REVIEW: Biological Activity of Hemoprotein Nitrosyl Complexes
A. N. Osipov1*, G. G. Borisenko2, and Yu. A. Vladimirov3
1Russian State Medical University, ul. Ostrovityanova 1, 117997 Moscow, Russia; fax: (495) 434-1174; E-mail: anosipov@yahoo.com2Research Institute for Physical and Chemical Medicine, Malaya Pirogovskaya ul. 1a, 119992 Moscow, Russia; fax: (495) 246-4401; E-mail: info@ripcm.org.ru
3Faculty of Basic Medicine, Lomonosov Moscow State University, Lomonosovsky pr. 31/5, 119192 Moscow, Russia; fax: (499) 932-8814; E-mail: info@fbm.msu.ru
* To whom correspondence should be addressed.
Received May 14, 2007
Chemical and biological functions of hemoprotein nitrosyl complexes as well as their photolysis products are discussed in this review. Chemical properties of nitric oxide are discussed, and major chemical reactions such as interaction with thiols, free radicals, and transition metals are considered. Specific attention is paid to the generation of hemoprotein nitrosyl complexes. The mechanisms of nitric oxide reactions with hemoglobin and cytochrome c and physicochemical properties of their nitrosyl complexes are discussed. A review of photochemical reactions of nitrosyl complexes with various ligands is given. Finally, we observe physiological effects of visible radiation on hemoprotein nitrosyl complexes: smooth muscle relaxation and reactivation of mitochondrial respiration.
KEY WORDS: hemoproteins, nitric oxide, laser radiationDOI: 10.1134/S0006297907130068
Abbreviations: cGMP) cycloGMP; cNOS) constitutive isoform of NO-synthase; cyt a32+-NO) nitrosocytochrome a3 reduced; cyt a33+-NO) nitrosocytochrome a3 oxidized; cyt c2+) reduced cytochrome c; cyt c2+-NO) reduced nitrosocytochrome c; cyt c3+) oxidized cytochrome c; cyt c3+-NO) oxidized nitrosocytochrome c; deoxyHb) deoxyhemoglobin; DETA-NONOate) a compound releasing free NO upon decomposition; DNIC) dinitrosyl iron complexes; EDRF) endothelium-derived relaxing factor; EPR) electron paramagnetic resonance; Hb) hemoglobin; Hb(I)*, Hb(II)*) hemoglobin excited states; Hb-4FeIV=O) ferryl hemoglobin; Hb4NO) nitrosyl hemoglobin; Hb4O2) oxyhemoglobin; HbCO) carboxyhemoglobin; HbNO) nitrosyl hemoglobin; HbO2) oxyhemoglobin; Hb-SNO) S-nitrosohemoglobin; heme-Fe2+) heme containing Fe2+; heme-Fe2+-NO) nitrosyl heme; HFS) hyperfine structure; iNOS) inducible isoform of NO-synthase; IP6) hexainositol phosphate; LPS) lipopolysaccharides; Mb-FeIV=O) ferryl myoglobin; metHb) methemoglobin; metMb) metmyoglobin; mtNOS) mitochondrial isoform of NO-synthase; NO) nitric oxide; NO-) nitroxyl-anion; NO+) nitrosonium-cation; NO2) nitrogen dioxide radical; NO2-) nitrite; NO3-) nitrate; OH) hydroxyl radical; O2-) superoxide radical; ONOO-) peroxynitrite anion; RSH(RS-)) thiols (protonated/deprotonated form); RS-NO) nitrosothiols; TNF) tumor necrosis factor.
The discovery by R. Furchgott of a factor that can induce smooth muscle
relaxation of blood vessel wall, produced in the endothelium and named
endothelium-derived relaxing factor (EDRF), prompted researchers to
reconsider the function of endothelial cells [1].
In the beginning of the 1990s, EDRF properties were studied enough to
suggest that NO is a part of EDRF [2]. Later, it
was experimentally proven that NO binding to the active site of
guanylate cyclase produces a complex with enzymatic activity much
greater than that of the original enzyme [3]. In
1998, a Nobel Prize was awarded to R. Furchgott, L. Ignarro, and F.
Murad for “... studies of the physiological function of nitric
oxide”.
Primary interest in NO was based on its ability to induce blood vessel relaxation, but soon the list of biological activities of NO was significantly expanded. Nitric oxide was found responsible for neuron signal transduction, immune reactions, reproductive functions, etc. A molecule previously considered toxic and carcinogenic demonstrated various physiological activities, and thus in 1992 the journal Science awarded nitric oxide the status of “molecule of the year”.
Not only nitric oxide, but also its numerous derivatives can manifest this activity. Among them peroxynitrite, nitrites, nitrates, nitrosothiols, and heme- and non-heme nitrosyl complexes occupy a specific place. Except for peroxynitrite, all of them can release free nitric oxide and thus play a role of nitric oxide depot. The aim of the present review is to discuss and clarify the generation and decay mechanisms of nitrosyl complexes and their biological activity.
CHEMICAL AND BIOLOGICAL PROPERTIES OF NO
NO production in vivo. There are several brilliant reviews on the chemical and biological activities of nitric oxide, for instance the paper of Wink and Mitchell [4]. Nevertheless, we decided to present here a short analysis of the properties of NO as they have a direct relationship to the main goal of this review. There are two major pathways of NO generation in the organism: enzymatic and non-enzymatic. The non-enzymatic one is the reduction of nitrite or nitrate to NO. Usually, it occurs as a dismutation of nitrite ions in acidic medium or as a direct reduction of nitrite (for instance with iron ions). Nitrite dismutation becomes detectable at pH < 6 [5]. Zweir proposed the following reaction scheme for this mechanism:
NO2- + H+ <--> HNO2, (1)
NO2- + HNO2 <--> N2O3 + OH-, (2)
N2O3 <--> NO2 + NO. (3)
However, this mechanism can take place in specific conditions when cells are acidified, in ischemia/reoxygenation or in inflammation. Direct reduction of nitrite by hemoproteins occurs at neutral pH [6].
Among hemoproteins demonstrating nitrite reductase activity, hemoglobin, myoglobin, cytochrome c reductase, and cytochrome P450 are the most active [7, 8]. An approximate reaction for hemoglobin is as follows [9]:
Hb + NO2- + 2H+ → metHb + NO + H2O. (4)
Enzymatic synthesis of NO in cells is catalyzed by proteins called NO-synthases [10, 11]. NO is synthesized through transformation of L-arginine into L-citrulline and acquiring of two oxygen atoms [12]. This means that NO synthesis is an oxygen-dependent reaction. There are two isoforms of the enzyme--constitutive and inducible NO-synthases [13]. Constitutive isoform is represented by neuronal and endothelial NO-synthases. Neuronal NO-synthase (nNOS) is produced in neurons, microglia, astrocytes, and skeletal muscle cells. Endothelial NO-synthase (eNOS) is localized in endothelial and smooth muscle cells [14, 15]. The presence of Ca2+ ions and calmodulin is necessary for enzyme activation. The eNOS maintains a constant level of NO equal to ~4 pM/min per mg of protein. Daily NO production by endothelium is 1.7 mM, and the steady state NO concentration in blood plasma is about 3 nM [16]. Inducible NO-synthase (iNOS) is present in neutrophils and macrophages, i.e. in cells participating in inflammation, and is synthesized in response to cytokines and lipopolysaccharides. In contrast to nNOS and eNOS, inducible NO-synthase does not need Ca2+ for activation.
Physical and chemical properties of nitric oxide. Nitric oxide exists in three forms that can be interconverted. Besides NO itself there are two more forms, nitrosonium cation (NO+) and nitroxyl anion (NO-), occurring as a result of oxidation or reduction of NO. The main reactions of NO are reactions with oxygen, free radicals (including O2- and *OH), thiols, and transition metals:
2NO + O2 → 2NO2, (5)
NO2 + NO <--> N2O3, (6)
N2O3 + H2O → 2NO2- + 2H+. (7)
NO decay in this process is a first order reaction with k = 0.124 sec-1 [17]. The rate of the reaction of NO with superoxide anion-radical is extremely high and is diffusion limited (6.7·10. M-1·sec-1). This reaction results in the production of a highly reactive compound--peroxynitrite [18]:
NO + O2- → ONOO-. (8)
This reaction accounts for cell injury in the ischemia/reoxygenation process, as reoxygenation is responsible for enhanced superoxide anion-radical production [19]. Interaction of NO with other radicals is effective too, for instance, in the reaction with hydroxyl radical a nitrite anion is generated:
NO + *OH → HNO2 <--> NO2- + H+. (9)
Reaction with peroxyl and alkoxyl radicals quenches lipid peroxidation process by reducing radicals to corresponding lipid hydroperoxides or oxy-derivatives [20, 21]. Thus, NO can effectively control the free radical lipid peroxidation processes.
NO can interact with hemoproteins in two ways: i) producing nitrosyl complex with heme iron in the active site (the rate constant is in the range 103-107 M-1·sec-1) or, ii) participating in oxidation-reduction reactions, mostly as a reductant. Thus, NO can inhibit cyclooxygenase by reducing compound I or II of the peroxidase cycle [22], or reduce ferryl hemoglobin to metHb [23].
Our experiments demonstrated that NO can interact with strong oxidants (compounds I or II of hemoglobin containing Fe5+ or Fe4+), reducing them to metHb (with Fe3+) without producing nitrosyl complexes. Due to the reduction of Mb and Hb oxoferryl radicals, NO plays an antioxidant role [23]. We have shown that oxoferryl radicals Mb-Fe+4=O and Hb-Fe+4=O, produced in the reaction with H2O2 or organic hydroperoxide (say, tert-butyl hydroperoxide), are strong oxidants. Addition of NO inhibits oxidative processes induced by Mb-Fe+4=O or Hb-Fe+4=O by reducing these radicals to metMb or metHb [23].
The chemical properties of NO+ are mainly reactions of addition and substitution with nucleophiles and aromatic compounds. Among a variety of organic compounds participating in these reactions, amines, carboxyls, and thiols can be listed. Under physiological conditions thiols are the most efficient NO acceptors--they can produce stable nitrosothiols [24, 25]:
NO+ + RS- <--> RS-NO. (10)
Transition metals can react with NO and its derivatives too. Cu and Fe ions can catalyze decomposition of nitrosothiols producing NO, NO+, and RSSR or RS- [26, 27]. For instance:
Cu+ + RS-NO <--> Cu2+ + NO + RS-. (11)
This reaction is reversible and its equilibrium can often be shifted to RS-NO production [28]:
RS-NO <--> RS- + NO+. (12)
Moreover, NO can produce non-heme mono- and dinitrosyl iron complexes [29, 30]. In this case, transition metals can form nitrosyl complexes and facilitate the transformation of NO redox forms.
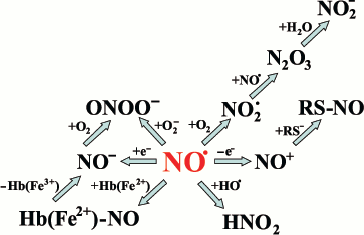
The lifespan of free NO in cells and tissues is rather short due to its fast interaction with oxygen, various radicals (such as superoxide and lipid radicals), and metals. Its lifespan is about 100 msec in blood plasma, and diffusion distance of NO from the point of generation does not exceed 200 µm [31, 32]. These properties of NO define its biological functions: i) NO activity takes place close to the point of production, and ii) NO can be stored in and released from stable intracellular complexes that belong to the NO pool. Major chemical reactions of NO are summarized in Fig. 1.
NO pool in the organism. Dinitrosyl iron complexes (DNIC) are considered to be rather stable forms of NO in cells. To produce DNIC, SH-groups of proteins or low molecular weight thiol and non-heme iron are necessary [33]. DNIC are formed in macrophages and endothelial cells and they are considered to be the main pool of NO in the organism [34]. Experimental evidence exists that NO can be released from cells not as free NO, but as DNIC. It is well known that low molecular weight thiols (such as cysteine or glutathione) can compete with proteins for NO, generating DNIC. Finally, DNIC have almost the same stability and physiological activity as EDRF [35].Fig. 1. Main chemical reactions of NO.
Dinitrosyl non-heme iron complexes are in dynamic equilibrium with S-nitrosothiols, non-heme iron, and thiols. Like DNIC, nitrosothiols possess EDRF-like activity, but their lifespan is much shorter [36]. Most stable are nitrosoproteins; that is why 95% of total nitrosothiols is nitrosoalbumin [2]. S-Nitrosothiol concentration in plasma is 3-4 orders of magnitude higher than NO concentration. Decomposition of nitrosoproteins is catalyzed by transition metal ions, and iron ions are the most efficient among them [27, 28].
Besides nitrosylation of albumin, NO can nitrosylate beta-chain Cys93 in hemoglobin producing S-nitrosohemoglobin (Hb-SNO) [37]. It was long considered that the main product of interaction of NO and oxyhemoglobin (90% in arteries and 70% in veins) is nitrate:
HbO2 + NO → metHb + NO3-. (13)
However, Stamler et al. detected 0.3-0.4 nM Hb-SNO in rat arterial blood; in venous blood Hb-SNO concentration was negligibly low. It was proposed that Hb-SNO is produced in the organism, and, moreover, it can serve as a NO donor. It was also found that if Hb-SNO in R-conformation is transformed into T-conformation, releasing free oxygen, then the nitrosothiol bond breaks and free NO is released. Stamler et al. demonstrated that Hb-SNO can dilate blood vessels and enhances blood flow. Opposite to Hb-SNO, HbO2 induces vessel contraction and converts NO into nitrate [38]. Thus, Hb-SNO both in solution and in erythrocytes shows EDRF-like properties.
Moreover, NO can react not only with Hb SH-groups, but with heme too, producing nitrosyl complexes (HbNO). HbNO was detected in human blood plasma in ischemia/reoxygenation [39], tumor necrosis [40], hyperthermia [41], and transplant seizure [42]. At the same time, it was found that Hb in erythrocytes reacts with NO 2-3 orders of magnitude more slowly than in solution. Up to the middle of 1990s, it was supposed that erythrocytes in vivo cannot accumulate NO [43]. Moreover, it was considered that oxyHb is more likely undergoing oxidation than nitrosyl complex formation in reaction with NO [44]. In 1996, Stamler et al. observed that both arterial and venous blood in rats contains HbNO (0.6 and 0.9 nM, correspondingly) [37]. It was experimentally proven in vitro that at physiological ratio NO and Hb (~1 : 1000) and oxygen saturation of Hb, approximately 10-40% of NO is engaged in nitrosyl complex formation [45, 46], and the rest in Hb-SNO formation. Hb conformation transitions (R-T transition) are accompanied with NO transfer from Cys SH-group to heme and its release in the free state [45].
Thus, Hb-SNO, HbNO, and non-heme iron nitrosyl complexes form the main pool of stored NO [8]. However, interaction of NO with thiols and heme and nitrosyl complex production can be considered both as means to accumulate NO and as enzyme nitrosylation, if thiols constitute the enzyme active site.
Regulatory functions of NO. NO can modify the activity of an enzyme, interacting with its functional groups, especially with heme Fe and thiols. The most impressive example is activation of guanylate cyclase. Addition of NO to heme iron of the guanylate cyclase regulatory subunit induces breaking of the bond between Fe and histidine imidazole group nitrogen and as a result protein conformational transition and change in active site structure [47]. Enzymatic activity in this case and cGMP level is increased a hundred times. If the target cells are platelets, then elevation of cGMP induces decrease in blood coagulation. If the target cell is a smooth muscle cell, then elevation of cGMP induces muscle relaxation. The mechanism described is the basic mechanism for blood vessel tension and intestine sphincter control.
In addition to guanylate cyclase, NO also forms nitrosyl complexes with CuB and heme of cytochrome a3 and in the active site of cytochrome c oxidase, a terminal acceptor in the mitochondrial respiratory chain. These complexes are unstable, and obviously are intermediates in the reduction of NO to NO- [48]. As a result of the interaction of NO with cytochrome c oxidase, cell respiration is inhibited irreversibly [49].
Another mechanism of NO-mediated modulation of enzyme activity is nitrosylation of an SH-group that is involved in the enzymatic activity. As a rule, reduction or oxidation of these groups can induce protein conformational transitions and as a result variation of enzymatic activity. A good example is the modulation of glutamate receptor activity. NO can nitrosylate glutamate receptor SH-groups of postsynaptic membranes, the conformation of this complex is changed, and it becomes inactive. This is the way that NO can modulate signal transduction in brain neurons [50].
Protein S-nitrosylation/denitrosylation reactions are one of the major mechanisms in intracellular signal transfer. This is the way that serine protease (caspase) activity participating in apoptosis can be controlled too [51]. One of the most likely mechanisms of induction of Fas-mediated apoptosis is activation of caspase 3. In a resting cell, caspase 3 is inactive (it exists as a procaspase 3). To activate procaspase 3 (to convert it into caspase 3), a catalytically active subunit should be removed. In some cases this is not enough, if the caspase 3 active site Cys is nitrosylated [52]. In this case denitrosylation is the second step of the activation process. Both activation steps are supported by Fas protein [51]. Caspase S-nitrosylation inhibits both caspase 3 activity and apoptosis. It is clear that induction of the apoptotic cascade is mediated by intracellular NO level.
Finally, NO can nitrosylate SH-groups in proteins participating in transition metal transport and deposition. These are copper metabolizing proteins and metallothioneins [53, 54]. An important characteristic of these proteins is a high cysteine content. Cysteine SH-groups produce stable complexes with Cu ions and maintain their enzymatic activity. Interaction of NO with cysteine and nitrosyl complex formation induces the release of free copper ions [55]. Released copper can catalyze oxidative processes in cells (first of all Fenton-like reactions) and cause necrosis or apoptosis [56]. Moreover, copper released from metallothioneins can be incorporated into superoxide dismutase and increase SOD activity [55, 57].
Controlling of enzyme activity by means of formation/decomposition of nitrosyl complexes in the active site is a very significant phenomenon not only in normal physiological conditions, but also in pathology.
Role of NO in pathology. NO is an active participant in pathogenesis of heart diseases, including hypertension and atherosclerosis; it controls blood vessel tension and blood pressure. A protective role of NO in the development of initial stages of ischemia and blood flow normalization and decreasing of tissue injury is well known [58]. The ability of NO to affect blood flow in the lungs is used in clinical practice [59]. S-Nitrosoglutamate (a natural NO metabolite), for instance, can control aerial resistance in bronchial tubes [36]. Being a neurotransmitter of the peripheral nervous system, NO controls reproductive functions in men and plays an important role in the treatment of impotence [60].
NO participates in inflammatory and immune processes. Macrophages stimulated with gamma-interferon, TNF, or LPS dramatically increase NO and ONOO- synthesis, damage bacterial cells, and in such a way manifests their antimicrobial activity. At the same time, NO production in excessive amounts (for instance, in sepsis) plays a negative role. NO-induced weakening of a vessel tension and subsequent decrease in blood pressure can develop a shocking status. Excessive synthesis of NO and ONOO- in ischemia/reoxygenation is a cause of tissue injury and cell death [18]. NO toxicity in cells is realized through hemoprotein nitrosyl complexes formation and/or protein S-nitrosylation. Inhibition of mitochondrial respiratory chain, Krebs cycle enzymes, and DNA synthesis reactions is the result of nitrosylation and nitrosyl complex formation. Moreover, the development of NO-dependent oxidative stress is mediated by the generation of the efficient oxidant ONOO-, which can irreversibly inhibit enzymes and oxidize lipids and DNA. NO can easily interact with free radicals, thus quenching lipid peroxidation or inhibiting its initialization [20, 21, 61].
Thus, on one hand, due to the production of ONOO-, NO plays a role of prooxidant, and on the other hand, being a scavenger of free radicals and a reducing agent it is an antioxidant.
INTERACTION OF HEMOPROTEINS WITH NO
Structure and properties of nitrosyl complexes. Specific interest of researchers in hemoprotein nitrosyl complexes was based, first, on their important biological functions and, second, on the fact that nitrosyl complex formation is followed by structural alterations of the heme environment. Impressive examples of this phenomenon are nitrosyl complexes of hemoglobin and cytochrome c oxidase [62-64].
It is well known that heme iron has six coordinating bonds. Four of them are bonds with nitrogen of the porphyrin ring, and they are located in the heme plane. The 5th and the 6th bonds have axial location, and in hemoproteins they are occupied by imidazole nitrogen atoms or sulfur atoms of the protein globule. Proteins with unoccupied 6th bond (hemoglobin, myoglobin, peroxidases) can interact with NO faster than those with entirely occupied coordinative bonds (cytochrome c). The latter will need additional energy to displace the 6th ligand.
Proteins in the ferric state with vacant 6th bond can have a water molecule in this position (as in metHb). The presence of a ligand in the 6th position hampers embedding of a NO molecule and formation of a nitrosyl complex if compared to the ferrous state [65], i.e. heme reacts with NO faster in ferrous state (107-108 M-1·sec-1) than in ferric state (102-107 M-1·sec-1) [66]. And vice versa, the dissociation rate of these complexes is higher for ferric state of hemoproteins (Fe3+-NO).
Complex formation between NO and heme affects the heme physical properties: the optical spectrum changes, and in the case of Fe2+-NO an ESR signal with g ~ 2.0 can be detected [63, 67]. In contrast to Fe2+-NO, Fe3+-NO complex is diamagnetic [68].
Nitrosyl hemoprotein complexes are rather stable. Hemoglobin has high affinity to NO, which is about 1012 M-1. NO and heme can participate in redox reactions. NO is neither a strong reducing agent nor a strong oxidant (ENO/NO+ = 380 mV, ENO/NO¯ = 1210 mV) [69], and the direction of these reactions depends, mostly on the redox properties of the protein:
heme-Fe2+ + NO <--> heme-Fe2+-NO <--> heme-Fe3+ + NO-, (14)
heme-Fe3+ + NO <--> heme-Fe3+-NO → heme-Fe2+ + NO+. (15)
In spite of the fact that heme redox reactions are mostly reversible, the reactions of the subsequent products can shift the equilibrium and make the process practically irreversible.
Hemoglobin nitrosyl complexes. Hemoglobin nitrosyl complexes first were described in the 1960s [64]: they are formed when NO is associated with heme iron, and the iron atom is most often in the reduced state (heme-Fe2+-NO). The paramagnetic properties of this complex are due to the presence of an unpaired electron that belongs to NO*. Both reduced (Hb(Fe+2)) and oxidized (Hb(Fe+3) - metHb) forms of hemoglobin can interact with NO. The reaction of metHb with NO is reversible, and the rates of the direct and reverse reactions are rather low (Table 1):
NO + metHb <--> metHb-NO. (16)
The reaction of Hb (deoxyHb) with NO is diffusion limited and practically irreversible. NO binding to Hb is not a cooperative process, in contrast to O2 binding. This phenomenon is confirmed by a saturation dependence of Hb on NO concentration and by the fact that NO binding to heme does not require structural changes in the protein globule [70, 71].
Table 1. Rate constant of the reaction of NO
and Hb
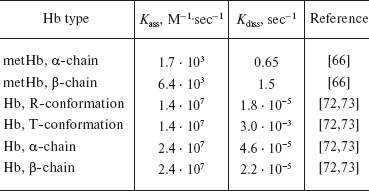
Due to the high affinity of deoxyHb for NO (K = 1012 M-1), the dissociation rate of Hb nitrosyl complexes is rather low. This affinity is three orders of magnitude greater than that to CO [44]. That is why NO can displace CO from carboxyhemoglobin (HbCO):
HbCO + NO → HbNO + CO. (17)
In contrast to CO, which is toxic, NO does not have any negative consequences and, moreover, can be used for treatment of lung hypertension in newborns [74]. This fact can be explained as following: the CO molecule, when bound to the deoxyHb subunit, can transfer the protein into R-state and decrease its affinity to oxygen. Binding a NO molecule to deoxyHb can also change the affinity to O2, but the rate constant of this process depends on pH. Decreasing the pH lowers the affinity of Hb to O2. At pH 7.4 the affinity of the partially nitrosylated Hb to O2 is higher than that of the native protein (Hb4NO binds O2 better than Hb4O2), but at pH 5.8 the opposite situation can be observed (Hb4NO(O2)3 better releases O2 than Hb4(O2)4). Thus, nitrosylation facilitates O2 transport to the tissues [71].
The reaction of Hb entirely saturated with O2 and NO has a rate constant of 3·107 M-1·sec-1 and produces metHb and nitrite ion [75]. However, under physiological conditions Hb has saturation of about 35% (in capillaries) to ~95% (in arterial blood) and the number of vacant binding sites is higher than NO concentration (<1 µM). This means, that in parallel with Hb oxidation formation of nitrosyl complexes should take place [45]. Moreover, HbNO can be produced in the reaction of S-nitrosothiols with HbO2 [76]. And finally, generation of HbNO was demonstrated not only in solution, but also in erythrocytes [37, 77]. HbNO can undergo decomposition producing metHb [46]:
HbNO → metHb-NO-, (18)
2 metHb-NO- + 2 H+ → 2 metHb + N2O + H2O. (19)
ESR spectroscopy is a very effective method for studying the structure of nitrosyl complexes. In most cases, the ESR signal of a hemoprotein nitrosyl complex is a single asymmetric line with g-factor 2.03. Alteration of the nitrosyl complex structure with hexainositol phosphate (IP6) or sodium dodecyl sulfate induces the formation of triple hyperfine structure in the ESR spectrum. The central line of this spectrum has g = 2.01 and splitting constant 16.5 G. The mechanism of this phenomenon is as following: it is known that the unpaired electron is localized mostly on the d-orbitals of the iron ion and slightly on the pi-orbitals of the nitrogen atom [78]. Addition of IP6 (or sodium dodecyl sulfate) cleaves the iron histidine bond, and 5-coordinated heme is produced. The heme iron is shifted to NO and hyperfine splitting structure can be observed [78, 79]. The ESR hyperfine splitting structure is specific for HbNO alpha-chains only (Fig. 2) [72].
Shifting of alpha-chain heme iron along the NO bond changes the shape of the plane heme into a dome-shaped form, and this complex becomes a high spin complex. This kind of heme-ligand interaction with proximal histidine is specific for Hb-NO reactions. If the heme ligand is CO or O2, then the Fe-His bond is not changed [71].Fig. 2. ESR spectra of Hb nitrosyl complexes [63]. X-Axis, magnetic field (H, Gauss); Y-axis, signal intensity (dX´´/dH). alphaNO, betaNO, and alpha-hyperfine are ESR signals of alpha-chain and beta-chain nitrosyl complexes (in 6-coordinated state) and alpha-hyperfine (alpha-chain in 5-coordinated state, hyperfine structure can be seen).
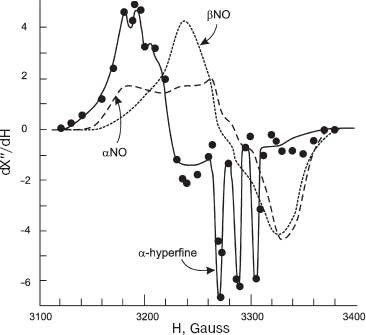
Hill et al. concluded that all the variety of ESR spectra observed in experiments is a combination of alpha- and beta-chain of 6-coordinated heme iron (Fe(alphaNO and betaNO)) and alpha-chain of 5-coordinated iron (Fe(alpha(5)NO)) (Fig. 1) [63]. The contribution of alpha(5)NO fraction depends on several factors: it increases with decreasing pH and lowering of Hb saturation [63, 71]. At pH = 6 HbNO, 10% saturated, has alpha(5)NO-signal, and at pH = 8 and complete saturation Hb with NO there is no hyperfine splitting structure at all. It is obvious that the pH effect can be explained by histidine pK value--when pH is increased it becomes more stable, and all the hemes are transferred into the 6-coordinated state. The increase of alpha(5)NO fraction at lower saturation values can be explained by the fact that the affinity of alpha-chain to NO is higher than that of beta-chain (Table 1) [71-73].
Cytochrome c nitrosyl complexes. Another interesting protein producing nitrosyl complexes is cytochrome c. Cytochrome c can produce nitrosyl complexes both in reduced and oxidized states (cyt c2+ and cyt c3+):
cyt c2+ + NO <--> cyt c2+-NO, (20)
cyt c3+ + NO <--> cyt c3+-NO. (21)
In contrast to Hb, these reactions are much slower (Table 2). The equilibrium rate constants for cytochrome c nitrosyl complexes in reduced and oxidized state (cyt c2+-NO and cyt c3+-NO) are 2.9·105 and 1.6·104 M-1, correspondingly. They are several orders of magnitude lower than that for Hb. This phenomenon can be explained by the fact that in native cytochrome c heme iron is in the 6-coordinated (and not 5-coordinated as in Hb) state. Both the reduced and oxidized protein states have two axial ligands besides the four that belong to heme. These ligands are histidine (His18) and methionine (Met80).
It was found that the reaction rate of a small ligands (say, NO or H2O2) with hemoproteins which have 6-coordinated heme can be altered. This phenomenon was studied on cytochrome c. This problem was also interesting because the interaction of cytochrome c and NO or H2O2 plays an important role in apoptosis in mitochondria. The main idea is that interaction of cytochrome c with some negatively charged lipids (especially cardiolipin and phosphatidylserine) increase peroxidase activity of cytochrome c complex. This phenomenon can be explained by the enhancement of the interaction of Fe and H2O2. Interaction of cytochrome c with cardiolipin modifies the structure and properties of the active site of cytochrome c. As it was shown by means of ESR [80], Raman [81], and 31P-NMR [82] spectroscopy, the cytochrome c heme channel becomes open and heme iron is transferred from low spin (6-coordinated) to high spin (5-coordinated) state. The NMR data demonstrates that this interaction affects the polypeptide chain conformation and destabilizes the alpha-helixes [82]. IR spectroscopic data shows that approximately 10% of the alpha-helix becomes unfolded [83]. The NMR spectroscopy data suggested that cardiolipin and cytochrome c binding increased the distance between Met80 and heme, as well as modifying the environment of Met65, which is close to the heme [84]. Finally, these intermolecular rearrangements increase the distance between Met80 and the heme and facilitate the access of H2O2 or NO to the heme iron. In the case of H2O2, these rearrangements result in a dramatic enhancement of cytochrome c peroxidase activity. In mitochondria, increase of cytochrome c peroxidase activity can destroy the mitochondrial membrane and cause cytochrome c release, which is observed in apoptosis. The presence of NO close to the iron atom can lead to production of the cytochrome c nitrosyl complex. If NO interacts with cytochrome c heme, then the Met80-heme bond is cleaved with replacement of amino acid by NO as the 6th ligand. Nitrosyl complex formation inhibits cytochrome c peroxidase activity due to the impediment of H2O2 access to heme Fe [85, 86].
Table 2. NO and cytochrome c rate
constants
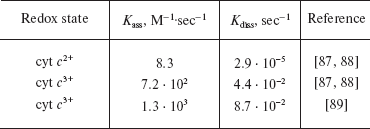
Thus, low rate and equilibrium constants are due to energy losses for 6th ligand replacement. In fact, cytochrome c with alkylated Met80 (and 5-coordinated heme) has two orders of magnitude higher affinity to NO (2·107 M-1) than that in the native protein [67].
Cyt c2+ nitrosyl complex (cyt c2+-NO), as in HbNO, has an ESR signal with g ~ 2.0. As in the case of Hb, the ESR spectrum corresponds to a rhombic symmetry iron center.
Besides nitrosyl complex formation, cytochrome c can participate in redox reactions with NO. Reduction of cytochrome c with NO is well known and is analogous to that of Hb [90]:
cyt c3+ + NO <--> cyt c3+-NO, (22)
cyt c3+-NO <--> cyt c2+-NO+, (23)
cyt c2+-NO+ + 2OH- <--> cyt c2+ + NO2- + H2O. (24)
Sharpe et al. demonstrated that cyt c3+-NO complex can be spontaneously destroyed, and in this case practically the entire NO is converted into nitrite [89]. Besides that, NO can be reduced to nitroxyl anion (NO-), and this reaction is characterized by rate constant 200 M-1·sec-1 [89]:
cyt c2+ + NO → cyt c3+ + NO-. (25)
Finally, we can say that both Hb and cytochrome c can produce rather stable nitrosyl complexes that serve as a NO depot in the organism, and NO can be released from this depot. Moreover, these nitrosyl complexes possess photosensitivity and can be destroyed upon irradiation with visible light.
PHOTOCHEMICAL REACTIONS OF LIGAND-BOUND HEMOPROTEINS
Mechanisms of photochemical reactions of nitrosyl complexes. Photolysis of hemoprotein complexes have been known for more than 100 years, since the publications of Holdane and Lorrain, who studied carboxyhemoglobin (HbCO) photolysis with visible light [91]. More than 60 years later Keilin and Hartree observed cyanomyoglobin photolysis [92]. Then, Gibson and Einsworth expanded the list of photolyzed hemoprotein complexes to O2 and NO hemoglobin complexes [93].
Initially, hemoprotein photochemistry was studied mainly by optical and IR spectroscopy. The most popular object in these studies was HbCO, as its photolytic effect is greatly expressed. In the 1950-60s the quantum yields of these Hb complexes were evaluated to be for HbCO = 0.4 [94], HbO2 = 0.01, and HbNO < 0.001 [95]. In parallel with improvement of spectroscopic techniques, these quantum yields were evaluated more precisely: HbCO = 0.7, HbO2 = 0.08, HbNO < 0.004 [96]. Many publications have appeared clarifying the mechanisms of the photodissociation process, to evaluate the rate constants, and to study the conformational changes [96-98]. Recently it was found that that the quantum yields for the variety of hemoproteins (Hb, Mb) and complexes (CO, O2, NO) are similar to each other [99]. Petrich et al. demonstrated that the kinetics consist of several general stages [98, 99]: (i) transformation of hemeprotein-ligand complex into the excited state (tau1/2 < 50 fsec); (ii) complex photodissociation and appearance of the absorption spectrum of the original protein (tau1/2 = 300 fsec); (iii) fast relaxation and conversion of the excited molecule to the ground state (tau1/2 = 2.5-3.2 psec); (iv) slow recombination of the photolysis products (say, heme-ligand association) [97]. This recombination has various half-life times for different ligands: 0.1 µsec for HbCO, 1-3 nsec for HbO2, and about 10 psec for HbNO.
The comparative study of Hb complex absorption spectra in the Soret band showed that Hb transfer into the excited state is realized within 50 fsec, independent of ligand type. After dissociation Hb can be found in one of the two excited states--Hb(I)* or Hb(II)*. From the state Hb(I)*, heme can relax to the ground state with tau1/2 = 300 fsec, and from Hb(II)* with tau1/2 = 2.5 fsec. The Hb(I)*/Hb(II)* ratio depends on ligand type. In the case of CO, Hb(I)* is mainly produced, and vice-versa, dissociation of HbNO most probably results in Hb(II)*. This fact can explain a fast recombination of HbNO (10 psec).
It is well known that Hb complex photodissociation induces conformational alterations both in protein globule and in heme. These alterations of HbCO are studied in detail [99]. These events can be explained as “iron atom shifting from the heme plane” or as “dome-shaped heme bending”. These transformations induce heme Fe transition from low spin into high spin state. The Hb(I)* excited state is a state with the dome-shaped heme. Relaxation of Hb(I)* to the ground state evokes structure changes inhibiting heme-ligand recombination. In Hb(II)* the conformation of the heme is not modified; it remains flat and can follow fast association with the ligand.
A similar phenomenon was detected by Zhu et al. as a consequence of irradiation of myoglobin nitrosyl complex [100]. It was shown that quantum absorption induces the cleavage of the heme-NO bond, alters the iron spin state, extends the Fe-His bond, and the heme acquires the dome shape.
Fast reassociation of the photolysis products does not allow suggesting more precise mechanisms of the heme-ligand complex photolysis rearrangements [98, 99]. It is known that recurring irradiation of these complexes results in the incomplete recovery of the protein absorption spectrum due to side reactions [98, 101]. In our case in the photolytic decomposition of HbNO we can expect the formation of free NO and its derivatives--nitroxyl anion and/or nitrosonium cation. Moreover, as Hb reacts with NO with a rate constant .107 M-1·sec-1 and has the affinity of .1015 M-1, we can expect that free NO, produced in the photolysis, can reassociate with Hb.
Cytochrome c nitrosyl complex photolysis is poorly studied, but there are some publications [87, 88]. According to Hoshino and Rose, cyt c3+-NO and cyt c2+-NO complexes are photosensitive and can be decomposed upon irradiation.
Photochemical reactions of Hb nitrosyl complex. We have obtained in our lab some experimental evidence of Hb nitrosyl complex photodissociation. It was shown that irradiation of Hb nitrosyl complexes with He-Cd (441 nm) results in the cleavage of the heme-NO bond [102]. The heme iron in this process remains reduced, and nitric oxide is released as a free radical (NO). The photodissociation of HbNO is reversible. It was proven in anaerobic experiments that: (i) HbNO complexes can be reduced in the dark incubation after irradiation; (ii) the intensity of the photolytic effect depends on the irradiation power, and (iii) the effective quantum yield is low, about <0.001. During aerobic irradiation of HbNO, besides the photolytic decomposition/reassociation of nitrosyl complexes, a reaction between the photolysis products (Hb and NO) and oxygen takes place. The reaction with oxygen make the photolytic effect irreversible: (i) this effect becomes independent of irradiation power, and (ii) the quantum yield of HbNO photolysis increases to ~0.01.
Thus, if the photolysis occurs in the absence of oxygen, then fast reassociation takes place. In the presence of oxygen, the photolysis products can interact with oxygen and the reassociation reaction becomes negligible [102]. In our experiments we have observed NO production under anaerobic conditions when a spin trap (nitronyl-nitroxyl radical) interacted with the photolysis products [102]. This was due to the high rate constant of the interaction of NO with the spin trap (104 M-1·sec-1). This rate constant for interaction with oxygen is approximately 106 M-1·sec-1, so under aerobic conditions the photolysis products cannot be seen. Oxygen is a good competitor for NO with heme.
Photolysis of HbNO takes place both in high spin (R) and in low spin (T) conformations. This observation is very important as the generation of R- and T-conformations occurs at low NO concentrations, close to physiological ones [45], and also in vivo [37, 39-41].
Moreover, we found that photolysis of Hb nitrosyl complex can be observed not only in solution but also in erythrocytes. Nitrosyl complexes in erythrocytes have even higher photosensitivity than in solution. HbNO decomposition in erythrocytes takes place at lower doses, and NO generation can be detected even in the presence of oxygen; moreover, its production remains after complete decomposition of HbNO. This fact can be explained by the interaction of free NO with thiols (especially with glutathione, as its concentration is ~1 mM) inside the erythrocytes and production of nitrosothiols. Nitrosothiols can undergo decomposition upon irradiation producing free NO. Thus, on one hand, GSH facilitates photolytic decomposition due to the removal of free NO, and on the other hand, it extends the lifespan of NO as the nitrosyl complexes are formed.
Independent of erythrocyte HbNO photolysis mechanism, Hb nitrosyl complexes can serve as a NO donors. It is obvious that laser irradiation induces decomposition of HbNO and release of free NO [103]. Thus, HbNO can participate in irradiation-mediated blood vessel relaxation. This mechanism occurs both in normal physiological conditions (HbNO level is about 1 nM [37]) and in pathological cases when tissue reoxygenation decreases and NO production rises [39].
We have experimentally tested the statement that HbNO can serve as a temporal NO depot and can be released to dilate blood vessels [104]. The experimental procedure was as follows: animals were anaesthetized; an aliquot of blood was taken from the femoral vein and saturated with NO to produce nitrosyl Hb complexes. Then blood containing HbNO was introduced back into the femoral vein and the circulation in a. epigastrica and its descending branches was monitored in the process of He-Cd laser irradiation. We observed that the laser irradiation of a. epigastrica induces significant enhancement of blood flow (.30%) compared to that in control (without HbNO). These results can be explained as follows: laser irradiation induces HbNO decomposition and increases generation of free NO, which activates guanylate cyclase and finally leads to the blood vessel relaxation. Thus, Hb nitrosyl complex photolysis can facilitate blood circulation. These experiments are very important from the point of view of improving circulation after organ transplantation or vessel embolism. The main reactions of Hb nitrosyl complexes are presented in Fig. 3.
Photochemical reactions of cytochrome c nitrosyl complex. Reactions of NO with cytochrome c play an important role in the functioning of the mitochondrial respiratory chain. Nitrosyl complex formation both of ferrous and ferric cytochrome c states can inhibit mitochondrial respiration. At the same time, reduction of NO to NO- by ferric cytochrome c removes free NO from the reaction sphere (cytochrome bc1, cytochrome a3, and ubiquinol) and facilitates mitochondrial respiration [105]. In this case, effects of visible light on cytochrome c and NO interaction look very attractive.Fig. 3. Scheme of generation and decay reactions of Hb nitrosyl complex.
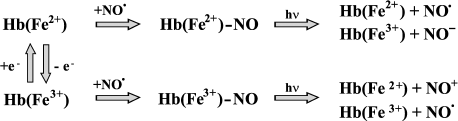
We have found experimental evidence of decomposition of both cyt c2+ and cyt c3+ nitrosyl complex upon He-Cd laser irradiation. We have seen that laser irradiation accelerates cyt c2+ nitrosyl complex decomposition, as detected by the decrease of the ESR signal, at least 20-fold compared to dark reaction. This phenomenon was in parallel with the decrease of the optical band of cyt c3+-NO at 560-570 nm. It is obvious that photolysis of nitrosyl complex generates reduced/oxidized cytochrome c and free NO. In other words, the irradiation shifts the equilibrium of the decay reaction to the increase of the initial compounds. We have detected cyt c2+-NO dissociated upon irradiation with He-Cd laser into cyt c2+ and NO, and cyt c3+-NO into cyt c2+ and NO+.
Based on our experimental results, we have suggested that He-Cd laser irradiation of mitochondria or native cells can reactivate respiration and ATP synthesis. One of the possible mechanisms of this reaction is the photodissociation of cytochrome c nitrosyl complexes.
It is appropriate to note the acceleration of cytochrome c-NO interaction in the presence of anionic lipids. We have found that not only H2O2, but also NO can easily react with cytochrome c if the cytochrome c-cardiolipin or cytochrome c-phosphatidylserine complex is formed. In the presence of cardiolipin, cytochrome c produces rather stable nitrosyl complex with NO [86]. Formation of this type of complexes decreases or even abolishes cytochrome c peroxidase activity. If this event takes place in mitochondria, then decrease in peroxidase activity will inhibit apoptotic reactions.
It was found that cytochrome c nitrosyl complexes produced in the presence of cardiolipin are photosensitive and can undergo decomposition upon irradiation with visible light. Our experiments showed that He-Cd laser irradiation (441 nm) induces nitrosyl complex decomposition and hence facilitates H2O2 access to the active site of the cytochrome c, i.e. restores the peroxidase activity of the cytochrome c. This means that laser irradiation can serve as a proapoptotic factor if nitrosyl complexes are present in the system. Thus, NO and laser radiation are the means to effect the cytochrome c peroxidase activity, and, probably, apoptosis [85].
It is obvious that cytochrome c is not the only place of NO application in the mitochondrial electron-transport chain. It was demonstrated that NO can produce nitrosyl complexes with cytochrome a3 heme in cytochrome c oxidase and induces its reversible inhibition [49]. Sarty et al. in the study of the cytochrome c oxidase inhibition showed that visible light reactivates the enzyme and enhances oxygen consumption in the system of cytochrome a3/NO [106]. Moreover, NO can irreversibly inhibit compound I by means of enzyme thiol nitrosylation, and UV irradiation of the nitrosylated compound I reactivates the enzyme. This effect is based on nitrosothiol photolysis [107]. Thus, we can conclude that not only the NO, but visible light too can modulate the mitochondrial electron transport chain activity. Further studies of the photochemical and photobiological reactions of NO can clarify the individual roles of the mitochondrial electron transport chain components in the inhibition of respiration. The major reactions of cytochrome c nitrosyl complex are presented in Fig. 4.
Fig. 4. Scheme of generation and decay reactions of cyt c nitrosyl complex.
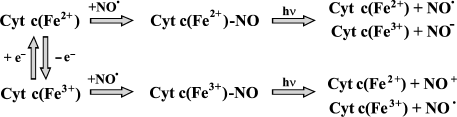
PHYSIOLOGICAL EFFECTS OF VISIBLE RADIATION
Relaxation of smooth muscle upon irradiation. Relaxation of rabbit aorta as a result of visible light irradiation was discovered by Furchgott in 1955 [108]. Later it became clear that light-induced vessel relaxation has very much in common with EDRF-mediated vessel response: both reactions elevated the amount of cGMP and both reactions can be inhibited by methylene blue [109]. Light-induced blood vessel dilation does not depend on endothelium, but it can be connected with the existence in the vessel wall of photosensitive compounds that can release NO on irradiation [110, 111]. This substance was consumed with increasing irradiation dose, but it can be restored by NO donors [103]. Venturini et al. demonstrated that photosensitive NO donors are S-nitrosothiols and S-nitrosoproteins. These substances are produced physiologically and can release free NO on irradiation with UV light [25, 103, 112].
Yu. A. Vladimirov suggested that nitrosyl Hb complexes can be one of the photosensitive chromophores participating in the mechanism of photorelaxation [113]. This hypothesis was based on the following experimental data: i) NO can easily bind Hb, and ii) nitrosyl Hb complexes were observed in vivo. Consequently, Hb can serve as NO transporter from the point of production to the biological targets. Finally, an important property of HbNO as well as a variety of hemoproteins is their photosensitivity. It is natural to suppose that light effect on HbNO will be the decomposition of HbNO and the release of free NO and subsequent blood vessel relaxation.
Photoreactivation of the enzymes of the mitochondrial electron transport chain. It is well known that visible light causes enhancement of mitochondria oxygen consumption and increases transmembrane potential and ATP production [114]. Similar effects were observed on intact cells [115]. Based on the action spectra, it is supposed that the primary acceptor of irradiation is cytochrome c oxidase [116]. Unfortunately, the mechanism of cytochrome c oxidase photoreactivation has not been studied in detail.
As mentioned above, among the various effects of NO the effect of inhibition of the mitochondrial electron-transport chain is well known [49]. This phenomenon was observed on isolated mitochondria [117] and intact cells [118]. Moreover, the possibility of controlling cell respiration by activated macrophages, producing NO, was shown [119]. Great interest in this phenomenon increased after the discovery of the mitochondrial NO-synthase (mtNOS) [120, 121]. These studies have showed that respiration can be controlled not only by exogenous NO, but also by endogenous NO through the modulation of mtNOS activity [122]. In this case, the consequence of respiration inhibition is the reduction of all elements of the electron transport chain and increase of O2*¯, H2O2, and peroxynitrite production [117, 123, 124]. Thus, NO--a free radical scavenger--becomes an inducer of free radical generation in mitochondria. Induction of oxidative stress initiated by NO leads to the oxidation of mitochondrial lipids, decrease of transmembrane potential, opening of membrane pores, and release of cytochrome c into the cytoplasm [123, 125]. The latter effect is the key event to initiate the apoptotic reaction cascade and cell death [126].
The attention of researchers studying the inhibition of respiration in mitochondria was attracted to the functioning of cytochrome c oxidase as the main target of NO. However, numerous experimental data demonstrate that practically all elements of the mitochondrial electron transport chain can interact with NO. Utilizing respiratory chain inhibitors, it was shown that suppressive effect of NO is revealed in all stages of the electron transport chain [105]. NO can inhibit cytochrome c oxidase [49] and decreases the activity of cytochrome bc1 [117, 124].
The inhibition of cytochrome c oxidase by mitochondrial transport chain inhibitors was demonstrated on the purified enzyme [106, 127], on mitochondria [105], and on native cells [128]. The specific characteristic of this phenomenon is its reversibility; decreasing the NO concentration corresponds to restoration of the enzyme activity. There are several explanations for this phenomenon. One is that NO can produce nitrosyl complexes with the heme iron of the cytochrome a3 (cyt a32+-NO) and with CuB+ and CuB2+ [48, 127-132]. However, besides the nitrosyl complex formation with cytochrome c oxidase heme, NO can reduce the iron in the active site and hence inhibit the enzyme. This is because during reduction of oxygen to water cytochrome c oxidase produces two strong oxidative states: compound P (Fe(a3)5+=O (oxoferryl, similar to compound I in the peroxidase cycle)) and compound F (Fe(a3)4+=O, (ferryl, similar to compound II in the peroxidase cycle)). NO can reduce any of these cytochrome c oxidase compounds (P→F, F→Fe(a3)3+, CuB2+→CuB+), redirecting the reaction [127, 130].
Sarti made an interesting observation while studying the inhibition of cytochrome c oxidase by NO [106]. It is known that nitrosyl complex of cytochrome c oxidase (cyt a32+-NO) is photosensitive and can be decomposed on irradiation with visible light [62]. Thus, if the main role of this process belongs to the generation of cyt a32+-NO, then the enzyme activity and mitochondria respiration should be sensitive to the presence of NO, and irradiation should reverse the inhibition induced by NO. In fact, it was observed that visible light practically completely restores the enzyme activity. These events can take place only in the presence of a reducing agent. If the concentration of a reducing agent was low, then oxygen consumption was low both in the light and dark. In other words, if the reducing ability of the media is high, the key step of inhibition is the production of cyt a32+-NO, but if the reducing ability of the media is low the main mechanism of inhibition is reduction of the cytochrome with NO or formation of light insensitive complexes cyt a33+-NO2- [106]. We suppose that in vivo, when the reducing agent concentration is rather high, the first mechanism works. In pathology, when the concentration of the reducing agents is low, the second mechanism is engaged. This mechanism can be observed in the process of inflammation.
Mitochondrial complex I can also serve as a NO acceptor. It was shown on mouse J774 line macrophage culture that complex I of the respiratory chain can be inhibited in the presence of NO donor--DETA-NONOate [107]. In contrast to cytochrome c oxidase, the inhibition of mitochondria respiration complex I by NO proceeds rather slowly (90% inhibition of activity takes approximately 3 h) and is irreversible. The mechanism of this phenomenon is probably due to the S-nitrosylation of a critical protein thiol group. The correctness of this supposition can be proved by two facts. First, etherified glutathione (glutathione analog that can easily cross the plasma membrane) restores cell respiration. Second, the enzyme is reactivated after irradiation with visible light. Glutathione can participate in transnitrosylation reaction and thus reduce the protein SH-groups. UV radiation induces the homolytic cleavage of RS-NO bond, and it is reasonable to suggest that S-nitrosylation is responsible for protein deactivation.
Thus, both cytochrome c oxidase and its compound I can be inactivated by NO. These mechanisms are fundamentally different. In one case, it is heme nitrosyl complex formation, and in the other case it is thiol group nitrosylation. Hence, visible light destroys both complexes and in both cases respiration and enzymatic activity are restored.
The mechanism of the reaction of cytochrome c and NO is complicated and is not entirely clear. However, as mentioned above, both reduced and oxidized forms of the cytochrome c can produce nitrosyl complexes [133, 134]. Moreover, ferrous cytochrome c can catalyze the reduction of NO to NO- [117], and ferric cytochrome c the oxidation of NO to nitrite [90]. Thus, we can conclude that both cytochrome c forms participate in NO metabolism. Moreover, in the presence of anionic phospholipids (cardiolipin and phosphatidylserine) the reaction of nitrosyl complex production is accelerated tenfold. Cytochrome c nitrosyl complexes formed are photosensitive and can undergo decomposition on illumination. The photochemical reactions described (nitrosyl complex photolysis or alteration of catalytic activity) can play an important role both in the regulation of mitochondrial respiration and development of apoptosis.
The experimental data discussed in this review prove the critical biological role of the hemoprotein nitrosyl complexes. It is also obvious that this role is based on the ability of nitrosyl complexes to serve as a nitric oxide depot. Irradiation is often used to facilitate the release of NO. Thus, laser irradiation can improve brain circulation. Laser therapy shows positive effects in shock cases. It was shown in animal models of hemorrhagic shock that laser irradiation in the acute phase makes their conditions better [135]. It was found that laser radiation of visible range enhances cell proliferation and tissue regeneration. For instance, in cardiology clinics laser therapy is used to improve transmural revascularization [136]. This approach significantly improves cardiologic status of the patients. Moreover, it was shown that He-Ne laser radiation facilitates wound and ulcer healing [137]. Proliferation activation upon laser irradiation was demonstrated if lasers of the various wavelengths were used: He-Cd (441.6 nm), argon (488 nm), Nd:YAG (760-830 nm), He-Ne (632.8 nm), ruby (694.3 nm) [116]. It was supposed that the sophisticated picture of biological laser light effects can be explained by the existence of numerous chromophores in cells. First of all, these are the enzymes of the respiratory chain [116, 137]. Finally, it was shown that laser irradiation can enhance NO production in cells. Klebanov et al. have demonstrated neutrophil priming in the process of laser irradiation: this means that a cell after irradiation can show many fold higher response to a stimuli resulting in greater activation degree and higher NO production [138]. He-Ne laser irradiation can activate T-lymphocytes. This phenomenon in parallel with increased NO synthesis can play an important role in the control of inflammation [139, 140]. Thus, it becomes clear that visible light radiation can be effectively applied as a therapeutic means if hemoprotein nitrosyl complexes are present in the system.
The authors are thankful for financial support from the Russian Foundation for Basic Research, grants 06-04-49296 and 05-04-49765.
REFERENCES
1.Furchgott, R. F., and Zawadzki, J. V. (1980)
Blood Vessels, 17, 151.
2.Furchgott, R. F. (1988) in Vasodilatation,
Vascular Smooth Muscle, Peptides and Endothelium (Vanhoutte, P. M.,
ed.) Raven Press, New York, pp. 401-414.
3.Ignarro, L., Lippton, H., Edwards, J. C., Baricos,
W. H., Hyman, A. L., Kadowitz, P. J., and Gruetter, C. A. (1981) J.
Pharmacol. Exp. Ther., 218, 739-749.
4.Wink, D. A., and Mitchell, J. B. (1998) Free
Rad. Biol. Med., 25, 434-456.
5.Zweier, J. L., Wang, P., Samouilov, A., and
Kuppusamy, P. (1995) Nat. Med., 1, 1103-1104.
6.Zweier, J. L., Samouilov, A., and Kuppusamy, P.
(1999) Biochim. Biophys. Acta, 1411, 250-262.
7.Kozlov, A. V., Staniek, K., and Nohl, H. (1999)
FEBS Lett., 454, 127-130.
8.Reutov, V. P., and Sorokina, E. G. (1998)
Biochemistry (Moscow), 63, 874-884.
9.Reutov, V. P., Azhipa, Ya. I., and Kayushin, L. P.
(1983) Izvestiya Akad. Nauk SSSR, Ser. Biol., 3,
408-418.
10.Bredt, D. S., Hwang, P. M., and Lowenstein, C.
(1991) Nature, 351, 714-718.
11.Stuehr, D. J., and Ikeda, S. M. (1992) J.
Biol. Chem., 267, 20547-20550.
12.Leone, A. M., Palmer, R. M., and Knowles, R. J.
(1991) J. Biol. Chem., 266, 23790-23795.
13.Gross, C. S., Jaffe, E. A., and Levi, R. (1991)
Biochem. Biophys. Res. Commun., 178, 823-829.
14.Nathan, C. (1992) FASEB J., 6,
3051-3064.
15.Vincent, S. R. (1994) Progr. Neurobiol.,
42, 129-160.
16.Kelm, M., Feelish, M., and Deussen, A. (1991)
Cardiovasc. Res., 25, 831-836.
17.Kelm, M., Feelish, M., and Spahr, R. (1988)
Biochem. Biophys. Res. Commun., 154, 236-244.
18.Beckman, J. S., Beckman, T. W., Chen, J., and
Marshall, P. A. (1990) Proc. Natl. Acad. Sci. USA, 87,
1620-1624.
19.Beckman, J. S. (1991) J. Dev. Physiol.,
15, 53-59.
20.Hogg, N., Kalyanaraman, B., Joseph, J., Struck,
A., and Parthasarathy, S. (1993) FEBS Lett., 334,
170-174.
21.Hogg, N., and Kalyanaraman, B. (1999) Biochim.
Biophys. Acta, 1411, 378-384.
22.Goodwin, D. C., Gunther, M. R., Hsi, L. C.,
Crews, B. C., Eling, T. E., Mason, R. P., and Marnett, L. J. (1998)
J. Biol. Chem., 273, 8903-8909.
23.Gorbunov, N. V., Osipov, A. N., Day, B. W.,
Betriz, Z., Kagan, V. E., and Elsayed, N. M. (1995)
Biochemistry, 34, 6689-6699.
24.Clancy, R. M., Levartovsky, D.,
Leszcynska-Piziak, J., Yegudin, J., and Abramson, S. B. (1994) Proc.
Natl. Acad. Sci. USA, 91, 3680-3684.
25.Stamler, J. S., Jaraki, O., Osborne, J., Simon,
D. I., Keaney, J., Vita, J., Singel, D., Valery, R., and Loscalzo, J.
(1992) Proc. Natl. Acad. Sci. USA, 89, 7674-7677.
26.Dicks, A. P., Swift, H. R., Williams, D. L. H.,
Butler, A. R., AlSadoni, H. H., and Cox, B. G. (1996) J. Chem. Soc.
Perkin Trans., 24, 481-487.
27.Vanin, A. F., Malenkova, I. V., and Serezhenkov,
V. A. (1997) NITRIC OXIDE: Biol. Chem., 1, 191-203.
28.Arnelle, D. R., and Stamler, J. S. (1995)
Arch. Biochem. Biophys., 318, 279-285.
29.Vanin, A. F. (1972) Candidate's dissertation
[in Russian], Institute of Chemical Physics, Academy of Sciences of
the USSR, Moscow.
30.Khrapova, N. V., Malenkova, I. V., and Vanin, A.
F. (1995) Biofizika, 40, 117-121.
31.Butler, A. R., Megson, I. L., and Wright, P. G.
(1998) Biochim. Biophys. Acta, 1425, 168-176.
32.Kelm, M., and Schrader, J. (1990) Circ.
Res., 66, 1561-1575.
33.Vanin, A. F., Mordvintcev, P. I., Hauschildt, S.,
and Mulsch, A. (1993) Biochim. Biophys. Acta, 1177,
37-42.
34.Vanin, A. F. (1991) FEBS Lett.,
289, 1-3.
35.Mulsch, A., Mordvintcev, P. I., Vanin, A. F., and
Busse, R. (1991) FEBS Lett., 294, 252-256.
36.Gaston, B., Reilly, J., and Drazen, J. M. (1993)
Proc. Natl. Acad. Sci. USA, 90, 10957-10961.
37.Bonaventura, J. L., Bonaventura, C. J., and
Stamler, J. S. (1996) Nature, 380, 221-226.
38.Stamler, J. S., Jia, L., Eu, J. P., McMahon, T.
J., Demchenko, I. T., Bonaventura, J., Gernert, K., and Piantadosi, C.
A. (1997) Science, 276, 2034-2037.
39.Kumura, E., Yoshimine, T., Tanaka, S., Hayakawa,
T., Shiga, T., and Kosaka, H. (1994) Neurosci. Lett.,
177, 165-167.
40.Symons, M. C., Rowland, I. J., Deighton, N.,
Shorrock, K., and West, K. P. (1994) Free Rad. Res., 21,
197-202.
41.Hall, D. M., Buettner, G. R., Matthes, R. D., and
Gisolfi, C. V. (1994) J. Appl. Physiol., 77, 548-553.
42.Stamler, J. S., Singel, D. J., and Loscalso, J.
(1992) Science, 258, 1898-1902.
43.Yoshida, K., Kasama, K., Kitabatake, M., and
Imai, M. (1980) Int. Arch. Occup. Environ. Health, 46,
71-77.
44.Kelm, M., and Yoshida, K. (1996) in Methods in
NO Research (Feelish, M., Stamler, J. S., and England, L., eds.)
Wiley & Sons Ltd, London.
45.Gow, A. J., Luchsinger, B. P., Pawloski, J. R.,
Singel, D. J., and Stamler, J. S. (1999) Proc. Natl. Acad. Sci.
USA, 96, 9027-9032.
46.Gow, A. J., and Stamler, J. S. (1998)
Nature, 391, 169-173.
47.Stone, J. R., Sands, R. H., Dunham, W. R., and
Marletta, M. A. (1995) Biochem. Biophys. Res. Commun.,
207, 572-575.
48.Zhao, X. J., Sampath, V., and Caughey, W. S.
(1994) Biochem. Biophys. Res. Commun., 204, 537-543.
49.Brown, G. C., and Cooper, C. E. (1994) FEBS
Lett., 356, 295-298.
50.Fujimori, H., and Pan Hou, H. (1991) Brain
Res., 554, 355-357.
51.Mannick, J. B., Hausladen, A., Liu, L., Hess, D.
T., Zeng, M., Miao, Q. X., Kane, L. S., Gow, A. J., and Stamler, J. S.
(1999) Science, 284, 651-654.
52.Rossig, L., Fichtlscherer, B., Breitschopf, K.,
Haendeler, J., Zeiher, A. M., Mulsch, A., and Dimmeler, S. (1999) J.
Biol. Chem., 274, 6823-6826.
53.Culotta, V. C., Klomp, L. W., Strain, J.,
Casareno, R. L., Krems, B., and Gitlin, J. D. (1997) J. Biol.
Chem., 272, 23469-23472.
54.Pufhal, R. A., Singer, C. P., Peariso, K. L.,
Lin, S.-J., Schmidt, P. J., Fahrni, C. J., Culotta, V., Penner-Hahn, J.
E., and O'Halloran, T. V. (1997) Science, 278,
853-856.
55.Liu, S., Fabisiak, J., Tyurin, V., Borisenko, G.,
Pitt, B., Lazo, J., and Kagan, V. (2000) The Toxicologist,
54, 167.
56.Kawai, K., Liu, S.-X., Tyurin, V. A., Tyurina, Y.
Y., Borisenko, G. G., Fabisiak, J. P., Pitt, B. R., and Kagan, V. E.
(2000) Chem. Res. Toxicol., 13, 1275-1286.
57.Liu, S.-X., Fabisiak, J. P., Tyurin, V. A.,
Borisenko, G. G., Pitt, B. R., Lazo, J. S., and Kagan, V. E. (2000)
Chem. Res. Toxicol., 13, 922-931.
58.Zhang, F., White, J. G., and Iadecola, C. (1994)
J. Cerebr. Blood Flow Metab., 14, 217-226.
59.Frostell, C., Fratacci, M., and Wain, J. C.
(1991) Circulation, 83, 2038-2047.
60.Koshland, D. E., and Culotta, E. (1992)
Science, 258, 1862-1865.
61.Brown, G. C. (1997) Mol. Cell. Biochem.,
174, 189-192.
62.Boelens, R., Rademaker, H., Pel, R., and Wever,
R. (1982) Biochim. Biophys. Acta, 679, 84-94.
63.Hill, R., Olson, J. S., and Palmer, G. (1979)
J. Biol. Chem., 254, 12110-12120.
64.Kon, H. J. (1968) J. Biol. Chem.,
242, 485.
65.Taylor, T. G., and Sharma, V. S. (1992)
Biochemistry, 31, 2847-2849.
66.Sharma, V. S., Taylor, T. G., and Gardiner, R.
(1987) Biochemistry, 26, 3837-3843.
67.Ascenzi, P., Coletta, M., Santucci, R., Polizio,
F., and Desideri, A. (1994) J. Inorg. Biochem., 53,
273-280.
68.Hori, H., Ikeda-Saito, M., Lang, G., and
Yonetani, T. (1990) J. Biol. Chem., 265, 15028-15033.
69.Koppenol, W. H., Moreno, J. J., Pryor, W. A.,
Ischiropoulos, H., and Beckman, J. S. (1992) Chem. Res.
Toxicol., 5, 834-842.
70.Nagai, K., Hori, H., Yoshida, S., Sakamoto, H.,
and Morimoto, H. (1978) Biochim. Biophys. Acta, 532,
17-28.
71.Yonetani, T., Tsuneshige, A., Zhou, Y., and Chen,
X. (1998) J. Biol. Chem., 273, 20323-20333.
72.Cassoly, R., and Gibson, Q. H. (1975) J. Mol.
Biol., 91, 301-313.
73.Moore, E. G., and Gibson, Q. H. (1976) J.
Biol. Chem., 251, 2788-2794.
74.Pepke-Zaba, J., Higenbottam, T. W., Dinh-Xuan, A.
T., Stone, D., and Wallwork, J. (1991) Lancet, 338,
1173-1174.
75.Doyle, M. P., Pickering, R. A., and Cook, B. R.
(1983) J. Inorg. Biochem., 19, 329-338.
76.Pietraforte, D., Mallozzi, C., Scorza, G., and
Minetti, M. (1995) Biochemistry, 34, 7177-7185.
77.Kosaka, H., and Seiyama, A. (1996) Biochem.
Biophys. Res. Commun., 218, 749-752.
78.Szabo, A., and Peruts, M. F. (1976)
Biochemistry, 15, 4427-4428.
79.Maxwell, J. C., and Caughey, W. S. (1976)
Biochemistry, 15, 388-396.
80.Vincent, J. S., Kon, H., and Levin, I. W. (1987)
Biochemistry, 26, 2312-2314.
81.Vincent, J. S., and Levin, I. W. (1988)
Biochemistry, 27, 3438-3446.
82.Spooner, P. J., and Watts, A. (1991)
Biochemistry, 30, 3380-3385.
83.Choi, S., and Swanson, J. M. (1995) Biophys.
Chem., 54, 271-278.
84.Spooner, P. J., and Watts, A. (1992)
Biochemistry, 31, 10129-10138.
85.Osipov, A. N., Stepanov, G. O., Vladimirov, Y.
A., Kozlov, A. V., and Kagan, V. E. (2006) Biochemistry
(Moscow), 71, 1128-1132.
86.Vlasova, I. I., Tyurin, V. A., Kapralov, A. A.,
Kurnikov, I. V., Osipov, A. N., Potapovich, M. V., Stoyanovsky, D. A.,
and Kagan, V. E. (2006) J. Biol. Chem., 281,
14554-14562.
87.Hoshino, M., Ozawa, K., Seki, H., and Ford, P. C.
(1993) J. Am. Chem. Soc., 115, 9568-9575.
88.Rose, E. J., and Hoffman, B. M. (1983) J. Am.
Chem. Soc., 105, 2866-2873.
89.Sharpe, M. A., and Cooper, C. E. (1998)
Biochem. J., 332, 9-19.
90.Orii, Y., and Shimada, H. (1978) J.
Biochem. (Tokyo), 84, 1542-1552.
91.Holdane, S., and Lorrain, J. (1896)
Physiol. (London), 20, 497-520.
92.Keilin, D., and Hartree, E. F. (1955) Biochem.
J., 16, 153-171.
93.Gibson, Q. H., and Ainsworth, S. (1957)
Nature, 180, 1416-1417.
94.Vladimirov, Yu. A. (1957) Ph. D. Dissertation [in
Russian], Moscow State University, Moscow.
95.Antonini, E., and Brunori, M. (1971)
Hemoglobin and Myoglobin in Their Reactions with Ligands,
North-Holland Publisher Corp., Amsterdam.
96.Saffran, W. A., and Gibson, Q. H. (1977) J.
Biol. Chem., 252, 7955-7962.
97.Hofrichter, J., Sommer, J. H., Henry, E. R., and
Eaton, W. (1983) Proc. Natl. Acad. Sci. USA, 80,
2235-2241.
98.Petrich, J. W., Martin, J. L., Houde, D., and
Poyart, C. (1987) Biochemistry, 26, 7914-7920.
99.Petrich, J. W., Poyart, C., and Martin, J. L.
(1988) Biochemistry, 49, 4049-4060.
100.Zhu, L., Sage, J. T., and Champion, P. M.
(1994) Science, 266, 629-632.
101.Tarasiev, M. Yu., and Ryl'kov, V. V. (1991)
Biokhimiya, 56, 273-279.
102.Vladimirov, Y. A., Borisenko, G. G., Kazarinov,
K. D., and Osipov, A. N. (2000) J. Photochem. Photobiol.,
59, 115-122.
103.Venturini, C. M., Palmer, R. M., and Moncada,
S. (1993) J. Pharmacol. Exp. Ther., 266, 1497-1500.
104.Mittermayr, R., Osipov, A., Piskernik, C.,
Haindl, S., Dungel, P., Weber, C., Vladimirov, Y. A., Redl, H., and
Kozlov, A. V. (2007) Mol. Med., 13, 22-29.
105.Poderoso, J. J., Lisdero, C., Schopfer, F.,
Riobo, N., Carreras, M. C., Cadenas, E., and Boveris, A. (1999) J.
Biol. Chem., 274, 37709-37716.
106.Sarti, P., Giuffre, A., Forte, E.,
Mastronicola, D., Barone, M. C., and Brunori, M. (2000) Biochem.
Biophys. Res. Commun., 274, 183-187.
107.Clementi, E., Brown, G. C., Feelisch, M., and
Moncada, S. (1998) Proc. Natl. Acad. Sci. USA, 95,
7631-7636.
108.Furchgott, R. F. (1955) Pharmacol. Rev.,
7, 183-265.
109.Furchgott, R. F., Martin, W., Cherry, P. D.,
Jothianandan, D., and Villani, G. (1985) in Vascular Neuroeffector
Mechanisms (Bevan, J. A., and Godfraind, T., eds.) Elsevier,
Amsterdam, pp. 105-114.
110.Chaudhry, H., Lynch, M., Schomacker, K.,
Birngruber, R., Gregory, K., and Kochevar, I. (1993) Photochem.
Photobiol., 58, 661-669.
111.Furchgott, R. F., and Jothianandan, D. (1991)
Blood Vessels, 28, 52-61.
112.Williams, D. L. H. (1985) Chem. Soc.
Rev., 14, 171-196.
113.Borisenko, G. G., Osipov, A. N., Kazarinov, K.
D., and Vladimirov, Yu. A. (1997) Biochemistry (Moscow),
62, 659-664.
114.Passarella, S., Casamassima, F., Molinari, S.,
Pastore, D., Quagliariello, E., Catalano, I. M., and Cingolani, A.
(1984) FEBS Lett., 175, 95-99.
115.Karu, T., Pyatibrat, L., and Kalendo, G. (1995)
J. Photochem. Photobiol. B, 27, 219-223.
116.Karu, T. (1999) J. Photochem. Photobiol.
B, 49, 1-17.
117.Poderoso, J. J., Carreras, M. C., Lisdero, C.,
Riobo, N., Schopfer, F., and Boveris, A. (1996) Arch. Biochem.
Biophys., 328, 8592.
118.Culotta, E., and Koshland, D. E. (1992)
Science, 258, 1862-1865.
119.Brown, G. C., Foxwell, N., and Moncada, S.
(1998) FEBS Lett., 439, 321-324.
120.Ghafourifar, P., and Richter, C. (1997) FEBS
Lett., 418, 291-296.
121.Tatoyan, A., and Giulivi, C. (1998) J. Biol.
Chem., 273, 11044-11048.
122.Giulivi, C. (1998) Biochem. J.,
32, 673-679.
123.Ghafourifar, P., Schenk, U., Klein, S. D., and
Richter, C. (1999) J. Biol. Chem., 274, 31185-31188.
124.Poderoso, J. J., Carreras, M. C., Schopfer, F.,
Lisdero, C. L., Riobo, N. A., Giulivi, C., Boveris, A. D., Boveris, A.,
and Cadenas, E. (1999) Free Rad. Biol. Med., 7/8,
925-935.
125.Borutaite, V., Morkuniene, R., and Brown, G. C.
(2000) FEBS Lett., 467, 155-159.
126.Hortelano, S., Dallaporta, B., Zamzami, N.,
Hirsch, T., Susin, S. A., Marzo, I., Bosca, L., and Kroemer, G. (1997)
FEBS Lett., 410, 373-377.
127.Torres, J., Darley-Usmar, V. M., and Wilson, M.
T. (1995) Biochem. J., 312, 169-173.
128.Brunori, M., Giuffre, A., Sarti, P., Stubauer,
G., and Wilson, M. T. (1999) Cell. Mol. Life Sci., 56,
549-557.
129.Stevens, T. H., Brudwig, G. W., Bocian, D. F.,
and Chan, S. I. (1979) Proc. Natl. Acad. Sci. USA, 76,
3320-3325.
130.Torres, J., Cooper, C. E., and Wilson, M. T.
(1998) J. Biol. Chem., 273, 8756-8766.
131.Giuffre, A., Sarti, P., D'Itri, E., Buse, G.,
Soulimane, T., and Brunori, M. (1996) J. Biol. Chem.,
271, 33404-33408.
132.Sharpe, M. A., and Cooper, C. E. (1998) J.
Biol. Chem., 273, 30961-30972.
133.Kon, H. (1969) Biochem. Biophys. Res.
Commun., 35, 423-427.
134.Yoshimura, T., and Suzuki, S. (1988) Inorg.
Chim. Acta, 152, 241-249.
135.Kozhura, V. L., Dvoretsky, S. B., and
Novoderzhkina, I. S. (1993) Anesteziol. Reanimatol., 4,
43-48.
136.Frasier, O. H., Cooly, D. A., and Kadipasaoglu,
K. A. (1995) Circulation, 92, 1158-1165.
137.Karu, T. J. (1989) Advances in Science and
Technology [in Russian], Vol. 4, VINITI, Moscow, pp. 44-84.
138.Klebanov, G. I., Teselkin, Y. O., Babenkova, I.
V., Bashkujeva, T. Y., Chichuk, T. V., and Vladimirov, Y. A. (1998)
Gen. Physiol. Biophys., 17, 365-376.
139.Delian, M., Abels, C., Kuhle, G. E., and Goets,
A. E. (1995) Br. J. Cancer, 72, 1125-1130.
140.Karu, T. (1992) Laser Ther., 4,
5-24.