REVIEW: Methods for Selection of Aptamers to Protein Targets
A. V. Kulbachinskiy
Institute of Molecular Genetics, Russian Academy of Sciences, pl. Kurchatova 2, 123182 Moscow, Russia; E-mail: akulb@img.ras.ru
Received February 9, 2006
Aptamers are synthetic single-stranded RNA or DNA molecules capable of specific binding to other target molecules. In this review, the main aptamer properties are considered and methods for selection of aptamers against various protein targets are described. Special attention is given to the methods for directed selection of aptamers, which allow one to obtain ligands with specified properties.
KEY WORDS: aptamers, aptazymes, SELEX, site-directed selectionDOI: 10.1134/S000629790713007X
Abbreviations: NES) nuclear export signal; nt) nucleotide; PCR) polymerase chain reaction; SELEX) systematic evolution of ligands by exponential enrichment; ssDNA) single-stranded DNA molecule.
FIELDS OF APPLICATION OF APTAMERS
For a long time nucleic acids were considered mainly as linear carriers of information, whereas most cell functions were ascribed to protein molecules possessing complex three-dimensional structure. However, more examples gradually appeared which showed that single-stranded molecules of nucleic acids can carry out numerous intracellular functions. It was found that single-stranded RNA molecules are able to form very different three-dimensional structures, which allows them to recognize specifically various molecular targets and even perform catalysis. As an example, one can mention tRNA and rRNA molecules playing key roles in protein synthesis. Analysis of transcription, splicing, and translation mechanisms has shown that specific interactions of proteins with certain RNA sites are of tremendous importance in the regulation of gene expression. It became clear that the immense combinatorial variety of nucleic acids and their ability to form diverse secondary and tertiary structures make possible the directed search for nucleic acid sequences exhibiting certain properties. This resulted in elaboration of methods for nucleic acid evolution in vitro, which can be used to obtain RNA or ssDNA molecules capable of recognition of other molecules or exhibiting catalytic activity [1, 2].
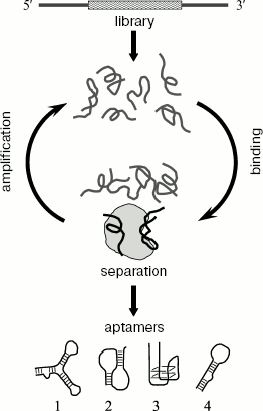
Synthetic RNA or ssDNA molecules, capable of specific binding to other target molecules, were called aptamers (derived from the Latin aptus - suitable, adjusted) [1]. Aptamers are obtained in vitro by directed selection from combinatorial oligonucleotide libraries. The method of aptamer selection was designated as SELEX (from the systematic evolution of ligands by exponential enrichment) [2]. The principle of this method is shown in Fig. 1. The experiment consists of several rounds of selection of sequences that bind to a target molecule. Each round includes three main stages: (I) the oligonucleotide library is incubated with the target molecule; (II) the oligonucleotide complexes with the target are separated from non-bound oligonucleotides; (III) the bound sequences are amplified by PCR. As a result, gradual enrichment of the oligonucleotide library with sequences exhibiting the increased affinity to the target molecule occurs. Numerous experiments on selection in vitro have shown that aptamers can be obtained to practically any targets including proteins, peptides, nucleic acids, polysaccharides, small organic molecules (amino acids, nucleotides, and other metabolites), virus particles, whole cells, and even tissues [3].
Aptamers are high-affinity and high specificity ligands [3, 4]. Aptamers to proteins often bind in functionally important parts of molecule and are inhibitors of their target proteins [5]. In most of their characteristics, aptamers are as good as monoclonal antibodies and can be used instead of the latter in many experiments [6]. At the same time, aptamers exhibit much higher stability. The method of aptamer selection is quite simple and makes possible obtaining new ligands during just one or two months. Recently methods of automatic aptamer selection have been developed which allow simultaneous selection of aptamers to a great number of different targets [7, 8]. Owing to such set of properties, aptamers are widely used in very different (fundamental and applied) fields of investigations. Some basic fields of aptamer usage are given below.Fig. 1. A general scheme for obtaining aptamers. The initial library of oligonucleotides contains a random region (marked by wavy hatching), surrounded by fixed sequences (shown in gray). Each selection round consists of three stages: (I) incubation of a random library, containing a large number of sequences capable of formation of different structures, with protein target; (II) separation of bound and unbound oligonucleotides; (III) elution and amplification of protein-bound oligonucleotides. The enriched library is used for next round of selection. After completion of selection, the enriched library is cloned and sequences of individual aptamers are determined. Different variants of secondary structure formed by aptamers are shown at the bottom: hairpins (1 and 4), pseudoknot (2), G-quartette (3).
1. Aptamers are used for investigation of mechanisms of interaction of proteins with nucleic acids. In the case of proteins, in vivo recognizing specific sequences of nucleic acids, SELEX allows one to reveal natural sequences of RNA or DNA recognized by the protein [2, 9-11]. Analysis of three-dimensional structure of aptamer-protein complexes is used for structural investigation of the RNA- and DNA-binding sites of target proteins [12-16].
2. Highly efficient and specific inhibitors of target proteins can be obtained on the basis of aptamers [3, 17-19]. Similar inhibitors can be used both in fundamental research (in particular, in functional studies of target proteins) and for creation of new drugs. For example, in this case different growth factors, hormones, enzymes, cell surface receptors, toxins, and proteins of viruses and pathogenic microorganisms can serve as targets [5, 20-24]. Several drugs on the basis of aptamers are presently under clinical tests, and the first drug has already appeared on the pharmaceutical market [22]. In addition to direct application of aptamers as specific inhibitors, they can be used in search for new inhibitors of different proteins. For this purpose, one can perform a directed search for molecules, competing with the aptamer-inhibitor for binding to the protein target and thus interacting with the same site of the protein as the original aptamer [25].
3. Aptamers are widely used for detection of different target molecules (in particular, for diagnostic purposes). It was shown that aptamers could successfully replace antibodies in methods of ELISA, fluorescent hybridization in situ, Western blotting, etc. [6]. Aptamers are used for measuring concentrations of various metabolites and protein factors, for detection of toxins, revealing specific types of cells and tissues, and cells of pathogenic microorganisms [5, 26-29]. Besides, aptamers like antibodies can be used for affinity purification and identification of target proteins [5, 30, 31].
The most promising for the purposes of diagnosis is the design of microchips incorporating a great number of aptamers that can be used for simultaneous analysis of many target molecules [32-34]. At the present time, there are microchips that make it possible to analyze some growth factors in human blood [35]. It is supposed to design in future microchips for analysis of expression of a great number of different human cell proteins, which should be of huge importance for development of methods for diagnosis of different diseases as well as for fundamental investigations [36-38].
Different experiments on aptamer selection as well as the possibility of practical application of aptamers in different fields of biology and medicine have been considered in detail in several recent reviews [3, 5, 20, 22, 28, 39]. Besides, the description of technology for obtaining aptamers and different modifications of the SELEX method can be found on the site of the SomaLogic Company (www.somalogic.com) as well as in descriptions of patents, references to which are given on this site. This review presents a brief consideration of basic methods for obtaining aptamers to different protein targets; most attention is paid to the possibilities of directed selection of aptamers exhibiting predicted functional characteristics.
METHODS FOR APTAMER SELECTION
General Scheme of Selection
For aptamer selection combinatorial oligonucleotide libraries are used which contain a central region with a random sequence, surrounded by two regions with fixed sequences (Fig. 1). Fixed sequences are necessary for library amplification during selection. The ssDNA library is obtained by standard methods of single-stranded oligonucleotide synthesis, in the case of the random region synthesis, the mixture of all four deoxyribonucleotide derivatives are added to reaction mixture. To obtain an RNA library, the promoter sequence for RNA polymerase of bacteriophage T7 is introduced into the 5´ terminal region of the ssDNA library, dsDNA is obtained by polymerase chain reaction (PCR), and in vitro transcription is carried out.
The key stage of aptamer selection is separation of complexes of oligonucleotides with the target molecule from unbound oligonucleotides. Several basic methods are used for this purpose in experiments on aptamer selection.
1. The protein target is immobilized on an affinity sorbent; not bound oligonucleotides are removed by washing with a buffer solution, and bound ones are used for further selection (cf. [17, 40]).
2. Protein complexes with oligonucleotides are sorbed on nitrocellulose filters (nitrocellulose binds proteins nonspecifically, but it does not interact with nucleic acids) [2, 10].
3. Protein complexes with oligonucleotides are separated from free oligonucleotides by gel electrophoresis under non-denaturing conditions (cf. [41]).
4. The oligonucleotide mixture with target protein is separated by capillary electrophoresis (without gels) [42-44].
5. When different macromolecular objects (cells, viruses) are used as targets, complexes with bound oligonucleotides are separated from non-bound sequences by centrifugation [29, 45, 46].
Sometimes during one SELEX procedure several selection methods are interchanged, which increases the selection efficiency and avoids selection of sequences interacting with the sorbent used for selection, or with contaminating proteins in the target protein preparation. In addition, in order to inhibit selection of sequences specifically binding to the sorbent, the library is incubated before each round with the sorbent in the absence of the target protein, which allows one to exclude these sequences from selection.
After each round of selection, sequences bound to the target molecule are amplified using primers corresponding to fixed regions of the library. In the case of RNA aptamers, this stage is preceded by reverse transcription. The amplified dsDNA is used for obtaining of an enriched library of single-stranded oligonucleotides, which is then used in the next selection round. In the case of the RNA aptamer selection, the enriched RNA library is obtained using in vitro transcription. ssDNA can be obtained by several methods: (i) a biotin residue is introduced into one of primers used for amplification, and DNA strands are separated under denaturing conditions on a column with streptavidin (cf. [47]); (ii) ssDNA is obtained in asymmetric PCR under a high excess of one of the primers [48]; (iii) some chemical groups (like several biotin residues, fluorescent dyes, etc.) are introduced to 5´ end of one of primers, which makes possible separation of two DNA strands by size using electrophoresis under denaturing conditions [31]; (iv) the phosphate group is introduced into 5´ end of one of primers and dsDNA obtained by amplification is treated with the phage lambda endonuclease that cleaves the phosphorylated strand of DNA [30].
The selection results in gradual enrichment of the library with sequences exhibiting the increased affinity to the target. Usually after successful selection, the affinity of the library to the target increases by several orders of magnitude. On average, from 5 to 15 rounds of selection is sufficient to obtain aptamers. However in some cases a significant enrichment of the library with sequences specific of the target protein becomes noticeable already after the first rounds of selection [30, 42, 49]. After cessation of affinity increase, the enriched library is cloned and sequences of individual aptamers are determined.
Properties of aptamers obtained during experiment are mainly defined by the specific selection conditions. In particular, selection conditions influence the affinity, specificity, and secondary structure of aptamers, define the dependence of aptamer binding on the presence of different ions and other low-molecular-weight compounds, etc. So, aptamers with specified properties can be obtained by changing the structure of oligonucleotide library, conditions of its incubation with the target protein, and the method of separation of bound and non-bound oligonucleotides.
Libraries Used for Selection
Classical libraries. Libraries containing a random region of 20-60 nt (in some cases from 8 to 220 nt) in length are used in most experiments on aptamer selection. It was supposed that libraries with a longer random region may stimulate more efficient aptamer selection, because longer sequences are able to form a larger variety of different structures [50]. At the same time the amount of DNA or RNA used in selection is usually 10-8-10-10 M or 1014-1016 molecules which corresponds to the total number of different sequence variants for oligonucleotides of 25 nucleotides in length (425 ~ 1015). So, libraries with the random region longer than 25 nt are much underrepresented and contain the same number of different sequence variants as shorter libraries. For example, aptamers of identical structure were obtained in the case of selection of aptamers to reverse transcriptase of HIV-1 virus, when RNA libraries with a random region of 32, 70, and 80 nt were used [19, 51]. Moreover, it was shown in an experiment on selection of aptamers to isoleucine that if the random region is longer than 70 nt, the efficiency of aptamer selection decreases [52]. Both RNA and ssDNA libraries are used in different SELEX experiments. Analysis of a great number of different aptamers shows that the ssDNA aptamers do not differ by affinity and specificity from RNA ligands [53]. The advantage of RNA aptamers is that they can be expressed inside cells, which may be of great importance in experiments in vivo. At the same time, DNA aptamers exhibit higher stability in a broad range of conditions. Owing to this, during recent years DNA aptamers have become more and more widespread [53].
Structurally constrained libraries. In some experiments, libraries with assigned secondary structure are used in selection. In this case, the random region is placed between fixed sequences able to form a certain secondary structure (hairpin, G-quartette, pseudoknot). This method is used when it is known what secondary structure of RNA or DNA is recognized by the target protein or for increase in general stability of selected aptamers (cf. [2, 54-57]).
Libraries on the basis of a known sequence. Libraries of this type contain a random region on the basis of a known sequence (for example, the sequence of an earlier obtained aptamer), but during synthesis of each position of this region a small amount of the other three nucleotides is added (from 1 to 30%). This makes it possible to obtain an oligonucleotide library to a variable extent resembling the initial sequence (so-called doped library). The selection using this library allows one to reveal different variants of the initial sequence able to interact with the target molecule. This approach is used for optimization of binding of earlier obtained sequences as well as for revealing the aptamer nucleotides that are most important for binding [51, 58, 59].
Libraries free of fixed sequences. These libraries are used for minimization of aptamer size (table). In many cases, parts of fixed sequences of an initial library are necessary for formation of aptamer structure, and their removal disturbs binding to the target molecule. To obtain aptamers free of fixed sequences, and as a result, smaller in size, the tailored SELEX method was proposed. In this case, a library with very short fixed regions (4-6 nt) on both sides of the random region is used in selection. After each selection round, adaptor sequences used for amplification of the library and removed before next round [60].
Different modifications of the SELEX method and their application
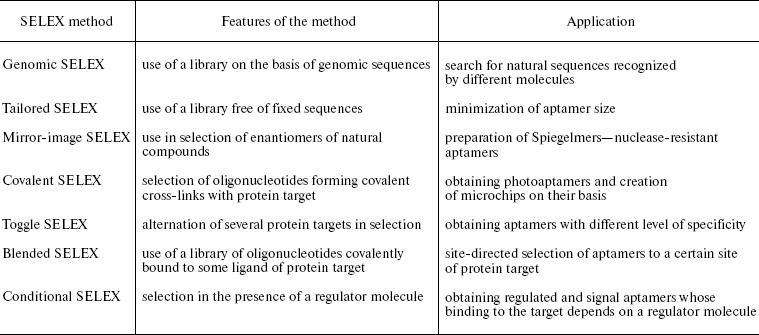
Libraries on the basis of genomic sequences (genomic SELEX). These libraries are used for searching of binding sites of different proteins in genomic DNA and RNA sequences (for example in the case of transcription factors, splicing, and translation regulators) (table) [11, 61]. To create such libraries, genomic DNA is fragmented to sufficiently short fragments (50-500 nt) and the fixed sequences are joined to them. The sequence of the T7 promoter is inserted into one of the fixed regions during analysis of RNA sequences. Further selection of sequences binding to the target protein is carried out by the usual methods.
Use of Modified Nucleotides
Selection in vitro allows the use of oligonucleotides containing chemically modified nucleotides. Modifications are introduced into 2´_position of all nucleotides, positions 5 of pyrimidines, 7 and 8 of purines, etc. (the detailed description of different modifications can be found in reviews [39, 62-64]). There are two alternative approaches to obtaining modified aptamers. In the first approach modified oligonucleotides are used directly during selection. The main restriction in this case is that modification should not significantly influence the ability of the nucleotide to serve as a substrate for RNA or DNA polymerase. In the second approach already obtained aptamers are modified. In this case, the variety of available modifications grows significantly (because there are no restrictions for their compatibility with enzymology of RNA and DNA syntheses), but many modifications may result in lowering the aptamer affinity to their targets. Owing to this, it is necessary to search for modifications not affecting the interaction of the aptamer with the target.
Modifications are introduced to attain the following.
1. Introduction of different modifications significantly enlarges the potential variety of oligonucleotides. It is also assumed that additional functional groups are able to provide more contacts between aptamers and their targets, which should stimulate obtaining of aptamers with higher affinity and specificity towards their targets [63, 65, 66].
2. Many modifications of aptamers (especially in the case of F or NH2-group introduced in 2´-position of ribose) make them nuclease-resistant, which is of great importance when applying aptamers for diagnosis and therapy [6, 22, 63]. One of the ways of obtaining nuclease-resistant aptamers is the use of mirror analogs of natural nucleotides (on the basis of L-ribose or L-deoxyribose). These aptamers were designated as Spiegelmers (from German spiegel - mirror) and the method of their selection was named as mirror-image SELEX (table) [67]. Since RNA and DNA polymerases are not able to carry out polymerization of L-nucleotides, selection in this case is performed by a standard method (using D-nucleotides), but an enantiomer of a natural compound is used as a target (like a peptide consisting of D-amino acids). After determination of primary structure of obtained aptamers, the oligonucleotide L-variant is synthesized which, according to the law of symmetry, is able to interact with natural target (such as L-peptide) [60, 67, 68]. Oligonucleotides, consisting of L-nucleotides, are characterized by very high stability because they are not recognized by natural nucleases.
3. The use in selection of modified oligonucleotides, containing functional groups that can be activated upon irradiation (such as 5-iodo-, 5-bromo-, and 4-thiouridine), makes possible obtaining aptamers capable of forming covalent cross-links with the protein target (so-called photoaptamers) [69, 70]. In this case, in each round of selection oligonucleotide complexes with protein target are irradiated by ultraviolet light and sequences, involved in the cross-link, are selected (for example, by electrophoresis under denaturing conditions). The method of photoaptamer selection was named covalent SELEX (table). The use of photoaptamers significantly enhances specificity of protein target recognition because the cross-link formation requires correct mutual arrangement of the nucleic acid and protein sites involved in the cross-link [37, 71]. The technology of obtaining aptamers capable of establishing covalent cross-links with protein targets is presently used in creation of microchips for analysis of expression of various human proteins [35, 69].
4. Introduction of fluorescent groups into an aptamer is used for analysis of its binding to the protein target [72]. This is most important in aptamer application for detection of various target molecules. In the simplest case, the binding can be detected by an alteration in the aptamer fluorescence, which is caused by changes in the fluorescent group environment upon complex formation. Besides, methods have been developed which allow directed selection of aptamers the fluorescence of which changes upon binding to a target molecule [72-75].
Analysis of Aptamers
Selection is followed by the investigation of characteristics of individual aptamers and their interaction with the target. The following scheme is suggested for a standard experiment.
1. Individual sequences obtained by selection are analyzed. They are combined in several classes by homology to each other. The sequences of each class are analyzed for ability to interact with the protein target. Sequences exhibiting the highest affinity to the target are selected for further investigations.
2. Analysis of conserved motifs of the aptamer sequence is used for prediction of the secondary structure characteristic of each class. At the present time specialized programs have been developed which allow prediction of aptamer secondary structures on the basis of analysis of related sequences within one class of aptamers [76]. The structure predictions are checked by the cleavage of the aptamers by different reagents (chemical compounds and nucleases) specific to single- or double-stranded sites of nucleic acids.
3. Minimal aptamer dimensions sufficient for interaction with the protein target are determined. For this aim, several shortened oligonucleotide variants are synthesized on the basis of predicted secondary structure and their ability to bind to protein is checked. Minimization of aptamer dimensions is especially important in the case of their application for diagnosis and therapy, because shortening of the aptamer length results in increased specificity of its activity and significant decrease in the cost of its production.
4. The structure of the aptamer complex with its protein target is investigated. The use of different footprinting techniques (most often footprinting by hydroxyl radicals and various nucleases) reveals which sites in the aptamer directly interact with the protein. The importance of individual nucleotides for interaction with the target is monitored by analysis of mutant variants of the aptamer. The aptamer contacts with the protein target may be also studied using the covalent cross-linking techniques, which reveals the protein site, and in some cases even concrete amino acid with which the aptamer interacts [77]. These data can be used to build a three-dimensional model of the aptamer-protein complex.
5. Finally, the effect of the aptamer on the properties of the protein target is studied. As a rule, aptamers inhibit activities of their targets. However, in some cases aptamers are able to stimulate protein activity (cf. [78, 79]).
APTAMER CHARACTERISTICS
Structural Features of Aptamers
A distinguishing feature of aptamers is their ability to form pronounced secondary structure. It has been shown in many cases that unpaired sites in the oligonucleotide are most important for the specific interaction with targets, whereas sites with stable secondary structure are necessary to maintain the proper spatial arrangement of the recognition elements [80, 81]. This is confirmed by the fact that many aptamer sites are non-structured in the free state (like loops in hairpins) and acquire a stable conformation only after binding to the target [12, 80, 82].
Usually one or two structural motifs comprise the basis of an aptamer. Most frequent are the following structural elements (Fig. 1).
1. Hairpins of different structure. It is one of the most widespread secondary structure motifs occurring both in RNA and DNA aptamers.
2. Pseudoknots. The pseudoknot structure is formed as a result of complementary interactions of the sequence located to the right or to the left of a hairpin with the sequence of the hairpin loop. Pseudoknots are most characteristic of RNA aptamers, but they occur in DNA sequences as well [19, 81, 83].
3. Four-stranded structures (quadruplexes). As a rule, the quadruplex cross section is formed by four guanine nucleotides (G-quartet). Each guanine base in the G-quartet forms hydrogen bonds with two adjacent bases. Usually in a quadruplex there are 2 or 3 G-quartets arranged in succession. Such structures exhibit very high stability compared to simpler structural motifs. The quadruplex is additionally stabilized in the presence of K+ ions coordinated between planes of two neighboring G-quartets [84]. The G-quartet structures appeared to be the most typical of DNA aptamers, although in some cases they were also found in RNA ligands [17, 40, 77, 85-88]. It is supposed that in all cases the structural motif of the G-quartet is the architectural basis of the aptamer, whereas nucleotides in variable loops between quadruplex strands are involved in protein recognition.
Affinity and Specificity
In most experiments selection produces aptamers that interact with their targets with very high affinity. Proteins are the best targets for aptamer selection, and dissociation constants (Kd) of aptamer complexes with protein targets usually lie in nanomolar and sub-nanomolar ranges (10-11-10-9 M). In the case of various low-molecular-weight targets Kd is usually 10-6-10-7 M [3]. The higher affinity of aptamers to protein targets is probably due to a larger area of aptamer contact with the target molecule in such complexes [4].
In addition to high affinity, aptamers also exhibit high specificity to the target molecule. It was supposed that selection for the high affinity to the target automatically results in identification of highly specific ligands [4]. This is so because the ligands with highest affinity as a rule have a larger area for interactions with the target and form with the latter very close “complementary” contacts. As a result, even minimal alterations in the target surface (in the case of closely related proteins) may significantly weaken interactions with the aptamer.
At the same time, the ability of aptamers to distinguish similar proteins in each particular case evidently depends on the site in the protein target in which binding takes place. If an aptamer binds in a variable region of a protein molecule, the interaction will be more specific compared to binding with conserved region, because in the first case the recognized epitope will differ more even in related proteins. Accordingly, the level of protein homology, which defines the limit of aptamer specificity, may be different in different cases. Thus, aptamers to Rev protein of HIV-1 virus also recognize Rev protein of HIV-2, although these proteins exhibit only 65% identity [89]. At the same time, aptamers to protein kinase C do not interact with the closely related isoform of the enzyme that differs by just 4% [90]. In the case of aptamers to thrombin [91], hepatitis C virus replicase [54], and reverse transcriptase of HIV-1 virus [92] point mutations in the target protein were obtained which resulted in a significant deterioration of aptamer binding to the target (Kd of complexes increased more than a hundred-fold). So, it can be concluded that aptamers are able to distinguish proteins that differ by a single amino acid substitution.
The aptamer specificity to a protein target can be regulated during selection. Thus, if it is required to obtain an aptamer of a very high specificity, it is necessary to select during SELEX only sequences that bind the protein target but do not interact with another closely related protein. On the contrary, if it is necessary to obtain an aptamer able to recognize several related proteins (such as proteins orthologs from different organisms) then sequences are selected which are able to bind to several protein targets (this procedure was named toggle-SELEX) (table) [93].
Epitopes Recognized by Aptamers
Aptamers to proteins, like antibodies, recognize specific sites on the target surface, which are called epitopes. It is assumed that most aptamers recognize structured epitopes on protein targets [3, 4, 37]. Since the full-size proteins in native state are usually used as targets in aptamer selection, the epitope recognized by aptamers may contain several protein sites brought together in the three-dimensional structure. This distinguishes aptamers from antibodies that are selected within an organism for binding to relatively short fragments of processed protein target. Nevertheless, there are examples of aptamers able to recognize short peptides [94] and denatured proteins [49]. In these cases, interaction follows the principle of induced affinity, and upon binding to aptamer, the peptide acquires a certain conformation [30, 82, 94].
Analysis of numerous experiments shows that in the course of selection one epitope of a protein target may become dominant in the presence of many other potential sites for oligonucleotide binding. It was found that even in the case of significant differences in several experiments on aptamer selection, the ligands obtained often have similar structures and bind to the same epitope of the protein target. One of the most prominent examples is selection of aptamers to reverse transcriptase of HIV-1 virus. Three different RNA libraries and different schemes of selection were used in three independent experiments [19, 51]. Nevertheless, in all cases the majority of selected sequences formed the same class of aptamers having pseudoknot structure and binding in the region of RNA-binding center of the enzyme. In another experiment it was found that the use as a target for aptamer selection of isolated ribosomal protein S1, the whole 30S ribosomal subparticle, as well as Qbeta bacteriophage replicase results in revealing the same class of aptamers interacting with S1 protein present in all three targets [81, 95].
It turned out that often even different aptamer classes, distinguished by sequence and three-dimensional structure, also bind to identical or overlapped epitopes of a protein target. As an example one can mention aptamers to reverse transcriptase of HIV-1 virus [83, 96], DNA polymerase of T. aquaticus [97], translation elongation factor EF-Tu [98], and RNA polymerase of E. coli [40]. Thus, it can be concluded that a single protein epitope is able to bind several aptamers with different sequence and, probably, secondary and tertiary structures. It should be noted that in the case of RNA and DNA binding proteins aptamers are usually bound in natural nucleic acid binding sites [19, 40, 97]. This phenomenon can probably be explained by asymmetric charge distribution on the protein surface: as a rule, the sites of the molecule interacting with RNA or DNA carry a partial positive charge, whereas the rest of the surface is neutral or negatively charged [19, 99].
The effect of predominance of a single (or a very small number of) epitopes is revealed most clearly upon selection of aptamers to the so-called complex targets containing a large number of different proteins. Examples of this are experiments on selection of aptamers to virus particles [100], Trypanosoma cells [29], erythrocyte membranes [31], cell cultures [46, 101], blood plasma [30], blood vessels [45], etc. In all these cases, just several classes of aptamers were obtained despite the presence of many thousand different protein targets. Thus, aptamers obtained to glioblastoma cells appear to be specific to the extracellular matrix protein TN-C (tenascin-C) [101]. The pigpen protein is the target for aptamers to transformed endothelial cells [45]. In the case of selection of aptamers to blood plasma proteins, most ligands appear to be specific to prothrombin [30]. During selection of aptamers to activated neutrophils, ligands were obtained which recognize elastase exposed on the neutrophil membrane; these ligands are structurally identical to aptamers previously obtained to isolated elastase [102].
The effect of domination of a limited number of different epitopes is evidently explained by the nature of in vitro selection, during which the prevalent amplification of a small number of sequences that most efficiently bind to the target occurs, whereas other sequences are gradually excluded from the library. In fact, it was found during analysis of dynamics of selection of aptamers to human blood plasma proteins that at initial stages of selection the enriched library contains aptamers to many plasma proteins, whereas during further selection the number of different classes of aptamers undergoes significant reduction [30]. Thus, reducing the number of selection rounds may result in revealing of a larger number of different aptamer classes. From this point of view, the most perspective is the recently developed method of aptamer selection using capillary electrophoresis, which can yield high affinity aptamers by a single step procedure without library amplification [42, 103].
SITE-DIRECTED SELECTION OF APTAMERS
In many cases the goal is to obtain aptamers to a certain epitope of a protein target, such as ligands to different protein domains, to the enzyme active site and substrate-binding centers, etc. In analysis of complex biological targets, the aims of investigations often include obtaining ligands to a large number of various proteins. However, owing to the domination of one or several target epitopes, the achievement of this aim during non-directed selection often appears to be impossible. In this section, different methods are considered briefly, which can be used to avoid selection of aptamers to an undesirable epitope or, on the contrary, can be used to obtain ligands to particular epitopes of protein targets.
To inhibit a dominating target epitope and obtain aptamers to other protein sites (or to other proteins in the case of complex targets) the following methods are used.
1. Libraries of different structure are used in selection. It was shown in several experiments that the use of RNA and DNA libraries may result in obtaining aptamers to different epitopes of the same target. Thus, RNA and DNA aptamers to thrombin were described which bind in different parts of the molecule and have different effects on functional properties of thrombin [17, 104].
2. The dominating site for nucleic acid binding can be masked by various polyanionic competitors of nucleic acid binding, such as heparin [18], poly(dA-dT), tRNA [105], non-amplified random library [70, 89], etc. In this case selection can be either directed to another epitope of the target, or result in identification of aptamers with higher affinity to the same binding site.
3. A previously obtained aptamer recognizing a dominant protein site can be used to mask the dominating epitope (cf. [106]).
4. To exclude any epitope of a protein target from selection, it is possible to use a modified protein form lacking this epitope.
5. A similar result can be achieved using the counter-selection procedure that excludes from the oligonucleotide library sequences specific for this epitope. According to this procedure, sequences that do not interact with the concrete epitope of a protein target are selected during some rounds. Thus, in the case of non-directed selection of aptamers to Qbeta replicase, aptamers recognizing S1 protein are selected. It is possible to change the direction of selection using counter-selection of aptamers to ribosomal 30S subparticle. In this case, sequences recognizing S1 protein interact with the ribosome and are excluded from further selection [95].
6. For removal of the dominating class sequences, the method of their directed degradation using RNase H was proposed. For this aim, the library is incubated with short oligonucleotides (8-12 nt) complementary to the site of the sequence of the dominating aptamer class. In the case of an RNA library, DNA oligonucleotides are used, and vice versa. RNase H specifically recognizes and cleaves sites of the RNA/DNA hybrid, which results in elimination of this class of sequences [107].
7. In selection of aptamers to targets consisting of a large number of proteins, the protein containing the dominating epitope can be removed before selection. For example, to avoid selection of aptamers to S1 protein during selection of aptamers to the small ribosomal subunit, 30S subparticles free of this protein were used as a target [81]. One of the ways of removing the dominating protein from complex targets containing a mixture of many proteins is its binding on an affinity column with an aptamer specific for this protein. The use of a depleted target in the subsequent experiment on selection can identify new classes of aptamers specific for different proteins. Successive application of this procedure can be used for identification of a large number of proteins and analysis of the compositions of complex targets [30].
To obtain aptamers to any particular epitope of a protein target, the following methods of directed selection are used.
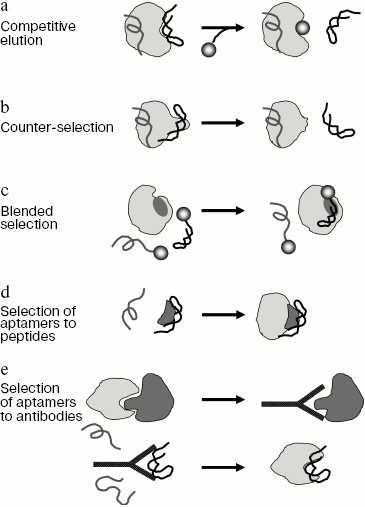
1. One of the simplest methods of directed aptamer selection is the competitive elution of sequences associated with the target molecule and other ligands specific for a particular site of the target (Fig. 2a). As examples, one can mention selection of aptamers to agglutinin (elution was carried out by the natural substrate analog triacetyl chitotriose [108]) and Ile-tRNA-synthetase (for selection of aptamers to the tRNA binding site elution was carried out by Ile-tRNA [79]). A similar method can be used for selection of aptamers-inhibitors interacting with binding sites of many known antibiotics. The drawback of the competitive elution technique is that only sequences that are in rapid equilibrium with solution are eluted, while aptamers forming the strongest complexes remain protein-bound and are lost during selection. It is possible to avoid this drawback if in each round sequences forming strong complexes with protein target are selected first, and then sequences not interacting with this protein in the presence of a competitive ligand. This approach can be used to select aptamers to bacterial RNA polymerase, which were strongly bound in the rifampicin-binding site and were inhibitors of RNA synthesis [40].
2. Aptamers to a certain domain of a protein target can be obtained using the counter-selection procedure (Fig. 2b). In this case, the library is first incubated with the whole protein target, and then oligonucleotides are selected that do not interact with the protein mutant form lacking the site to which aptamers should be obtained. For example, a similar method was used in selection of aptamers to the RNase H domain of HIV-1 reverse transcriptase [85]. This procedure can be also used in the case of complex targets. Thus, a similar approach was used to obtain aptamers specifically interacting with transformed glioblastoma cells and not recognizing normal cells [45]. In this case whole cells were used as target and “counter-target”, which allowed the authors to obtain aptamers specific for a protein present in membranes of transformed cells. It was shown that the obtained aptamers could be used as histological markers for identification of specific cell types.
3. Selection of aptamers to a particular epitope can be directed using the blended SELEX technique (Fig. 2c and table). In this case, a low-molecular-weight ligand interacting with this protein epitope is joined to the random library oligonucleotides. This can be done either by a direct covalent cross-linking between the ligand and oligonucleotide (at 5´ or 3´ end), or due to complementary interactions between the library fixed region and another oligonucleotide modified with the ligand. For example, this approach was used in selection of aptamer-inhibitors of human neutrophil elastase [102]. A relatively low-activity phosphonate inhibitor of serine proteases inhibiting the enzyme by formation of a stable covalent bond with serine of the active center was used as a “directing” ligand. Selection produced new highly specific elastase inhibitors on the basis of aptamers whose efficiency exceeded that of the original inhibitor 100-fold.
In some cases a low-molecular-weight inhibitor can be attached to aptamers after selection is over. For instance, covalent attachment of a low-affinity tetrapeptide inhibitor of elastase to the high-affinity DNA aptamers, that were not inhibitors of the enzyme, greatly increased the efficiency of inhibition. So, in this case the aptamer served as an “anchor” for fixation of another ligand in its own binding site [109].Fig. 2. Basic methods of site-directed aptamer selection. a) Method of competitive elution of aptamers using another ligand binding in the same site of the protein. b) Method of counter-selection. In the course of the experiment sequences are selected which interact with the full-size protein but do not bind to the mutant protein devoid of this epitope. c) Method of blended selection. Oligonucleotides used in selection carry a known ligand specific for this protein. The selection provides aptamers that interact with a site (shown in dark gray) near the binding site of this ligand. d) Selection of aptamers to the peptide corresponding to any epitope of protein target (shown in dark gray). The resulting aptamers recognize this epitope within the full-size protein. e) Selection of aptamers using the anti-idiotypic approach. At the first stage antibodies specific for a protein partner (shown in dark gray) of the target protein are generated (a short peptide corresponding to a concrete epitope can be used as an antigen). At the second stage, aptamers interacting with the obtained antibodies are selected. Due to complementarity of sites for interaction of aptamers, antibodies, protein partner, and protein target the obtained aptamers can recognize protein target.
A similar method can be used for selection of higher affinity aptamers interacting near the same target site as already obtained aptamers of the “first generation”. This is done by SELEX using the library into constant regions of which sequences of already known aptamers were introduced [57].
4. Not a full-size protein, but a separate protein domain (cf. [110]) or even a short peptide corresponding to the desired epitope (Fig. 2d) can be used as a target for aptamer selection. Thus, it was shown that aptamers to a peptide of protein Rev interact with the full-size protein and inhibit replication of HIV-1 virus [94]. Aptamers to a non-structured epitope of human prion protein PrP recognize the full-size protein PrP and inhibit formation of the infectious form of the protein [111]. This method makes possible obtaining aptamers to practically any protein with a known sequence (like different hypothetical proteins identified by genome sequencing) [112]. The efficiency of this method can be significantly increased by using as a target the whole protein after several rounds of selection against an isolated peptide fragment, which allows selection of only sequences that recognize the required epitope in the native conformation. If the purified preparation of the full-size protein is inaccessible, complex targets (like cell lysates) containing this protein can be used in some selection rounds [49, 112]. Since in the course of selection against peptide the library becomes enriched with ligands to this protein, the presence of different proteins in the complex target is not an obstacle to obtaining highly specific aptamers.
5. Finally, the directed selection of aptamers can be based on the anti-idiotypic approach (Fig. 2e). This method is used for obtaining aptamers mimicking any other ligand to the protein target (for example, another protein, short peptide, or a low-molecular-weight compound). At the first stage of the experiment antibodies to this ligand are obtained. Then aptamers to the antibodies are selected. It was found that like in the case of anti-idiotypic antibodies, the obtained aptamer appears to be able to interact with the original protein. Thus, aptamers to antibodies against a peptide of CREB protein, a substrate for protein kinase MSK1, interact with the substrate-binding center of this protein kinase and are efficient and specific inhibitors of its activity [57]. Aptamers to antibodies specific for the nuclear export signal (NES) of Rev protein interact with CRM1 protein of the nucleic export system and block the Rev-dependent RNA export from the cell nucleus into the cytoplasm [56, 113]. Moreover, these aptamers are themselves exported from the nucleus, which is indicative of a high similarity between their three-dimensional structure and that of native NES.
REGULATED AND SIGNAL APTAMERS. APTAZYMES
The above-described methods of aptamer selection provide high-affinity and specific aptamers to different proteins, but they do not give the possibility to control aptamer binding to their targets. However, in many investigations it is desirable to use ligands whose binding to protein targets could be regulated. In particular, such aptamers may be of great importance in studying functions of various proteins at different stages of the cell cycle and during embryonic development [114]. The group of selection methods that provides aptamers whose binding to the target molecule changes depending on the presence of another molecule-regulator was named conditional SELEX (table) [115]. Depending on the aim of the experiment, it is possible to obtain aptamers that require any compound for binding to a protein target, or on the contrary, aptamers that are not able to interact with the target in the presence of such compound. In the first case sequences are selected which form complex with protein target only in the presence of a regulator molecule and do not interact with the target in its absence (Fig. 3a). In the second case, everything is done in the opposite way (Fig. 3b). Thus, for instance, in selection of aptamers to formamidopyrimidine glycosylase (an enzyme of DNA repair) protein-bound oligonucleotides were eluted by neomycin [114]. This resulted in aptamers not able to bind the protein target in the presence of neomycin.
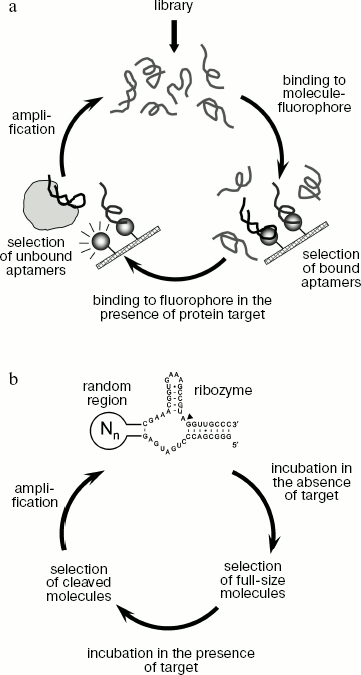
A similar approach can be used to obtain signal aptamers that generate an easily detected signal upon binding to the protein target [28, 115]. Signal aptamers can be used for detection of various target molecules. For instance, this method makes possible obtaining aptamers that bind to a fluorophore molecule in the absence of protein target but change their conformation upon interaction with the target, which results in fluorophore dissociation and enhancement of fluorescence signal. A possible way of obtaining signal aptamers is shown in Fig. 4a.Fig. 3. Methods for selection of regulated aptamers. a) Obtaining aptamers that bind to protein target only in the presence of another molecule-regulator. Each selection round is carried out in two stages. In the first stage, the library is incubated with protein target in the presence of molecule-regulator and bound oligonucleotides are selected. Then the obtained sequences are incubated with protein in the absence of the molecule-regulator and unbound oligonucleotides are selected. The selected sequences are amplified and used in the next round of selection. b) Obtaining aptamers that do not bind to protein target in the presence of a molecule-regulator. In the first stage, oligonucleotides interacting with the target in the absence of molecule-regulator are selected, whereas at the second stage the sequences that do not bind in the presence of this molecule are selected.
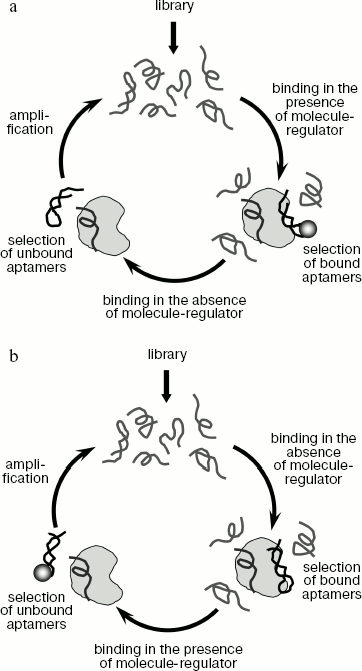
The SELEX technique can be used to obtain aptamers that exhibit catalytic activity activated upon binding to a target molecule. Such aptamers were named aptazymes [28, 116]. A scheme for aptazyme generation is shown in Fig. 4b. The library used for aptazyme selection contains a ribozyme sequence (like hammerhead-ribozyme able to cleave specifically its own sequence), connected to the random sequence. Selected sequences do not exhibit catalytic activity in the absence of target molecule, but carry out the catalytic reaction when they are complexed with the target. Various metabolites or short peptides are usually used as effector molecules in aptazyme selection [28, 117]. It should be noted that this method makes possible obtaining both RNA and DNA aptazymes. Aptamers of this type can be also used for detection of target molecules [28]. For instance, if during activation of a ribozyme containing a fluorescent group and fluorescence quencher, the latter is split off, binding of the target molecule can be detected by the enhancement of fluorescence signal [25]. Besides, aptazymes can be used for creation of artificial systems of in vivo regulation of gene expression. It was shown that if the sequence of the theophylline-regulated aptazyme is inserted in the 5´-untranslated region of mRNA, then after exogenous addition of theophylline to the cells expressing chimeric mRNA the aptazyme activation and mRNA cleavage take place [118, 119].Fig. 4. Method of obtaining signal aptamers and aptazymes. a) A scheme for obtaining signal aptamers that generate fluorescent signal upon interaction with protein target. Each round of selection includes two stages. In the first stage, sequences able to interact with a fluorescent molecule are selected. A sorbent containing covalently cross-linked fluorophore molecules can be used for selection of such sequences. In the second stage, the obtained sequences are incubated with protein target and then oligonucleotides not bound to the sorbent containing fluorophore molecules are selected. The use of such scheme of selection can produce ligands that bind to fluorophore in the absence of protein target and dissociate in its presence, which results in enhancement of fluorescence signal. b) A scheme of aptazyme generation. In the course of library synthesis, a random region is joined to the sequence of a ribozyme able to carry out the RNA cleavage reaction (the point of RNA cleavage is shown by a black arrow). Joining of the random region results in different catalytic activity of different RNA molecules. During selection sequences that are inactive in the absence of the target molecule but carry out RNA cleavage upon binding to the target are obtained.
The recently developed methods of oligonucleotide evolution in vitro can be used for the directed selection of aptamers exhibiting very different properties. Some modifications of the SELEX method described in this review are listed in the table. Different experiments on aptamer selection clearly demonstrate that nucleic acids are capable of specific recognition of very different molecules. Although aptamers are synthetic sequences obtained in vitro, this feature of single-stranded nucleic acid molecules is, evidently, of enormous importance in living systems as well. It has long been known that many cellular processes are regulated by proteins specifically interacting with certain RNA sequences. As two examples, splicing regulation by the spliceosome proteins, specifically interacting with different splicing enhancers in exon sequences [120], and translation regulation in bacteria due to binding of protein regulators with translation operators in 5´ terminal region of mRNA can be mentioned [121, 122]. Sequences specifically recognized by such proteins can be considered as natural aptamers. The directed selection of sequences interacting with very different intracellular molecules can probably take place during evolution. A clear confirmation of this hypothesis is the recent detection in 5´ terminal regions of many bacterial RNA of sequences that are able to interact specifically with different cellular metabolites (amino acids, coenzymes) [123-125]. Depending on the presence or absence of metabolites-regulators, these sequences form different secondary structure, which makes possible regulation of transcription or translation of this gene. It can be supposed that similar specific interactions of different cellular proteins and metabolites with nucleic acids are much more widespread in nature than it was previously assumed. If it is really so, then many of such interactions can be revealed experimentally using genomic SELEX techniques that allow a search for natural aptamers in genomic sequences [61, 126-128].
The author is grateful to A. Sevostyanova and N. Barinova for their help in writing this article.
This work was supported by the Russian Foundation for Basic Research (grant No. 07-04-00247), a grant of the President of the Russian Federation (MK-1017.2007.4), and a grant of the Russian Science Support Foundation.
REFERENCES
1.Ellington, A. D., and Szostak, J. W. (1990)
Nature, 346, 818-822.
2.Tuerk, C., and Gold, L. (1990) Science,
249, 505-510.
3.Gold, L., Polisky, B., Uhlenbeck, O., and Yarus, M.
(1995) Annu. Rev. Biochem., 64, 763-797.
4.Eaton, B. E., Gold, L., and Zichi, D. A. (1995)
Chem. Biol., 2, 633-638.
5.Proske, D., Blank, M., Buhmann, R., and Resch, A.
(2005) Appl. Microbiol. Biotechnol., 69, 367-374.
6.Jayasena, S. D. (1999) Clin. Chem.,
45, 1628-1650.
7.Cox, J. C., Hayhurst, A., Hesselberth, J., Bayer,
T. S., Georgiou, G., and Ellington, A. D. (2002) Nucleic Acids
Res., 30, e108.
8.Eulberg, D., Buchner, K., Maasch, C., and
Klussmann, S. (2005) Nucleic Acids Res., 33, e45.
9.Giver, L., Bartel, D., Zapp, M., Pawul, A., Green,
M., and Ellington, A. D. (1993) Nucleic Acids Res., 21,
5509-5516.
10.Schneider, D., Tuerk, C., and Gold, L. (1992)
J. Mol. Biol., 228, 862-869.
11.Shtatland, T., Gill, S. C., Javornik, B. E.,
Johansson, H. E., Singer, B. S., Uhlenbeck, O. C., Zichi, D. A., and
Gold, L. (2000) Nucleic Acids Res., 28, E93.
12.Convery, M. A., Rowsell, S., Stonehouse, N. J.,
Ellington, A. D., Hirao, I., Murray, J. B., Peabody, D. S., Phillips,
S. E., and Stockley, P. G. (1998) Nat. Struct. Biol., 5,
133-139.
13.Huang, D. B., Vu, D., Cassiday, L. A., Zimmerman,
J. M., Maher, L. J., 3rd, and Ghosh, G. (2003) Proc. Natl. Acad.
Sci. USA, 100, 9268-9273.
14.Jaeger, J., Restle, T., and Steitz, T. A. (1998)
EMBO J., 17, 4535-4542.
15.Kettenberger, H., Eisenfuhr, A., Brueckner, F.,
Theis, M., Famulok, M., and Cramer, P. (2006) Nat. Struct. Mol.
Biol., 13, 44-48.
16.Rowsell, S., Stonehouse, N. J., Convery, M. A.,
Adams, C. J., Ellington, A. D., Hirao, I., Peabody, D. S., Stockley, P.
G., and Phillips, S. E. (1998) Nat. Struct. Biol., 5,
970-975.
17.Bock, L. C., Griffin, L. C., Latham, J. A.,
Vermaas, E. H., and Toole, J. J. (1992) Nature, 355,
564-566.
18.Thomas, M., Chedin, S., Carles, C., Riva, M.,
Famulok, M., and Sentenac, A. (1997) J. Biol. Chem., 272,
27980-27986.
19.Tuerk, C., MacDougal, S., and Gold, L. (1992)
Proc. Natl. Acad. Sci. USA, 89, 6988-6992.
20.Brody, E. N., and Gold, L. (2000) J.
Biotechnol., 74, 5-13.
21.Cerchia, L., Hamm, J., Libri, D., Tavitian, B.,
and de Franciscis, V. (2002) FEBS Lett., 528, 12-16.
22.Nimjee, S. M., Rusconi, C. P., and Sullenger, B.
A. (2005) Annu. Rev. Med., 56, 555-583.
23.Pestourie, C., Tavitian, B., and Duconge, F.
(2005) Biochimie, 87, 921-930.
24.Zhang, Z., Blank, M., and Schluesener, H. J.
(2004) Arch. Immunol. Ther. Exp. (Warsz.), 52,
307-315.
25.Burgstaller, P., Jenne, A., and Blind, M. (2002)
Curr. Opin. Drug Discov. Devel., 5, 690-700.
26.Bruno, J. G., and Kiel, J. L. (1999) Biosens.
Bioelectron., 14, 457-464.
27.Charlton, J., Sennello, J., and Smith, D. (1997)
Chem. Biol., 4, 809-816.
28.Hesselberth, J., Robertson, M. P., Jhaveri, S.,
and Ellington, A. D. (2000) J. Biotechnol., 74,
15-25.
29.Homann, M., and Goringer, H. U. (1999) Nucleic
Acids Res., 27, 2006-2014.
30.Fitter, S., and James, R. (2005) J. Biol.
Chem., 280, 34193-34201.
31.Morris, K. N., Jensen, K. B., Julin, C. M., Weil,
M., and Gold, L. (1998) Proc. Natl. Acad. Sci. USA, 95,
2902-2907.
32.Brody, E. N., Willis, M. C., Smith, J. D.,
Jayasena, S., Zichi, D., and Gold, L. (1999) Mol. Diagn.,
4, 381-388.
33.Collett, J. R., Cho, E. J., and Ellington, A. D.
(2005) Methods, 37, 4-15.
34.McCauley, T. G., Hamaguchi, N., and Stanton, M.
(2003) Analyt. Biochem., 319, 244-250.
35.Bock, C., Coleman, M., Collins, B., Davis, J.,
Foulds, G., Gold, L., Greef, C., Heil, J., Heilig, J. S., Hicke, B.,
Hurst, M. N., Husar, G. M., Miller, D., Ostroff, R., Petach, H.,
Schneider, D., Vant-Hull, B., Waugh, S., Weiss, A., Wilcox, S. K., and
Zichi, D. (2004) Proteomics, 4, 609-618.
36.Gander, T. R., and Brody, E. N. (2005) Expert
Rev. Mol. Diagn., 5, 1-3.
37.Petach, H., and Gold, L. (2002) Curr. Opin.
Biotechnol., 13, 309-314.
38.Walter, G., Bussow, K., Lueking, A., and Glokler,
J. (2002) Trends Mol. Med., 8, 250-253.
39.Kopylov, A. M., and Spiridonova, V. A. (2000)
Mol. Biol. (Moscow), 34, 1097-1113.
40.Kulbachinskiy, A., Feklistov, A., Krasheninnikov,
I., Goldfarb, A., and Nikiforov, V. (2004) Eur. J. Biochem.,
271, 4921-4931.
41.Triqueneaux, G., Velten, M., Franzon, P., Dautry,
F., and Jacquemin-Sablon, H. (1999) Nucleic Acids Res.,
27, 1926-1934.
42.Berezovski, M., Drabovich, A., Krylova, S. M.,
Musheev, M., Okhonin, V., Petrov, A., and Krylov, S. N. (2005) J.
Am. Chem. Soc., 127, 3165-3171.
43.Drabovich, A., Berezovski, M., and Krylov, S. N.
(2005) J. Am. Chem. Soc., 127, 11224-11225.
44.Mendonsa, S. D., and Bowser, M. T. (2004) J.
Am. Chem. Soc., 126, 20-21.
45.Blank, M., Weinschenk, T., Priemer, M., and
Schluesener, H. (2001) J. Biol. Chem., 276,
16464-16468.
46.Cerchia, L., Duconge, F., Pestourie, C., Boulay,
J., Aissouni, Y., Gombert, K., Tavitian, B., de Franciscis, V., and
Libri, D. (2005) PLoS Biol., 3, e123.
47.Murphy, M. B., Fuller, S. T., Richardson, P. M.,
and Doyle, S. A. (2003) Nucleic Acids Res., 31, e110.
48.Ellington, A. D., and Szostak, J. W. (1992)
Nature, 355, 850-852.
49.Bianchini, M., Radrizzani, M., Brocardo, M. G.,
Reyes, G. B., Gonzalez Solveyra, C., and Santa-Coloma, T. A. (2001)
J. Immunol. Meth., 252, 191-197.
50.Conrad, R. C., Giver, L., Tian, Y., and
Ellington, A. D. (1996) Meth. Enzymol., 267, 336-367.
51.Burke, D. H., Scates, L., Andrews, K., and Gold,
L. (1996) J. Mol. Biol., 264, 650-666.
52.Legiewicz, M., Lozupone, C., Knight, R., and
Yarus, M. (2005) RNA, 11, 1701-1709.
53.Breaker, R. R. (1997) Curr. Opin. Chem.
Biol., 1, 26-31.
54.Biroccio, A., Hamm, J., Incitti, I., de
Francesco, R., and Tomei, L. (2002) J. Virol., 76,
3688-3696.
55.Hamm, J. (1996) Nucleic Acids Res.,
24, 2220-2227.
56.Hamm, J., Huber, J., and Luhrmann, R. (1997)
Proc. Natl. Acad. Sci. USA, 94, 12839-12844.
57.Hamm, J., Alessi, D. R., and Biondi, R. M. (2002)
J. Biol. Chem., 277, 45793-45802.
58.Hirao, I., Harada, Y., Nojima, T., Osawa, Y.,
Masaki, H., and Yokoyama, S. (2004) Biochemistry, 43,
3214-3221.
59.Knight, R., and Yarus, M. (2003) Nucleic Acids
Res., 31, e30.
60.Vater, A., Jarosch, F., Buchner, K., and
Klussmann, S. (2003) Nucleic Acids Res., 31, e130.
61.Gold, L., Brown, D., He, Y., Shtatland, T.,
Singer, B. S., and Wu, Y. (1997) Proc. Natl. Acad. Sci. USA,
94, 59-64.
62.Eaton, B. E., and Pieken, W. A. (1995) Annu.
Rev. Biochem., 64, 837-863.
63.Eaton, B. E. (1997) Curr. Opin. Chem.
Biol., 1, 10-16.
64.Kusser, W. (2000) J. Biotechnol.,
74, 27-38.
65.Uphoff, K. W., Bell, S. D., and Ellington, A. D.
(1996) Curr. Opin. Struct. Biol., 6, 281-288.
66.Wilson, D. S., and Szostak, J. W. (1999) Annu.
Rev. Biochem., 68, 611-647.
67.Vater, A., and Klussmann, S. (2003) Curr.
Opin. Drug Discov. Devel., 6, 253-261.
68.Wlotzka, B., Leva, S., Eschgfaller, B.,
Burmeister, J., Kleinjung, F., Kaduk, C., Muhn, P., Hess-Stumpp, H.,
and Klussmann, S. (2002) Proc. Natl. Acad. Sci. USA, 99,
8898-8902.
69.Golden, M. C., Collins, B. D., Willis, M. C., and
Koch, T. H. (2000) J. Biotechnol., 81, 167-178.
70.Jensen, K. B., Atkinson, B. L., Willis, M. C.,
Koch, T. H., and Gold, L. (1995) Proc. Natl. Acad. Sci. USA,
92, 12220-12224.
71.Smith, D., Collins, B. D., Heil, J., and Koch, T.
H. (2003) Mol. Cell. Proteomics, 2, 11-18.
72.Nutiu, R., and Li, Y. (2005) Methods,
37, 16-25.
73.Hamaguchi, N., Ellington, A., and Stanton, M.
(2001) Analyt. Biochem., 294, 126-131.
74.Stojanovic, M. N., de Prada, P., and Landry, D.
W. (2001) J. Am. Chem. Soc., 123, 4928-4931.
75.Yamamoto, R., Baba, T., and Kumar, P. K. (2000)
Genes Cells, 5, 389-396.
76.Davis, J. P., Janji, N., Javornik, B. E., and
Zichi, D. (1996) Meth. Enzymol., 267, 302-306.
77.Tasset, D. M., Kubik, M. F., and Steiner, W.
(1997) J. Mol. Biol., 272, 688-698.
78.Cui, Y., Rajasethupathy, P., and Hess, G. P.
(2004) Biochemistry, 43, 16442-16449.
79.Hale, S. P., and Schimmel, P. (1996) Proc.
Natl. Acad. Sci. USA, 93, 2755-2758.
80.Patel, D. J., Suri, A. K., Jiang, F., Jiang, L.,
Fan, P., Kumar, R. A., and Nonin, S. (1997) J. Mol. Biol.,
272, 645-664.
81.Ringquist, S., Jones, T., Snyder, E. E., Gibson,
T., Boni, I., and Gold, L. (1995) Biochemistry, 34,
3640-3648.
82.Hermann, T., and Patel, D. J. (2000)
Science, 287, 820-825.
83.Schneider, D. J., Feigon, J., Hostomsky, Z., and
Gold, L. (1995) Biochemistry, 34, 9599-9610.
84.Jing, N., Rando, R. F., Pommier, Y., and Hogan,
M. E. (1997) Biochemistry, 36, 12498-12505.
85.Andreola, M. L., Pileur, F., Calmels, C.,
Ventura, M., Tarrago-Litvak, L., Toulme, J. J., and Litvak, S. (2001)
Biochemistry, 40, 10087-10094.
86.Shi, H., Hoffman, B. E., and Lis, J. T. (1997)
Mol. Cell. Biol., 17, 2649-2657.
87.Weiss, S., Proske, D., Neumann, M., Groschup, M.
H., Kretzschmar, H. A., Famulok, M., and Winnacker, E. L. (1997) J.
Virol., 71, 8790-8797.
88.Wen, J. D., and Gray, D. M. (2002)
Biochemistry, 41, 11438-11448.
89.Yamamoto, R., Katahira, M., Nishikawa, S., Baba,
T., Taira, K., and Kumar, P. K. (2000) Genes Cells, 5,
371-388.
90.Conrad, R., Keranen, L. M., Ellington, A. D., and
Newton, A. C. (1994) J. Biol. Chem., 269,
32051-32054.
91.Tsiang, M., Jain, A. K., Dunn, K. E., Rojas, M.
E., Leung, L. L., and Gibbs, C. S. (1995) J. Biol. Chem.,
270, 16854-16863.
92.Fisher, T. S., Joshi, P., and Prasad, V. R.
(2002) J. Virol., 76, 4068-4072.
93.White, R., Rusconi, C., Scardino, E., Wolberg,
A., Lawson, J., Hoffman, M., and Sullenger, B. (2001) Mol.
Ther., 4, 567-573.
94.Xu, W., and Ellington, A. D. (1996) Proc.
Natl. Acad. Sci. USA, 93, 7475-7480.
95.Brown, D., and Gold, L. (1995)
Biochemistry, 34, 14765-14774.
96.Chen, H., and Gold, L. (1994)
Biochemistry, 33, 8746-8756.
97.Dang, C., and Jayasena, S. D. (1996) J. Mol.
Biol., 264, 268-278.
98.Hornung, V., Hofmann, H. P., and Sprinzl, M.
(1998) Biochemistry, 37, 7260-7267.
99.Darst, S. A. (2001) Curr. Opin. Struct.
Biol., 11, 155-162.
100.Pan, W., Craven, R. C., Qiu, Q., Wilson, C. B.,
Wills, J. W., Golovine, S., and Wang, J. F. (1995) Proc. Natl. Acad.
Sci. USA, 92, 11509-11513.
101.Hicke, B. J., Marion, C., Chang, Y. F., Gould,
T., Lynott, C. K., Parma, D., Schmidt, P. G., and Warren, S. (2001)
J. Biol. Chem., 276, 48644-48654.
102.Charlton, J., Kirschenheuter, G. P., and Smith,
D. (1997) Biochemistry, 36, 3018-3026.
103.Berezovski, M., Musheev, M., Drabovich, A., and
Krylov, S. N. (2006) J. Am. Chem. Soc., 128,
1410-1411.
104.Kubik, M. F., Stephens, A. W., Schneider, D.,
Marlar, R. A., and Tasset, D. (1994) Nucleic Acids Res.,
22, 2619-2626.
105.Allen, P., Worland, S., and Gold, L. (1995)
Virology, 209, 327-336.
106.Kumar, P. K., Machida, K., Urvil, P. T.,
Kakiuchi, N., Vishnuvardhan, D., Shimotohno, K., Taira, K., and
Nishikawa, S. (1997) Virology, 237, 270-282.
107.Shi, H., Fan, X., Ni, Z., and Lis, J. T. (2002)
RNA, 8, 1461-1470.
108.Bridonneau, P., Chang, Y. F., Buvoli, A. V.,
O'Connell, D., and Parma, D. (1999) Antisense Nucleic Acid Drug
Dev., 9, 1-11.
109.Lin, Y., Padmapriya, A., Morden, K. M., and
Jayasena, S. D. (1995) Proc. Natl. Acad. Sci. USA, 92,
11044-11048.
110.Fukuda, K., Vishinuvardhan, D., Sekiya, S.,
Kakiuchi, N., Shimotohno, K., Kumar, P. K., and Nishikawa, S. (1997)
Nucleic Acids Symp. Ser., 237-238.
111.Proske, D., Gilch, S., Wopfner, F., Schatzl, H.
M., Winnacker, E. L., and Famulok, M. (2002) Chembiochem,
3, 717-725.
112.Gold, L., Zichi, D., and Smith, J. D. (2002)
USA Patent 6,376,190.
113.Hamm, J., and Fornerod, M. (2000) Chem.
Biol., 7, 345-354.
114.Vuyisich, M., and Beal, P. A. (2002) Chem.
Biol., 9, 907-913.
115.Smith, J. D., and Gold, L. (2004) USA Patent
6,706,482.
116.Famulok, M. (2005) Curr. Opin. Mol.
Ther., 7, 137-143.
117.Robertson, M. P., Knudsen, S. M., and
Ellington, A. D. (2004) RNA, 10, 114-127.
118.Davidson, E. A., and Ellington, A. D. (2005)
Trends Biotechnol., 23, 109-112.
119.Thompson, K. M., Syrett, H. A., Knudsen, S. M.,
and Ellington, A. D. (2002) BMC Biotechnol., 2, 21.
120.Liu, H. X., Zhang, M., and Krainer, A. R.
(1998) Genes Dev., 12, 1998-2012.
121.Nomura, M., Gourse, R., and Baughman, G. (1984)
Annu. Rev. Biochem., 53, 75-117.
122.Winter, R. B., Morrissey, L., Gauss, P., Gold,
L., Hsu, T., and Karam, J. (1987) Proc. Natl. Acad. Sci. USA,
84, 7822-7826.
123.Lai, E. C. (2003) Curr. Biol.,
13, R285-291.
124.Mandal, M., Boese, B., Barrick, J. E., Winkler,
W. C., and Breaker, R. R. (2003) Cell, 113, 577-586.
125.Nudler, E., and Mironov, A. S. (2004) Trends
Biochem. Sci., 29, 11-17.
126.Gold, L., Singer, B., He, Y. Y., and Brody, E.
(1997) Curr. Opin. Genet. Dev., 7, 848-851.
127.Gold, L., Brody, E., Heilig, J., and Singer, B.
(2002) Chem. Biol., 9, 1259-1264.
128.Huttenhofer, A., and Vogel, J. (2006)
Nucleic Acids Res., 34, 635-646.