying protein, and HDA6 is histone deacetylase. This process is independent of RNAi. The asymmetric sites should be de novo methylated each cell cycle, and the process is close to de novo methylation of introduced artificial transgenes.
The de novo methylation system includes DNA methyltransferase (DRM2), siRNA corresponding to a regulated region, and components of RNAi machinery. RNApol IVa apparently transcribes direct repeats in the upstream region of FWA. The resulting RNA molecules are transported to Cajal bodies, and then undergo reverse transcription (RDR2) and cleavage (DCL3) to produce siRNA. The siRNA binds to protein complex containing AGO4 and NRPD1b. After that, siRNA-containing complex moves to the FWA gene and there the methylation and repression processes commence. Additional components, DRM2 methyltransferase and DRD1 chromatin protein, are required at this step. The siRNA component seems to perform precise targeting of repressing activity of the protein complex.Fig. 6. RNA-dependent DNA methylation in plants (FWA case). De novo methylation of artificial transgenic constructions and maintenance methylation of asymmetrical sites (CpHpH) after round of DNA synthesis requires RNAi system components RDR2, DCL3, and AGO4, as well as siRNA corresponding to direct repeats from 5´-upstream region of FWA gene. See full description in text.
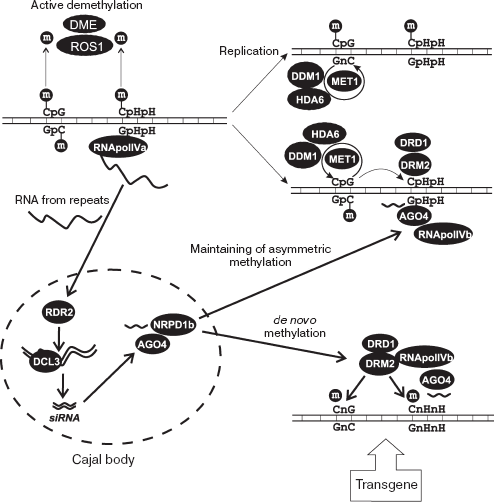
It is assumed that siRNA performs its targeting activity via recognition of complementary sequences, in the case of FWA the 5´-end direct repeats. This process suggests the single-strand state of target. The role of RNApol IVb in repression may be either local DNA unwinding or transcription of direct repeats. Resulting RNA may be the target of siRNA-containing complex. During the transformation of plants with artificial transgenic construction, transgene DNA exists in single-strand state at some stage. This may be the reason for full methylation and repression of transgenes after transformation.
FWA repression is the best-studied example of RdDM in plants: a number of involved proteins have been disclosed and the biochemical properties of some of them have been studied. However, little is known about the arrangement of repressing complex on the target gene. Moreover, the studies of plants bearing stable fwa epimutation show that it contains a siRNA corresponding to the 5´-end direct repeats [149]. As mentioned above, fwa is a non-methylated and actively transcribed FWA gene. It is a stable epimutation: it can be inherited in a number of generations and behave as a normal allele in plants containing all the necessary components of the RdDM system. So, the presence of corresponding siRNA is not enough for gene repression, the gene itself should be “competent” for inactivation message. In the case of FWA, “competence” is achieved during the process of transgene transformation and integration, and maintenance methylation of asymmetric sites depends on methylation of symmetric sites [149].
Finally, there is a mechanism of active demethylation and activation of FWA in endosperm, a tissue where this gene is transcribed [150]. DNA demethylation occurs as a result of DNA-glycosylase activity. In Arabidopsis, two glycosylases, DEMETER and ROS1 (Repressor of silencing 1) have been characterized [150-154]. After demethylation, FWA starts to express in endosperm, but this state is not inherited by progeny since endosperm does not become a part of the adult plant.
During the last two decades our understanding of transcription regulation in eukaryotes has significantly changed. According to the classic model, the level of expression is predetermined by transcription factors binding to regulatory regions of a gene. The factors deal directly with components of the transcription apparatus and initiate mRNA synthesis. According to this hypothesis, chromatin, as a totality of DNA-related histones and structural non-histone proteins, played merely a passive role for compact DNA packaging in the nucleus.
Later, the active role of chromatin in transcription regulation was revealed. The so-called histone code--the set of covalent modifications of histones, dissimilar in different regions of the genome (e.g. in heterochromatin and active genes)--was discovered. The histone code is interpreted by protein complexes that are able to change the chromatin structure and make genome regions either accessible for transcription factors or transform them into a non-active state. A complex of heterochromatin proteins is an example. Its assembly leads to gene repression and compactization of the chromatin in the area.
It was recently found that noncoding RNA can participate in the assembly and maintenance of chromatin protein complexes. Xist is needed for heterochromatinization of X-chromosome in mammals in case of dosage compensation; roX is needed for the twofold increase of transcription from X-chromosome of males of drosophila. In yeast, siRNA produced from pericentromeric direct repeats participate in heterochromatin formation. In some genes of plants, repression is also started and kept by siRNA.
In all cases, the function of noncoding RNA is usually targeting, i.e. revealing genome areas with which the complex of chromatin-modifying proteins should bind. Specific recognition is due to homologous pairing between RNA of the complex and DNA or RNA sequences of the target. Later, histones are modified, DNA is methylated, and chromatin is compacted--some changes predetermined by the properties of the RNA-associated protein complex occur. In cases of X-chromosome inactivation (mammals) and RdDM and heterochromatin assembly (yeast) the result is repression; in case of drosophila X-chromosome in males--transcriptional hyperactivation.
If RNA (e.g. siRNA in plants) participates in repression of corresponding DNA sequence, a contradiction appears: efficient repression needs active transcription. Several solutions are conceivable. In plants, an additional RNA-polymerase IV (in comparison to three polymerases in other eukaryotes) exists, which transcribes repeats, and there is a possibility that the heterochromatinized state does not prevent its activity. In plants, repressed state of the gene can also be preserved via maintaining methylation of symmetric repeats. Finally, the participation of RNA-dependent RNA-polymerase in siRNA-mediated repression in both plants and yeast suggests the existence of “amplification contour”, a system reproducing siRNA without transcription from DNA.
This review has discussed only four example of influence of RNA on chromatin structure. However, dosage compensation alone deals with hundreds and thousands of genes. To date it is impossible to estimate the true scopes of modifications related with noncoding RNA, but they are undoubtedly very high. Thus, there are hundreds of targets of Polycomb proteins and heterochromatin components in drosophila, and both types of chromatin-modifying complexes are apparently linked someway to siRNA. Further studies will provide new information about intricate regulations of gene expression in eukaryotes.
This work was supported by the Russian Academy of Sciences program for Molecular and Cell Biology, by grants from the Russian Federation President for Leading Scientific Schools (SS 6113.2006.4), and by the Russian Foundation for Basic Research (grant No. 07-04-07054).
REFERENCES
1.Brown, C. J., Ballabio, A., Rupert, J. L.,
Lafreniere, R. G., Grompe, M., Tonlorenzi, R., and Willard, H. F.
(1991) Nature, 349, 38-44.
2.Yao, M. C., Fuller, P., and Xi, X. (2003)
Science, 300, 1581-1584.
3.Selker, E. U. (2003) Science, 300,
1517-1518.
4.Morris, K. V., Chan, S. W., Jacobsen, S. E., and
Looney, D. J. (2004) Science, 305, 1289-1292.
5.Kawasaki, H., and Taira, K. (2004) Nature,
431, 211-217.
6.Fukagawa, T., Nogami, M., Yoshikawa, M., Ikeno, M.,
Okazaki, T., Takami, Y., Nakayama, T., and Oshimura, M. (2004) Nat.
Cell Biol., 6, 784-791.
7.Pal-Bhadra, M., Leibovitch, B. A., Gandhi, S. G.,
Rao, M., Bhadra, U., Birchler, J. A., and Elgin, S. C. (2004)
Science, 303, 669-672.
8.Wolffe, A. P. (1994) Trends Biochem. Sci.,
19, 240-244.
9.Gregory, P. D., and Horz, W. (1998) Eur. J.
Biochem., 251, 9-18.
10.Marenda, D. R., Zraly, C. B., and Dingwall, A. K.
(2004) Dev. Biol., 267, 279-293.
11.Peterson, C. L. (1996) Curr. Opin. Genet.
Dev., 6, 171-175.
12.Wade, P. A., Gegonne, A., Jones, P. L.,
Ballestar, E., Aubry, F., and Wolffe, A. P. (1999) Nat. Genet.,
23, 62-66.
13.Katsani, K. R., Mahmoudi, T., and Verrijzer, C.
P. (2003) Curr. Top. Microbiol. Immunol., 274,
113-141.
14.Jenuwein, T., and Allis, C. D. (2001)
Science, 293, 1074-1080.
15.Thiagalingam, S., Cheng, K. H., Lee, H. J.,
Mineva, N., Thiagalingam, A., and Ponte, J. F. (2003) Ann. N. Y.
Acad. Sci., 983, 84-100.
16.Turner, B. M. (2002) Cell, 111,
285-291.
17.Braunstein, M., Sobel, R. E., Allis, C. D.,
Turner, B. M., and Broach, J. R. (1996) Mol. Cell Biol.,
16, 4349-4356.
18.Tsukada, Y., Fang, J., Erdjument-Bromage, H.,
Warren, M. E., Borchers, C. H., Tempst, P., and Zhang, Y. (2006)
Nature, 439, 811-816.
19.Ebert, A., Schotta, G., Lein, S., Kubicek, S.,
Krauss, V., Jenuwein, T., and Reuter, G. (2004) Genes Dev.,
18, 2973-2983.
20.Schotta, G., Ebert, A., Dorn, R., and Reuter, G.
(2003) Semin. Cell Dev. Biol., 14, 67-75.
21.Weiler, K. S., and Wakimoto, B. T. (2002) Mol.
Genet. Genom., 266, 922-932.
22.Czermin, B., Schotta, G., Hulsmann, B. B., Brehm,
A., Becker, P. B., Reuter, G., and Imhof, A. (2001) EMBO Rep.,
2, 915-919.
23.Schotta, G., Ebert, A., Krauss, V., Fischer, A.,
Hoffmann, J., Rea, S., Jenuwein, T., Dorn, R., and Reuter, G. (2002)
Embo J., 21, 1121-1131.
24.Min, J., Zhang, Y., and Xu, R. M. (2003) Genes
Dev., 17, 1823-1828.
25.Fischle, W., Wang, Y., Jacobs, S. A., Kim, Y.,
Allis, C. D., and Khorasanizadeh, S. (2003) Genes Dev.,
17, 1870-1881.
26.Brehm, A., Tufteland, K. R., Aasland, R., and
Becker, P. B. (2004) Bioessays, 26, 133-140.
27.Akhtar, A., Zink, D., and Becker, P. B. (2000)
Nature, 407, 405-409.
28.Bannister, A. J., Zegerman, P., Partridge, J. F.,
Miska, E. A., Thomas, J. O., Allshire, R. C., and Kouzarides, T. (2001)
Nature, 410, 120-124.
29.Shareef, M. M., Badugu, R., and Kellum, R. (2003)
Genetica, 117, 127-134.
30.Seum, C., Pauli, D., Delattre, M., Jaquet, Y.,
Spierer, A., and Spierer, P. (2002) Genetics, 161,
1125-1136.
31.Pirrotta, V., Poux, S., Melfi, R., and Pilyugin,
M. (2003) Genetica, 117, 191-197.
32.Simon, J. A. (2003) Curr. Biol.,
13, R79-80.
33.Pirrotta, V. (1995) Curr. Opin. Genet.
Dev., 5, 466-472.
34.Chanas, G., Lavrov, S., Iral, F., Cavalli, G.,
and Maschat, F. (2004) Dev. Biol., 272, 522-535.
35.Ringrose, L., Rehmsmeier, M., Dura, J. M., and
Paro, R. (2003) Dev. Cell, 5, 759-771.
36.Negre, N., Hennetin, J., Sun, L. V., Lavrov, S.,
Bellis, M., White, K. P., and Cavalli, G. (2006) PLoS Biol.,
4, e170.
37.Sewalt, R. G., Kwaks, T. H., Hamer, K., and Otte,
A. P. (2004) Meth. Enzymol., 377, 282-296.
38.Pasini, D., Bracken, A. P., and Helin, K. (2004)
Cell Cycle, 3, 396-400.
39.Zhang, H., Azevedo, R. B., Lints, R., Doyle, C.,
Teng, Y., Haber, D., and Emmons, S. W. (2003) Dev. Cell,
4, 903-915.
40.Reyes, J. C., and Grossniklaus, U. (2003)
Semin. Cell Dev. Biol., 14, 77-84.
41.Weissmann, F., Muyrers-Chen, I., Musch, T.,
Stach, D., Wiessler, M., Paro, R., and Lyko, F. (2003) Mol. Cell
Biol., 23, 2577-2586.
42.Chan, S. W., Henderson, I. R., and Jacobsen, S.
E. (2005) Nat. Rev. Genet., 6, 351-360.
43.Liu, K., Wang, Y. F., Cantemir, C., and Muller,
M. T. (2003) Mol. Cell Biol., 23, 2709-2719.
44.Zemach, A., and Grafi, G. (2007) Trends Plant
Sci., 12, 80-85.
45.Fatemi, M., and Wade, P. A. (2006) J. Cell
Sci., 119, 3033-3037.
46.Wade, P. A. (2001) Bioessays, 23,
1131-1137.
47.Huynh, K. D., and Lee, J. T. (2003)
Nature, 426, 857-862.
48.Okamoto, I., Otte, A. P., Allis, C. D., Reinberg,
D., and Heard, E. (2004) Science, 303, 644-649.
49.Avner, P., and Heard, E. (2001) Nat. Rev.
Genet., 2, 59-67.
50.Penny, G. D., Kay, G. F., Sheardown, S. A.,
Rastan, S., and Brockdorff, N. (1996) Nature, 379,
131-137.
51.Marahrens, Y., Panning, B., Dausman, J., Strauss,
W., and Jaenisch, R. (1997) Genes Dev., 11, 156-166.
52.Lee, J. T., and Jaenisch, R. (1997)
Nature, 386, 275-279.
53.Brockdorff, N., Ashworth, A., Kay, G. F., Cooper,
P., Smith, S., Mccabe, V. M., Norris, D. P., Penny, G. D., Patel, D.,
and Rastan, S. (1991) Nature, 351, 329-331.
54.Lee, J. T., Davidow, L. S., and Warshawsky, D.
(1999) Nat. Genet., 21, 400-404.
55.Sado, T., Wang, Z., Sasaki, H., and Li, E. (2001)
Development, 128, 1275-1286.
56.Herzing, L. B., Romer, J. T., Horn, J. M., and
Ashworth, A. (1997) Nature, 386, 272-275.
57.Sun, B. K., Deaton, A. M., and Lee, J. T. (2006)
Mol. Cell, 21, 617-628.
58.Marahrens, Y., Loring, J., and Jaenisch, R.
(1998) Cell, 92, 657-664.
59.Boggs, B. A., Cheung, P., Heard, E., Spector, D.
L., Chinault, A. C., and Allis, C. D. (2002) Nat. Genet.,
30, 73-76.
60.Silva, J., Mak, W., Zvetkova, I., Appanah, R.,
Nesterova, T. B., Webster, Z., Peters, A. H., Jenuwein, T., Otte, A.
P., and Brockdorff, N. (2003) Dev. Cell, 4, 481-495.
61.Chadwick, B. P., and Willard, H. F. (2003)
Hum. Mol. Genet., 12, 2167-2178.
62.Rasmussen, T. P., Mastrangelo, M. A., Eden, A.,
Pehrson, J. R., and Jaenisch, R. (2000) J. Cell Biol.,
150, 1189-1198.
63.Csankovszki, G., Nagy, A., and Jaenisch, R.
(2001) J. Cell Biol., 153, 773-784.
64.Mak, W., Baxter, J., Silva, J., Newall, A. E.,
Otte, A. P., and Brockdorff, N. (2002) Curr. Biol., 12,
1016-1020.
65.Plath, K., Fang, J., Mlynarczyk-Evans, S. K.,
Cao, R., Worringer, K. A., Wang, H., de la Cruz, C. C., Otte, A. P.,
Panning, B., and Zhang, Y. (2003) Science, 300,
131-135.
66.Csankovszki, G., Panning, B., Bates, B., Pehrson,
J. R., and Jaenisch, R. (1999) Nat. Genet., 22,
323-324.
67.Wutz, A., Rasmussen, T. P., and Jaenisch, R.
(2002) Nat. Genet., 30, 167-174.
68.Wutz, A., and Jaenisch, R. (2000) Mol.
Cell, 5, 695-705.
69.Hendrich, B. D., Brown, C. J., and Willard, H. F.
(1993) Hum. Mol. Genet., 2, 663-672.
70.Nesterova, T. B., Slobodyanyuk, S. Y.,
Elisaphenko, E. A., Shevchenko, A. I., Johnston, C., Pavlova, M. E.,
Rogozin, I. B., Kolesnikov, N. N., Brockdorff, N., and Zakian, S. M.
(2001) Genome Res., 11, 833-849.
71.Mak, W., Nesterova, T. B., de Napoles, M.,
Appanah, R., Yamanaka, S., Otte, A. P., and Brockdorff, N. (2004)
Science, 303, 666-669.
72.Luikenhuis, S., Wutz, A., and Jaenisch, R. (2001)
Mol. Cell Biol., 21, 8512-8520.
73.Stavropoulos, N., Lu, N., and Lee, J. T. (2001)
Proc. Natl. Acad. Sci. USA, 98, 10232-10237.
74.Tada, T., Obata, Y., Tada, M., Goto, Y.,
Nakatsuji, N., Tan, S., Kono, T., and Takagi, N. (2000)
Development, 127, 3101-3105.
75.Norris, D. P., Patel, D., Kay, G. F., Penny, G.
D., Brockdorff, N., Sheardown, S. A., and Rastan, S. (1994)
Cell, 77, 41-51.
76.Ariel, M., Robinson, E., Mccarrey, J. R., and
Cedar, H. (1995) Nat. Genet., 9, 312-315.
77.Zuccotti, M., and Monk, M. (1995) Nat.
Genet., 9, 316-320.
78.Mcdonald, L. E., and Kay, G. F. (1997)
Biotechniques, 22, 272-274.
79.Sado, T., Okano, M., Li, E., and Sasaki, H.
(2004) Development, 131, 975-982.
80.Chen, T., Ueda, Y., Dodge, J. E., Wang, Z., and
Li, E. (2003) Mol. Cell Biol., 23, 5594-5605.
81.Mcdonald, L. E., Paterson, C. A., and Kay, G. F.
(1998) Genomics, 54, 379-386.
82.Sado, T., Tada, T., and Takagi, N. (1996) Dev.
Dyn., 205, 421-434.
83.Lee, J. T. (2002) Nat. Genet., 32,
195-200.
84.Shibata, S., and Lee, J. T. (2003) Hum. Mol.
Genet., 12, 125-136.
85.Migeon, B. R., Chowdhury, A. K., Dunston, J. A.,
and Mcintosh, I. (2001) Am. J. Hum. Genet., 69,
951-960.
86.Chow, J. C., Hall, L. L., Clemson, C. M.,
Lawrence, J. B., and Brown, C. J. (2003) Genomics, 82,
309-322.
87.Ogawa, Y., and Lee, J. T. (2003) Mol.
Cell, 11, 731-743.
88.Stavropoulos, N., Rowntree, R. K., and Lee, J. T.
(2005) Mol. Cell Biol., 25, 2757-2769.
89.Jin, Y., Wang, Y., Johansen, J., and Johansen, K.
M. (2000) J. Cell Biol., 149, 1005-1010.
90.Smith, E. R., Pannuti, A., Gu, W., Steurnagel,
A., Cook, R. G., Allis, C. D., and Lucchesi, J. C. (2000) Mol. Cell
Biol., 20, 312-318.
91.Akhtar, A., and Becker, P. B. (2000) Mol.
Cell, 5, 367-375.
92.Jin, Y., Wang, Y., Walker, D. L., Dong, H.,
Conley, C., Johansen, J., and Johansen, K. M. (1999) Mol. Cell,
4, 129-135.
93.Wang, Y., Zhang, W., Jin, Y., Johansen, J., and
Johansen, K. M. (2001) Cell, 105, 433-443.
94.Kelley, R. L., and Kuroda, M. I. (2000)
Cell, 103, 9-12.
95.Meller, V. H., Wu, K. H., Roman, G., Kuroda, M.
I., and Davis, R. L. (1997) Cell, 88, 445-457.
96.Meller, V. H., and Rattner, B. P. (2002) EMBO
J., 21, 1084-1091.
97.Kelley, R. L., Meller, V. H., Gordadze, P. R.,
Roman, G., Davis, R. L., and Kuroda, M. I. (1999) Cell,
98, 513-522.
98.Franke, A., Dernburg, A., Bashaw, G. J., and
Baker, B. S. (1996) Development, 122, 2751-2760.
99.Rastelli, L., Richman, R., and Kuroda, M. I.
(1995) Mech. Dev., 53, 223-233.
100.Gebauer, F., Grskovic, M., and Hentze, M. W.
(2003) Mol. Cell, 11, 1397-1404.
101.Palmer, M. J., Richman, R., Richter, L., and
Kuroda, M. I. (1994) Genes Dev., 8, 698-706.
102.Kelley, R. L., Solovyeva, I., Lyman, L. M.,
Richman, R., Solovyev, V., and Kuroda, M. I. (1995) Cell,
81, 867-877.
103.Lyman, L. M., Copps, K., Rastelli, L., Kelley,
R. L., and Kuroda, M. I. (1997) Genetics, 147,
1743-1753.
104.Meller, V. H. (2003) Mech. Dev.,
120, 759-767.
105.Meller, V. H., Gordadze, P. R., Park, Y., Chu,
X., Stuckenholz, C., Kelley, R. L., and Kuroda, M. I. (2000) Curr.
Biol., 10, 136-143.
106.Franke, A., and Baker, B. S. (1999) Mol.
Cell, 4, 117-122.
107.Stuckenholz, C., Meller, V. H., and Kuroda, M.
I. (2003) Genetics, 164, 1003-1014.
108.Kageyama, Y., Mengus, G., Gilfillan, G.,
Kennedy, H. G., Stuckenholz, C., Kelley, R. L., Becker, P. B., and
Kuroda, M. I. (2001) EMBO J., 20, 2236-2245.
109.Carthew, R. W. (2001) Curr. Opin. Cell
Biol., 13, 244-248.
110.Tijsterman, M., and Plasterk, R. H. (2004)
Cell, 117, 1-3.
111.Meister, G., and Tuschl, T. (2004)
Nature, 431, 343-349.
112.Matzke, M. A., and Birchler, J. A. (2005)
Nat. Rev. Genet., 6, 24-35.
113.Shankaranarayana, G. D., Motamedi, M. R.,
Moazed, D., and Grewal, S. I. (2003) Curr. Biol., 13,
1240-1246.
114.Grewal, S. I., Bonaduce, M. J., and Klar, A. J.
(1998) Genetics, 150, 563-576.
115.Nakayama, J., Rice, J. C., Strahl, B. D.,
Allis, C. D., and Grewal, S. I. (2001) Science, 292,
110-113.
116.Partridge, J. F., Borgstrom, B., and Allshire,
R. C. (2000) Genes Dev., 14, 783-791.
117.Grewal, S. I., and Moazed, D. (2003)
Science, 301, 798-802.
118.Volpe, T. A., Kidner, C., Hall, I. M., Teng,
G., Grewal, S. I., and Martienssen, R. A. (2002) Science,
297, 1833-1837.
119.Verdel, A., Jia, S., Gerber, S., Sugiyama, T.,
Gygi, S., Grewal, S. I., and Moazed, D. (2004) Science,
303, 672-676.
120.Motamedi, M. R., Verdel, A., Colmenares, S. U.,
Gerber, S. A., Gygi, S. P., and Moazed, D. (2004) Cell,
119, 789-802.
121.Sugiyama, T., Cam, H., Verdel, A., Moazed, D.,
and Grewal, S. I. (2005) Proc. Natl. Acad. Sci. USA, 102,
152-157.
122.Schramke, V., Sheedy, D. M., Denli, A. M.,
Bonila, C., Ekwall, K., Hannon, G. J., and Allshire, R. C. (2005)
Nature, 435, 1275-1279.
123.Buhler, M., Verdel, A., and Moazed, D. (2006)
Cell, 125, 873-886.
124.Iida, T., Kawaguchi, R., and Nakayama, J.
(2006) Curr. Biol., 16, 1459-1464.
125.Lindroth, A. M., Shultis, D., Jasencakova, Z.,
Fuchs, J., Johnson, L., Schubert, D., Patnaik, D., Pradhan, S.,
Goodrich, J., Schubert, I., Jenuwein, T., Khorasanizadeh, S., and
Jacobsen, S. E. (2004) Embo J., 23, 4286-4296.
126.Tran, R. K., Zilberman, D., de Bustos, C.,
Ditt, R. F., Henikoff, J. G., Lindroth, A. M., Delrow, J., Boyle, T.,
Kwong, S., Bryson, T. D., Jacobsen, S. E., and Henikoff, S. (2005)
Genome Biol., 6, R90.
127.Wassenegger, M., Heimes, S., Riedel, L., and
Sanger, H. L. (1994) Cell, 76, 567-576.
128.Mette, M. F., Aufsatz, W., van der Winden, J.,
Matzke, M. A., and Matzke, A. J. (2000) EMBO J., 19,
5194-5201.
129.Aufsatz, W., Mette, M. F., van der Winden, J.,
Matzke, A. J., and Matzke, M. (2002) Proc. Natl. Acad. Sci. USA,
99 (Suppl. 4), 16499-16506.
130.Beclin, C., Boutet, S., Waterhouse, P., and
Vaucheret, H. (2002) Curr. Biol., 12, 684-688.
131.Jones, L., Ratcliff, F., and Baulcombe, D. C.
(2001) Curr. Biol., 11, 747-757.
132.Kawasaki, H., and Taira, K. (2005) Curr.
Opin. Mol. Ther., 7, 125-131.
133.Lippman, Z., May, B., Yordan, C., Singer, T.,
and Martienssen, R. (2003) PLoS Biol., 1, E67.
134.May, B. P., Lippman, Z. B., Fang, Y., Spector,
D. L., and Martienssen, R. A. (2005) PLoS Genet., 1,
e79.
135.Melquist, S., and Bender, J. (2003) Genes
Dev., 17, 2036-2047.
136.Pelissier, T., and Wassenegger, M. (2000)
RNA, 6, 55-65.
137.Qin, H., Dong, Y., and von Arnim, A. G. (2003)
Plant Mol. Biol., 52, 217-231.
138.Sijen, T., Vijn, I., Rebocho, A., van Blokland,
R., Roelofs, D., Mol, J. N., and Kooter, J. M. (2001) Curr.
Biol., 11, 436-440.
139.Steimer, A., Amedeo, P., Afsar, K., Fransz, P.,
Mittelsten Scheid, O., and Paszkowski, J. (2000) Plant Cell,
12, 1165-1178.
140.Vaucheret, H., and Fagard, M. (2001) Trends
Genet., 17, 29-35.
141.Herr, A. J., Jensen, M. B., Dalmay, T., and
Baulcombe, D. C. (2005) Science, 308, 118-120.
142.Soppe, W. J., Jacobsen, S. E., Alonso-Blanco,
C., Jackson, J. P., Kakutani, T., Koornneef, M., and Peeters, A. J.
(2000) Mol. Cell, 6, 791-802.
143.Lippman, Z., Gendrel, A. V., Black, M., Vaughn,
M. W., Dedhia, N., Mccombie, W. R., Lavine, K., Mittal, V., May, B.,
Kasschau, K. D., Carrington, J. C., Doerge, R. W., Colot, V., and
Martienssen, R. (2004) Nature, 430, 471-476.
144.Jeddeloh, J. A., Stokes, T. L., and Richards,
E. J. (1999) Nat. Genet., 22, 94-97.
145.Kakutani, T., Jeddeloh, J. A., Flowers, S. K.,
Munakata, K., and Richards, E. J. (1996) Proc. Natl. Acad. Sci.
USA, 93, 12406-12411.
146.Lu, C., Tej, S. S., Luo, S., Haudenschild, C.
D., Meyers, B. C., and Green, P. J. (2005) Science, 309,
1567-1569.
147.Chan, S. W., Zilberman, D., Xie, Z., Johansen,
L. K., Carrington, J. C., and Jacobsen, S. E. (2004) Science,
303, 1336.
148.Zilberman, D., Cao, X., and Jacobsen, S. E.
(2003) Science, 299, 716-719.
149.Chan, S. W.-L., Zhang, X., Bernatavichute, Y.
V., and Jacobsen, S. E. (2006) PLoS Biol., 4, e363.
150.Kinoshita, T., Miura, A., Choi, Y., Kinoshita,
Y., Cao, X., Jacobsen, S. E., Fischer, R. L., and Kakutani, T. (2004)
Science, 303, 521-523.
151.Kapoor, A., Agarwal, M., Andreucci, A., Zheng,
X., Gong, Z., Hasegawa, P. M., Bressan, R. A., and Zhu, J. K. (2005)
Curr. Biol., 15, 1912-1918.
152.Gong, Z., Morales-Ruiz, T., Ariza, R. R.,
Roldan-Arjona, T., David, L., and Zhu, J. K. (2002) Cell,
111, 803-814.
153.Choi, Y., Gehring, M., Johnson, L., Hannon, M.,
Harada, J. J., Goldberg, R. B., Jacobsen, S. E., and Fischer, R. L.
(2002) Cell, 110, 33-42.
154.Choi, Y., Harada, J. J., Goldberg, R. B., and
Fischer, R. L. (2004) Proc. Natl. Acad. Sci. USA, 101,
7481-7486.
155.Jackson, J. P., Lindroth, A. M., Cao, X., and
Jacobsen, S. E. (2002) Nature, 416, 556-560.
156.Aufsatz, W., Mette, M. F., van der Winden, J.,
Matzke, M., and Matzke, A. J. (2002) EMBO J., 21,
6832-6841.
157.Kanno, T., Mette, M. F., Kreil, D. P., Aufsatz,
W., Matzke, M., and Matzke, A. J. (2004) Curr. Biol., 14,
801-805.
158.Verdel, A., and Moazed, D. (2005) FEBS
Lett., 579, 5872-5878.
159.Petrie, V. J., Wuitschick, J. D., Givens, C.
D., Kosinski, A. M., and Partridge, J. F. (2005) Mol. Cell
Biol., 25, 2331-2346.