REVIEW: Prions
I. S. Shkundina and M. D. Ter-Avanesyan*
Russian Cardiology Research-Industrial Center, 3-ya Cherepkovskaya ul. 15A, 121552 Moscow, Russia; E-mail: mdter@cardio.ru* To whom correspondence should be addressed.
Received February 9, 2006
Prions were originally defined as infectious agents of protein nature, which caused neurodegenerative diseases in animals and humans. The prion concept implies that the infectious agent is a protein in special conformation that can be transmitted to the normal molecules of the same protein through protein-protein interactions. Until the 1990s, the prion phenomenon was associated with the single protein named PrP. Discovery of prions in lower eukaryotes, the yeast Saccharomyces cerevisiae and fungus Podospora anserina, suggests that prions have wider significance. Prions of lower eukaryotes are not related to diseases; their propagation caused by aggregation of prion-like proteins underlies the inheritance of phenotypic traits and most likely has adaptive significance. This review covers prions of mammals and lower eukaryotes, mechanisms of their appearance de novo and maintenance, structure of prion particles, and prospects for the treatment of prion diseases. Recent data concerning the search for new prion-like proteins is included. The paper focuses on the [PSI+] prion of S. cerevisiae, since at present it is the most investigated one. The biological significance of prions is discussed.
KEY WORDS: prion, PrP, neurodegenerative disease, non-chromosomal inheritance, conformational rearrangementDOI: 10.1134/S0006297907130081
Abbreviations: aa) amino acid; GuHCl) guanidine hydrochloride; GFP) green fluorescent protein; ORF) open reading frame; PrD) prion-forming domain; prion) proteinaceous infectious particle; PrP) prion protein; PrPC) normal form of prion protein; PrPSc) infectious form of prion protein; PrPres) in vitro created infectious form of prion protein; Prnp) gene encoding PrP; SDS) sodium dodecyl sulfate.
Animal and human prion diseases are classified as
“conformational” ones. Diseases of this type are caused by
alterations in three-dimensional structure of certain proteins leading
to changes in cell physiology. Along with prion diseases, amyloid
diseases like Alzheimer's, Huntington's, and Parkinson's diseases are
also considered as “conformational”. All these diseases are
characterized by extra- or intracellular accumulation of fibrillar
protein aggregates consisting of cellular proteins that are normally
soluble.
The term “prion” appeared at the end of the XX century, but prion diseases such as sheep scrapie were known already in the middle of XVIII century. Among prion diseases, there are mammalian spongiform encephalopathies like bovine spongiform encephalopathy or mad cow disease, sheep scrapie, and some human neurodegenerative diseases such as Creutzfeldt-Jakob and Gerstmann-Straussler-Scheinker diseases, familial fatal insomnia, and kuru. At present all known prion diseases are fatal. They may be hereditary (~15%), acquired (<1%), and sporadic (~85%), but independently of the disease nature they can be infectious. The infectivity of prion diseases was demonstrated for the first time by R. Chandler [1] who infected laboratory mice with a sheep scrapie, and later by Gajdusek who infected a chimpanzee with the human disease kuru [2]. The infection with human and animal prions usually happens after consuming meat and especially brain or by an iatrogenic pathway, i.e. via not properly sterilized neurosurgical instruments. Experimental infection is carried out by intraperitoneal or intracerebral injection of brain homogenate from a sick animal to the healthy one.
PRIONS ARE A NEW TYPE OF INFECTIOUS AGENTS
The nature of the infectious agent causing prion diseases remained unknown for a long time. It was found in 1966 that the pathogen of scrapie exhibited unusual properties, namely resistance to ionizing radiation and UV light [3]. This cast doubt on the hypothesis popular at that time that scrapie was caused by a virus. In 1967, D. Griffith [4] supposed that the infectious agent did not contain genetic material, but was an altered form of a cellular protein, self-maintained by an autocatalytic mechanism. In the beginning of the 1980s, S. Prusiner et al. isolated the scrapie-causing agent from the brain of sick animals and described its properties. It turned out that the scrapie-causing agent was resistant to heating, retained its activity after treatment with proteinase K, urea, chaotropic salts, SDS (sodium dodecyl sulfate), DNA-damaging agents such as nucleases and psoralens. It was also found that this infectious agent was sensitive to ionizing radiation in the presence of oxygen, i.e. exhibited properties typical for lipid-bound hydrophobic proteins [5].
The pathogen of scrapie was named “prion” (proteinaceous infectious particle). This agent was a protein named PrP (Prion Protein). After determination of its primary structure, the PrP encoding gene was identified and named Prnp [6, 7]. The Prnp is present in the genome of all mammals as well as in birds [8] and fishes [9].
PrP is a membrane protein expressed mainly in cells of the central nervous system and lymphoreticular tissue. The normal form of PrP protein was designated as PrPC. The pathological form of this protein responsible for its infectivity was named PrPSc (the PrP form associated with scrapie). PrPSc is indistinguishable from PrPC in amino acid sequence [10] but has a different conformation. The three-dimensional structure of recombinant PrPC was determined for the first time by nuclear magnetic resonance [11]. The amino-terminal region of PrPC in solution is not structured; its C-terminal part forms a globule and consists of three alpha-helices and a short region with beta-structure. It was found that PrPC contains 42% alpha-helix and 3% beta-structure, whereas PrPSc contains 30% alpha-helix and 43% beta-structure [12]. Owing to this, it was supposed that the acquisition of infectious properties by PrP is caused by the conformational rearrangement in which formation of beta-sheets takes place. Unlike the normal PrP form, its pathological form is resistant to proteinase K. The treatment of PrPSc with proteinase K generates a protease-resistant fragment [13] with molecular mass of 27-30 kD (molecular mass of PrP varies from 33 to 35 kD depending on the extent of glycosylation). Detection of the PrPSc protease-resistant 27-30 kD fragment after treatment with proteinase K of the amygdaloid gland tissue is still used in the diagnostics of prion diseases.
Based on experimental data available by 1982, S. Prusiner formulated the prion concept [14]. This concept implied the following:
- the PrPSc protein is the infectious agent;
- infectious agent PrPSc can replicate itself in the absence of nucleic acid;
- the transition of the protein from normal form (PrPC) to the infectious one (PrpSc) happens via conformational conversion;
- conformational conversion from PrPC to PrPSc can be spontaneous, resulting in sporadic forms of prion diseases. It can be caused by entering an organism of a pathological PrPSc form from the outside (acquired forms of prion diseases). Finally, the transition can be due to mutations in the Prnp gene stimulating the generation of PrPSc from PrPC (hereditary forms of prion diseases).
The prion concept has been convincingly confirmed experimentally. If the propagation of PrPSc after entering an organism proceeds by means of induction of pathological conformation on PrPC, then organisms devoid of PrPC should be resistant to prion infection. Just this was shown using transgenic mice homozygous for deletion of Prnp gene (Prnp0/0). Injection of mouse brain homogenate from animals with scrapie to transgenic Prnp0/0 mice did not result in the development of the disease due to the absence of normal PrPC [15]. Moreover, it turned out that in the absence of PrPC neither prion replication nor damage of nervous tissue takes place [16]. PrPC is also necessary for the transport of the pathogen by peripheral nerves to the central nervous system [17, 18].
Final proof of the prion concept was delayed for a long time by the impossibility to obtain a significant amount of PrPres, in vitro generated form of PrPSc, which is resistant to partial proteolysis and is able to cause the disease upon injection into experimental animals. It has been recently shown that the 89 to 231 aa fragment of mouse recombinant PrP, expressed in Escherichia coli, forms amyloid fibrils in vitro which cause the appearance of neurological symptoms of prion disease upon injection into transgenic mice expressing the same PrP fragment [19].
The elaboration of a system for cyclic amplification of the PrP prion form [20], which can be used for in vitro production of a significant amount of PrPres, allowed Soto et al. [21] to obtain and demonstrate its infectivity. The original template for the formation of PrPres was PrPSc, a pathological protein from the brain homogenate of hamsters infected with scrapie. Incubation of a minimal amount of PrPSc (brain homogenate of hamsters suffering of scrapie was diluted 104 times) with an excess of PrPC resulted in formation of PrPres aggregates. The PrPres aggregates were disintegrated by sonication to smaller ones, diluted tenfold by a suspension containing the PrPC excess, and incubated again. The many times repeated cyclic process including incubation of PrPres with PrPC, disintegration of aggregates by sonication, and following dilution resulted in the decrease of the initial infectious agent concentration in the reaction mixture from 104 times (in the first cycle) to 1055 times (in the final one). The formation of PrPres in the first cycle proceeded on the PrPSc template, whereas in following cycles the conversion of PrPC to the infectious form was stimulated by PrPres obtained in vitro. The biochemical and structural characteristics of PrPres were identical to those of PrPSc isolated from the brain of sick animals. Intracerebral injection of PrPres to healthy hamsters caused disease and death. Histological analysis of the brain of dead animals showed the spongiform degeneration of brain tissue indistinguishable from that in animals infected with PrPSc, formed in vivo. However, it turned out that PrPres is far less infectious than the pathological protein produced in vivo. The reasons for such differences in infectivity are still unclear. The method of cyclic amplification of the prion form of PrP is efficient for diagnostics of human spongiform encephalopathies, because it allows the detection of PrPSc in human tissues and biological fluids at early stages of the disease progression.
The transmission of prion infection between mammalian species is restricted by interspecies barriers [22]. Spongiform encephalopathies are transmitted between members of the same or closely related species. For example, Creutzfeldt-Jakob disease is transmitted from one human to another and to a chimpanzee, whereas scrapie is transmitted between sheep and goats, but is not transmitted to chimpanzee. Also, no cases of human infection with scrapie are known [23]. However, interspecies barriers are not absolute. Thus, the infection of hamsters with scrapie and goats with Creutzfeldt-Jakob disease is possible. Interspecies barriers can be exhibited not only in the impossibility of transmission of disease between unrelated species and increased incubation period, but also in emergence of disease only in a portion of infected animals [24]. It is assumed that interspecies barriers are caused by differences in the primary structure of PrP in mammals of different species. The following observations confirmed this. Unlike the wild-type mice, transgenic mice expressing hamster PrP appeared to be highly susceptible to infection with hamster prions [25]. Transmission of Creutzfeldt-Jakob disease from human to mouse is restricted by interspecies barrier, but transgenic mice expressing human PrP are susceptible to this infection [26]. Later it became clear that transmission of prion infection is restricted not only by differences in the PrP primary structure but also depends on the prion strain [27, 28].
PRION STRAINS
Strain variability is a fundamental property of prions. Strain variability is related to the ability of prion protein to acquire and induce different prion conformations. The amino acid sequence of PrP defines a set of conformations that it can acquire. If sets of possible prion conformations in organisms of two different species intersect, then the interspecies barrier can be crossed [28].
Propagation of PrP with different conformations causes the differences in the course of prion diseases: different incubation periods, clinical symptoms, and lesions of different brain regions are possible. The existence of different conformational states of prions was first supposed during investigation of laboratory hamsters infected with two strains of the mink spongiform encephalopathy, HY and DY. Upon injection into hamsters, these two strains caused different incubation periods and clinical symptoms of the disease. After partial proteolysis of PrPSc, isolated from the brain of hamsters infected with HY and DY strains, by proteinase K, it turned out that the molecular mass of the protease-resistant fragment of HY prion was 2 kD larger than the molecular mass of the corresponding fragment of DY prion [29]. Hence in HY and DY prions, different parts of PrP polypeptide chain were accessible for proteolysis, and therefore the difference between strains represents the different three-dimensional structure of PrPSc. It was shown that the PrP conformations corresponding to HY and DY strains are reliably reproduced in vitro [30]. The incubation of PrPC with PrPSc preparations corresponding to HY and DY strains resulted in conversion of PrPC to PrPSc with two different conformations typical of HY and DY strains. Analysis of secondary structure of HY and DY strains of PrPSc has shown that they differ in character of beta-structures [31].
Several PrPSc strains and corresponding phenotypes were identified for Creutzfeldt-Jakob disease [32, 33]. Prion strains are stably maintained in vivo. In other words, laboratory animal infected with a particular PrPSc strain will propagate only the strain used for its infection [34].
Recently data have appeared concerning a possible role of PrP glycosylation in the acquisition of strain specificity by a prion. PrP is a sialoglycoprotein with two sites of N-glycosylation in the C-terminal region. Along with unglycosylated form, there are mono- and diglycosylated PrP forms. Analysis of many cases of human Creutzfeldt-Jakob disease has shown that the prion strains can differ by the extent of PrPSc glycosylation [32]. However, it is still unknown how glycosylation influences the PrPSc conformation and pathological symptoms.
MECHANISMS OF PRION CONVERSION
The prion hypothesis can now be considered as proved. However, the mechanism of PrPC →_PrPSc conversion is still unclear. Several models of prion conversion have been proposed.
According to the heterodimer model [35], prion state is inherent to the PrP monomer, and the physical interaction of PrPSc with PrPC catalyzes the PrPC → PrPSc conversion (Fig. 1a). Spontaneous transition PrPC → PrPSc is unlikely due to a high energy barrier. As a result of PrPC → PrPSc conversion, homodimers PrPSc/PrPSc are formed, which may dissociate, initiating new rounds of conformational rearrangement, or aggregate. Formation of an aggregated form of the protein is not obligatory for prion conversion and is considered as a secondary process, not associated with conformational rearrangement as such. There are experimental data consistent with this model [36], but it cannot be considered as proved.
An alternative mechanism of prion conversion is discussed in the polymerization model [37] according to which prion conversion is inseparable from aggregation, because only PrP oligomer or multimer can reliably support prion conformation. The rate-limiting step of the PrPC → PrPSc conversion is the formation of a “seed”--the PrPSc oligomer that is an intermediate of prion conversion (Fig. 1b). This model assumes the existence of two possible variants for the mechanism of prion conversion. The first variant of prion conversion suggests coexistence of PrPC and PrPSc in thermodynamic equilibrium shifted towards PrPC, and PrPSc is formed before PrP monomer joins the “seed”. Stabilization of the PrPSc state takes place upon monomer attachment to the PrPSc “seed”, which results in the PrPSc incorporation into PrPSc polymer. If PrPSc monomer does not join the PrPSc “seed”, then the reverse conversion happens--PrPSc → PrPC conversion. The second variant of polymerization model suggests that conformational rearrangement takes place not before but at the moment of the PrPC monomer attachment to PrPSc oligomer. The polymerization model is supported by experiments showing that the converting activity is associated just with PrPSc polymers [38].Fig. 1. Models of prion conversion: a) heterodimer model; b) two variants of polymerization model. Formation of PrPSc “seed” proceeds slowly and polymerization process proceeds quickly.
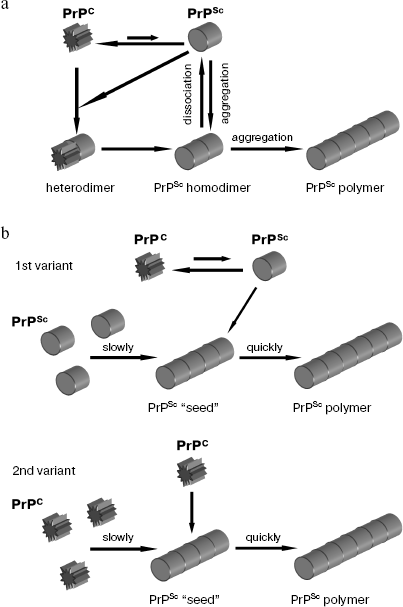
Another model of prion conversion was proposed later which is the second variant of the polymerization model with additional assumptions [39]. Oligomer intermediates which are less structured than prion fibrils and resemble micelles were found. To be able to catalyze prion conversion, such oligomer complex has to form a stable “seed” with prion conformation. Both monomer and oligomer complexes can undergo conformational conversion upon attachment to the stable “seed” serving as a template for formation of prion conformation.
PRIONS OF LOWER EUKARYOTES
In 1994, R. Wickner used the prion concept to explain the nature of the two cytoplasmically inherited determinants of the yeast Saccharomyces cerevisiae: [URE3] and [PSI+] [40]. He called yeast prions “proteins acting as genes”, pointing to the ability of prions to store and transmit conformational information. Several genetic criteria were proposed for evaluation of prion properties of the cytoplasmically inherited determinants. First, the prion determinant should be reversibly curable, i.e. conditions causing prion loss should exist. However, unlike irreversible virus elimination, a prion determinant can appear again, because its “encoding” protein is always present in the cell. Second, overproduction of prion protein should increase the frequency of the prion determinant appearance de novo, because the increase in the intracellular protein concentration promotes its incorrect folding. Third, the maintenance of the prion state should depend on the presence of the wild-type prion protein encoding gene in the genome.
Unlike mammalian prions, yeast prions do not cause cell death; on the contrary, they can increase cell viability under unfavorable conditions [41]. The discovery of [Het-s] prion of the fungus Podospora anserina [42] resulted in the notion that prions may fulfill physiological functions. A possible biological significance of prions is also implied by their broad natural occurrence. Rather recently the prion [PIN+] of S. cerevisiae, necessary for de novo [PSI+] induction, was discovered [43, 44], and data appeared pointing to the existence of prion-like determinants [ISP+] [45], [GAR+] [46] of S. cerevisiae, and [cif] determinant of Schizosaccharomyces pombe [47, 48].
PRION [URE3] OF S. cerevisiae
[URE3] was discovered in the 1970s as a dominant genetic element inherited in a non-chromosomal manner [49]. In 1994, a hypothesis was proposed according to which [URE3] determinant is maintained due to the autocatalytic propagation of alternative states of the Ure2 protein [40]. [URE3] is fully consistent with criteria of yeast prions. The determinant [URE3] can be eliminated using guanidine hydrochloride (GuHCl)--a protein-denaturing agent. In this case, GuHCl is used in a low concentration (5 mM) that is far from enough for protein denaturation [50]. [URE3] loss is reversible, because after loss [URE3] can appear again at the same frequency as in the original strain. Overproduction of Ure2 protein results in 20-200-fold increase in the [URE3] induction frequency. The presence of URE2 gene is necessary to maintain determinant [URE3]. [URE3] is transmitted by cytoduction (the method of crossing using karyogamy-deficient mutants in which fusion of the cell takes place without nuclear fusion), which confirms its cytoplasmic localization [51].
URE2 gene responsible for the maintenance of [URE3] determinant is not essential. The URE2 gene product, Ure2 protein, is a transcription regulator involved in nitrogen catabolite repression. In the presence of “rich” nitrogen sources like ammonium salts and glutamine, transcription of genes responsible for import of “poor” sources of nitrogen like allantoin is repressed [52, 53]. The allantoin import into the cell requires synthesis of its transporter protein Dal5 [54]. Transcription of the Dal5 encoding gene is under positive control of factor Gln3 [55], activity of which is under negative influence of protein Ure2 [56]. In the presence of ammonium salts cytoplasmic protein Ure2 binds to the transcription factor Gln3 and prevents its transport into the nucleus, thus inhibiting activation of many genes, including Dal5, which results in cessation of allantoin import into the cell. In the absence of ammonium salts both Dal5 synthesis and allantoin transport into the cell are activated. Along with allantoin, Dal5 is able to import into the cell ureidosuccinate resembling allantoin in chemical structure [57]. Prion [URE3] was discovered due to detection of mutants capable of ureidosuccinate uptake from medium rich with ammonium salts [49]. The bulk of mutations were recessive and one “mutation”, [URE3] was dominant. Besides, it was inherited in non-Mendelian fashion [51]. It was supposed that [URE3] is the prion form of Ure2 protein. Accordingly Ure2 may exist in two inherited states--native, able to inactivate transcription factor, and inactive prion state [40]. Ure2 in prion state does not prevent Gln3 transport into the nucleus, and as a result transcription of the allantoin/ureidosuccinate transporter is activated and yeast cells are able to take up ureidosuccinate from the medium independently of the presence of ammonia. The assumption about two different Ure2 conformations corresponding to [URE3] and [ure3] (the absence of prion determinant) phenotypes was confirmed later. It was found that Ure2 protein from lysates of the [URE3] containing cells was more resistant to proteinase K treatment than Ure2 from lysates of [ure3] cells [58]. In addition, protein Ure2 was aggregated in the [URE3] carrying cells [59].
The prion domain of Ure2 protein is its amino-terminal domain, rich in asparagine and glutamine residues and including amino acids from 1 to 94 [60]. The C-terminal catalytic domain (94-354 aa) is responsible for catabolite repression [61]. The conversion of the N-terminal domain to the prion state inactivates Ure2 protein. There are regions in the C-terminal domain that influence the ability of the amino-terminal domain to undergo prion rearrangement [62].
The prion nature of [URE3] is now considered as proven. The conversion of Ure2 to the prion state can be modeled in vitro. Ure2 is capable of oligomerization and formation of amyloid fibrils in vitro. Yeast cells can be “infected” with [URE3] by transformation with Ure2 fibrils formed in vitro [63].
THE [Het-s] PRION OF P. anserina
Unlike Ure2 protein of S. cerevisiae, conversion of the HET-s protein of the filamentous fungus P. anserina to prion form is not associated with its inactivation. On the contrary, only in prion form protein HET-s is able to cause the reaction of vegetative incompatibility revealed in the death of heterokaryotic cells formed upon parasexual process [42].
The colony of P. anserina is a syncytium in which cells can exchange cytoplasm and even nuclei. Hyphae of two fungal colonies can fuse with each other, and this allows exchange of the cytoplasm and formation of heterokaryons. The fusion of hyphae is potentially unsafe, because it can result in rapid dissemination of fungal viruses from one colony to another. Thus fusion of hyphae is genetically regulated in such a way that two colonies can fuse only when they have at least nine identical het loci [64]. If hyphae fusion happened between two colonies differing by at least a single het locus, then the reaction of programmed cell death takes place.
One of these loci, het-s, appeared to have unusual properties. The locus is represented by alleles het-s and het-S, products of which (proteins HET-s and HET-S) differ by 14 amino acids. Cells expressing HET-s protein can exist in two states: [Het-s] in which they are incompatible with fungal cells carrying the het-S allele (HET-S protein), and the [Het-s*] state when there is no incompatibility with het-S strains. It was shown that [Het-s] behaved as a non-chromosomal genetic element, and [Het-s*] as its absence [42]. Maintenance of [Het-s] requires the presence of the het-s gene and overexpression of the latter increases the frequency of [Het-s] appearance de novo. [Het-s] is inherited via the cytoplasm because fusion of [Het-s*] mycelium with mycelium of [Het-s] transforms the [Het-s*] to the [Het-s] state independently of nucleus transfer. Deletion of the het-s gene results in formation of colonies compatible with the het-s and het-S partners, thus showing that the prion form of the protein is responsible for incompatibility.
Prion properties of HET-s protein were also confirmed by biochemical experiments. Overproduction of HET-s protein in [Het-s] cells results in the aggregation of the protein [65]. The prion domain of HET-s is localized in the C-terminal region and in aggregated form HET-s is resistant to proteinase K [66]. Protein HET-s forms amyloid fibrils in vitro [67], and the [Het-s] state in fungus P. anserina can be achieved through “infection” with the HET-s amyloid polymers obtained in vitro [68].
THE [PSI+] PRION OF S. cerevisiae
[PSI+] determinant was described for the first time as a factor leading to an increase in the efficiency of a weak ochre-suppressor SUQ5, which encodes serine-specific tRNA containing anticodon complementary to nonsense-codon UAA [69]. Later it became clear that [PSI+] increases the efficiency of read-through of all three nonsense-codons [70]. Suppressor mutations were found in the SUP35 gene, which like [PSI+] caused omnipotent suppression (suppression of three types of nonsense mutations) [71, 72]. However, unlike recessive suppressor mutations in the SUP35 gene, [PSI+] determinant was dominant and inherited in non-Mendelian fashion. [PSI+] was transmitted via cytoduction and therefore was localized in the cytoplasm. It was assumed for a long time that [PSI+] is encoded by nucleic acid, although it was known that [PSI+] was independent of mitochondrial DNA and of 2 µm DNA [73]. It was supposed that similarly to [URE3], [PSI+] phenotype exists due to the ability of Sup35 protein to switch to the self-maintaining prion state [40]. [PSI+] is the most studied yeast prion, and for this reason it will be considered here in more detail.
The Sup35 protein consists of three regions (Fig. 2) [74]. Its amino-terminal region designated as N (1-123 aa) has an unusual amino acid composition, because it contains more than 55% of asparagine and glutamine residues. This domain is necessary to maintain Sup35 prion state and therefore it is often called PrD (Prion forming Domain). Deletion alleles of the SUP35 gene, which do not encode amino-terminal sequence, do not maintain [PSI+] [75]. Besides, all known mutations in the SUP35 gene, leading to the loss of [PSI+], are localized in Sup35 PrD [76, 77]. The role of this Sup35 region for cell physiology is still not clear, but recently its interaction with the poly(A)-binding protein PABP [78, 79] resulting in mRNA degradation [80] has been shown.
Sup35 middle region designated as M (124-253 aa) is rich with charged amino acids (42%), namely with lysine and glutamic acid. The function of this region is not clear, but its involvement in [PSI+] stability was shown [81, 82].Fig. 2. Structural organization of the Sup35 protein. The protein consists of N and C regions separated by M region. Within N region, NR and NQ domains are distinguished, which are essential for the prion properties of Sup35. Numbers designate positions of amino acid residues used for distinguishing different protein domains and regions.

It was shown in 1995 that the yeast Sup35 protein is an ortholog of the translation termination factor eRF3 of higher eukaryotes, which interacts with the Sup45 protein (an ortholog of translation termination factor eRF1) thus forming the translation termination complex [83, 84]. The essential C-terminal region of Sup35, designated as C (254-685 aa), is responsible for function of the yeast translation termination factor eRF3. The sequence of this C-terminal region is highly conserved and homologous to the translation elongation factor eEF1A [85]. The stop-codon recognition by protein Sup45 results in the polypeptide chain release [83, 84, 86]. Sup35 is a GTP-binding protein that stimulates translation termination. The mechanism of this process is not clear. Conversion of the Sup35 amino-terminal domain to the prion form results in Sup35 aggregation and inhibition of its termination function, which in turn causes the nonsense-codon readthrough and can be detected by suppression of nonsense mutations. Most likely, the Sup35 aggregation creates steric hindrances for involvement of its C domain in translation termination.
The role of Sup35 is not restricted to its participation in the translation process. The interaction of the N region of Sup35 protein with the Sla1 protein, involved in formation of actin microfilaments, was shown [87]. It is notable that this interaction could be violated by factors lowering the stability of [PSI+]. The role of Sup35 in formation of actin cytoskeleton was also shown [88]. Repression of the SUP35 gene resulted in actin depolymerization, defect of mitotic spindle formation, and as a result in abnormalities of cyto- and karyokinesis.
[PSI+] determinant completely fits all genetic criteria of a yeast prion. In fact, [PSI+] is lost at a high frequency in the presence of non-mutagenic agents like GuHCl and methanol [50]. [PSI+] can appear again in the strains in which it existed previously and was lost [89]. Normally [PSI+] appears de novo at a frequency of 1·10-5. Overproduction of Sup35 or its prion domain increases the frequency of [PSI+] appearance by a factor of 100 or more [90-92]. The maintenance of [PSI+] depends on the presence of the 5´-terminal part of SUP35 gene.
[PSI+] is dominant and is inherited in non-Mendelian fashion [69], which is easily explained in the framework of the prion concept. If [PSI+] appears, it is constantly maintained due to continuous transfer of prion conformation from Sup35 prion form to its normal molecules. Crossing of [PSI+] cell with the [psi-] cell, devoid of the [PSI+] determinant, causes cytoplasm mixing and the hybrid cells became [PSI+]. All mitotic and meiotic progeny of such hybrid receives a certain amount of Sup35 in the prion form. Therefore [PSI+] is inherited in a non-Mendelian fashion and is transmitted between cells by cytoduction.
Prion properties of Sup35 are confirmed by biochemical experiments. It was shown that Sup35 isolated from the [PSI+] strains is characterized by enhanced resistance to proteinase K [93, 94]. Sup35 in [PSI+] cells is within large aggregates, whereas in [psi-] cells most Sup35 is soluble [93, 94]. The Sup35 aggregates can be visualized in cells using green fluorescent protein (GFP) [93]. To achieve this it is necessary to construct a chimeric gene encoding the Sup35 fused to GFP sequence. Such protein may contain not the complete Sup35 sequence but only its fragment including N and M regions (Sup35NM). Introduction of plasmids encoding Sup35-GFP or Sup35NM-GFP into [PSI+] cells results in incorporation of these proteins into the Sup35 prion aggregates, owing to which visualization of the aggregates becomes possible. In [psi-] cells diffuse fluorescence of Sup35-GFP or Sup35NM-GFP proteins is observed. It should be noted that it is possible to observe only large Sup35 aggregates in such a way, whereas fluorescence of small aggregates is indistinguishable from diffuse fluorescence of GFP.
The Sup35 polymerization can be modeled in vitro [95, 96]. The purified full-size recombinant Sup35 protein and its NM fragment are capable of spontaneous formation of amyloid fibrils in vitro. The structure of Sup35 fibrils is analogous to that of fibrils of Abeta-peptide involved in pathogenesis of Alzheimer's disease. Amyloid fibrils have beta-structure in which beta-sheets are perpendicular to the fibril axis [97]. Such structure was detected in the crystallized peptide GNNQQNY of the prion-forming domain of Sup35 [98]. This peptide, like the whole domain of Sup35, forms amyloid fibrils in vitro. The X-ray diffraction pattern of these fibrils is similar to that of amyloid fibrils formed by other proteins. Sup35 amyloid fibril formation is a process extended in time and can last up to 60 h [95]. It is dependent on temperature and protein concentration and has a lag-phase from 10 to 30 h. It is assumed that the lag-phase is the period necessary for spontaneous formation of polymerization “nuclei” [39]. The lag-phase can be reduced to zero if preformed fibrils or lysates of [PSI+] strains are added to the purified Sup35 or Sup35NM [95, 99].
Quite recently infectious properties of the Sup35 amyloid fibrils have been demonstrated. A method was elaborated for “infection” of [psi-] cell spheroplasts with in vitro obtained amyloid fibrils of recombinant Sup35 [100, 101]. Incubation of the [psi-] cell spheroplasts with such fibrils resulted in [PSI+] appearance in these cells, and the efficiency of such “infection” depended on the amount of the Sup35 fibrils used.
[PSI+] de novo APPEARANCE, PRION
[PIN+]
[PSI+] can appear de novo not in every strain of S. cerevisiae upon Sup35 overproduction. A necessary condition for the appearance of [PSI+] de novo is the presence of an epigenetic element called [PIN+] ([PSI+] inducibility) [44]. However, if [PSI+] already exists, [PIN+] is not required for its maintenance [102]. The appearance of [PIN+] in a cell is associated with conversion of the Rnq1 protein with an unknown function to the aggregated state [43]. [PIN+] is characterized by dominant manifestation, is inherited in non-Mendelian fashion, is reversibly cured in presence of GuHCl, appears upon Rnq1 overexpression, and disappears upon deletion of the RNQ1 gene. All the above-mentioned properties show that [PIN+] is the prion form of Rnq1 protein [43, 102, 103]. The existence of inherited [PIN+] variants (strains) (see about prion variants in section “Variants of [PSI+]”) differing from each other by the efficiency of [PSI+] induction [104] was shown.
Two hypotheses explaining how the prion form of Rnq1 induces [PSI+] prion generation were put forward [102]. According to the first hypothesis, soluble protein Rnq1 is an inhibitor of [PSI+] appearance de novo and conversion of this protein to the aggregated prion form reduces the efficiency of such inhibition. The second hypothesis suggests the possibility of formation of Sup35 prion form on the template of Rnq1 aggregates. This hypothesis is supported by co-localization of Sup35NM-GFP aggregates with aggregates of Rnq1 during [PSI+] induction de novo [105]. Besides, the Sup35NM polymers contain Rnq1 protein [106], and Rnq1 fibrils formed in vitro accelerate conversion of Sup35NM into the prion form in vitro [105].
It was shown that [PIN+] increases the frequency of induction of both [PSI+] and [URE3] [104], whereas [PSI+] and [URE3] stimulate prionization of Rnq1 [102]. The [PIN+] phenotype can be defined not only by the prion state of Rnq1 protein. Overexpression of eleven different proteins, including Ure2, New1, as well as Lsm4, that controls mRNA degradation, and Ste18, involved in the pheromone signal pathway, was accompanied by their aggregation and resulted in appearance of the [PIN+] phenotype [43].
ROLE OF CHAPERONS IN YEAST PRION APPEARANCE AND
INHERITANCE
A surprising feature of yeast prions is their ability to be reliably maintained in dividing cells. It is known that the aggregated state of Sup35 protein corresponds to the presence of [PSI+] determinant [93, 94]. Here questions arise how such aggregates are inherited and whether they are units of [PSI+] inheritance. It is evident that the stable maintenance of a prion requires that each mitosis is accompanied by doubling of units of prion inheritance.
It has been shown that [PSI+] can only exist in the cells where Hsp104 chaperone, a member of Hsp100 protein family, is present [107]. It is notable that not only absence of Hsp104 protein, but also Hsp104 overproduction causes [PSI+] loss. The fact that all known yeast prions are unable to be propagated in the absence of Hsp104 supports the notion of a common mechanism of their inheritance.
The yeast Hsp104 and its bacterial ortholog ClpB are the main heat shock proteins providing the possibility of survival under stress conditions such as elevated temperature and high ethanol concentration in the medium. Hsp104/ClpB is a hexamer that does not prevent denaturation of cellular proteins caused by elevated temperature, but disintegrates large aggregates of already denatured proteins and thus stimulates their refolding and functional recovery [108]. It was shown that Hsp104 in a complex with Hsp40 and Hsp70 completely restores activity of denatured luciferase in vitro [109].
There are two models considering the mechanism of [PSI+] maintenance. One of them suggests that Hsp104 facilitates the process of prion conversion interacting with Sup35 molecules and stimulating the monomer to acquire some intermediate conformation [110]. It should be noted that Sup35 conversion to the prion form in vitro does not require the presence of Hsp104 [95]. However, it was shown that Hsp104 at a low concentration (the ratio of Hsp104 hexamers to the Sup35 monomers is 1 : 250) eliminated the lag phase of fibril formation and increased the rate of Sup35NM polymerization in vitro [111].
The other model (Fig. 3), maintained by the bulk of experimental data, suggests that Hsp104 is necessary not for prion conversion but rather for shearing large Sup35 aggregates into smaller particles, which is necessary for the stable [PSI+] inheritance [94, 112].
The mechanism of Hsp104 action in the maintenance of the yeast prion was studied in parallel with the mechanism of prion curing in presence of GuHCl. [PSI+] curing by GuHCl occurs only in dividing yeast cells [113], and the appearance of [psi-] cells is preceded by a lag-phase equal to approximately four or five cell generations. Studying the kinetics of [PSI+] loss suggested that GuHCl blocks replication of [PSI+] “seeds”, i.e. units of prion inheritance called also propagons. If prion “seeds” are not replicated, then the culture growth is accompanied by gradual decrease in their number in each cell. Tracing the process of [PSI+] loss in the presence of GuHCl during 20-30 h, Eaglestone et al. [113] determined the mean number of propagons present in the cell before the action of this substance. It was equal to 62 ± 10. It became clear later [114] that the number of propagons can vary from 30 to 1000 depending on the [PSI+] strain (variant) (see section “Variants of [PSI+]” below). It was shown that the cell growth in the presence of GuHCl does not cause destruction of already existing prion aggregates and does not result in proteolysis of Sup35 that forms these aggregates. Also, GuHCl does not block further polymerization of Sup35 catalyzed by “seeds” present in the cell. Only the number of intracellular propagons gradually decreases [115].Fig. 3. Role of the Hsp104 chaperone in [PSI+] maintenance. Hsp104 fragments prion polymers of Sup35, thus increasing the number of polymer ends involved in polymerization (figure taken from [112]).
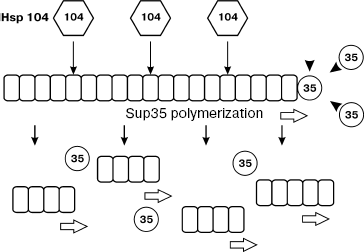
At the same time, it was found that growing of cells in the presence of GuHCl results in inactivation of Hsp104 [116]. It was supposed from these data that PSI+] curing under the influence of GuHCl was the result of the inactivation of this chaperon. This assumption was confirmed by obtaining mutations in the HSP104 gene conferring the resistance of [PSI+] to GuHCl [117] and also by demonstration of the inhibitory effect of GuHCl on the Hsp104 ATPase activity in vitro [118]. It was also shown in vitro that Hsp104 disaggregates Sup35 fibrils into smaller ones (at the ratio of Hsp104 hexamers to Sup35 monomers of approximately 1 : 50) [111]. The decrease in intracellular Hsp104 level or inhibiting its activity result in a decreased number of Sup35 aggregates and increase of their size [119], whereas overexpression of Hsp104 reduces the size of prion aggregates [120]. Stronger evidence in favor of the ability of Hsp104 to fragment Sup35 aggregates was obtained later [121]. It turned out that Sup35 aggregates consist of SDS-resistant polymers, each of which, in turn, contains from approximately 10 to 50 Sup35 molecules. In the case of cell growth in the presence of GuHCl, the mean size of the Sup35 polymers was doubled during a single cell generation, which can be explained only by the block of their fragmentation. After transferring cells into GuHCl-free medium, the polymer size gradually returned to the initial level. Gradual decrease of Hsp104 amount also increased the size of polymers. These data are in favor of the model according to which Hsp104 fragments prion polymers, providing the stability of their inheritance [112]. Thus, the stable maintenance of prions requires the balance of two processes: conversion of monomers into polymers (polymerization) and polymer breakage into smaller ones (fragmentation). Efficient fragmentation of polymers provides the necessary number of free polymer ends involved in polymerization.
Chaperons of the Hsp70 and Hsp40 families were also found to take part in the process of [PSI+] maintenance. Heat shock proteins of the Hsp70 family are the main chaperons required for the protein folding in yeast cells. In addition to the protein folding, Hsp70 carry out various functions such as protein stabilization upon heat shock, polypeptide chain translocation through membranes [122], and assembling and dissociation of macromolecular complexes [123]. The chaperons of Hsp40 family are cofactors of Hsp70 [124]. The family of Hsp70 chaperons of S. cerevisiae includes SSA and SSB subfamilies.
Overexpression of the Ssa1 protein prevents [PSI+] loss upon Hsp104 overexpression [125] and enhances [PSI+] induction de novo approximately tenfold [126]. The increase in intracellular amount of Ssa1 increases the size of prion polymers and simultaneously increases the level of Sup35 monomer. Other proteins of the SSA subfamily, namely Ssa2, Ssa3, and Ssa4, exert the same influence on [PSI+] as Ssa1 [126]. Physical interaction of Ssa and Ssb proteins with Sup35 was shown in vitro and in vivo. Thus, the family of SSA chaperons evidently carries out the function of [PSI+] “helper”. A possible mechanism of such action of Ssa proteins is the stabilization of intermediate conformation of Sup35 molecules. Stabilization of such partially folded intermediate state increases the probability of prion conversion during interaction of Sup35 molecule with a prion “seed”.
In contrast to the SSA subfamily, the subfamily of Hsp70 SSB chaperons behaves as [PSI+] antagonist [120, 127]. Overproduction of Ssb1 protein enhances the probability of [PSI+] loss observed at increased level of Hsp104. Deletion of the non-essential SSB1 and SSB2 genes makes the [PSI+] curing effect caused by overproduction of Hsp104 weaker [127]. Efficiency of [PSI+] induction increases in the strains with deletions of the SSB1 and SSB2 genes. Data concerning the effect of Ssb1 on the protein proteasomal degradation [128] suggest that Ssb1 interacts with Sup35 molecules and thus stabilizes their in the native state, but if this impossible then Ssb1 facilitates their degradation.
The maintenance of [PIN+] depends not only on Hsp104 [103], but on the Sis1 chaperon of the Hsp40 family as well [129, 130]. However, unlike [PSI+], overproduction of Hsp104 chaperon does not result in the loss of [PIN+] [44]. At the same time, it was found that overproduction of the Ydj chaperon of the Hsp40 family results in the loss of some [PIN+] variants [104].
Stable maintenance of [URE3] requires the Hsp104 chaperon and Ssa2 chaperon of Hsp70 family [131]. Deletion of the HSP104 gene results in the loss of [URE3], whereas overproduction of Hsp104 does not destabilize it [132]. The loss of [URE3] is also caused by overproduction of another member of Hsp70 family, Ssa1 protein [131], as well as of Ydj1 chaperone from the Hsp40 family [132].
INTERSPECIES BARRIERS OF TRANSMISSION OF Sup35 PRION PROPERTIES
AND MECHANISM OF [PSI+] CURING
The prion domain Sup35 can be divided into two regions with different structures and functions. The region designated as NQ (from 1 to 40 aa) is enriched with asparagine and glutamine residues. The second region, NR (from 41 to 97 aa) contains five complete and one incomplete copies of oligopeptide repeats with consensus sequence PQGGYQQ-YN.
Mutations in SUP35 gene causing [PSI+] loss (PNM mutations) or those interfering with its phenotypic manifestation result mainly in amino acid substitutions in the PrD region from 8 to 26 aa [76]. Later it became clear that this protein region is the determinant of the prion species specificity [133]. Like barriers restricting the prion transmission between unrelated mammalian species, there are also barriers of [PSI+] transmission between distantly related yeast species such as S. cerevisiae and Candida albicans. The transmission of [PSI+] between unrelated yeast species was modeled using chimeric proteins in which prion domain of Sup35 of S. cerevisiae was replaced by amino-terminal regions of Sup35 of C. albicans, Kluyveromyces lactis, and Pichia methanolica. Chimeric proteins containing prion domains of C. albicans, K. lactis, and P. methanolica are capable of aggregation in vivo and formation of amyloid fibrils in vitro, but they cannot incorporate into the prion aggregates of S. cerevisiae Sup35. Overproduction of Sup35 of S. cerevisiae did not result in aggregation and induction of prion state of chimeric proteins, and vice versa, overproduction of chimeric proteins did not cause induction of S. cerevisiae Sup35 prion state. Thus, the transmission of prion state between Sup35 proteins from different yeast species did not take place. At the same time, replacement of the region from 8 to 26 aa in chimeric protein, carrying prion domain of Sup35 from C. albicans and MC region from Sup35 of S. cerevisiae, for the same region of S. cerevisiae made the chimeric protein compatible with prion state of Sup35 from S. cerevisiae [133]. In this work, a model of prion polymer was proposed, according to which the region from 8 to 26 aa is located on the polymer surface and thus provides the polymerization of only those Sup35 molecules that contain a region of homology with this region. Recent data are in favor of this model: the Sup35NM molecules are oriented along an amyloid fibril in such a way that N-terminal parts of prion domains of adjacent molecules interact with each other [134].
Another PNM mutation was found in a site of the SUP35 gene corresponding to the second repeat of the NR region. This mutation, resulting in the replacement of glycine by aspartic acid in position 58, was dominant [77]. The mutant protein was capable of conversion to prion state, but the rate of such conversion in vitro was approximately two times lower [92]. The most probable mechanism of [PSI+] elimination in the presence of mutant Sup35 is the following: mutant protein molecules are able to join the growing Sup35 polymer and retard its further growth. This hypothesis is confirmed by the decrease in size of prion polymers of Sup35, which is observed in the case of simultaneous expression of mutant and the wild-type Sup35 proteins (D. Kryndushkin, unpublished).
Deletion analysis of the Sup35 prion domain [135] has shown that the PrD region from 1 to 93 aa, which includes the NQ region and five repeats of the NR region, is necessary for the maintenance of [PSI+]. Removal of the sixth incomplete repeat (R6) of NR region resulted in a slight weakening of the [PSI+] suppressor phenotype and decrease in its mitotic stability. Replacement of the wild-type allele SUP35 by the allele carrying deletion of the fifth (R5) and sixth (R6) repeats resulted in the loss of [PSI+]. Nevertheless, successive removal of repeats R6, R5, R4, and R3 had no effect on the abilities of mutant Sup35 to co-aggregate with the full-length Sup35 protein in [PSI+] cells. Overproduction of mutant chimeric proteins, in which GFP sequence was fused with Sup35NM with deletions of a part of amino acid repeats in prion domain, could induce [PSI+] de novo in the cells expressing the wild-type Sup35 [136]. Thus, Sup35 molecules with reduced number of amino acid repeats may co-aggregate with the full-length Sup35 and even stimulate [PSI+] induction de novo, but they are not able to maintain [PSI+] (except Sup35 containing five repeats in the prion domain).
Based on these observations, a model was proposed according to which the region including the NQ and the beginning of the NR of Sup35 prion domain is necessary and sufficient for aggregation and growth of prion polymers. Meanwhile the largest part of the NR region provides the interaction of Sup35 polymers with chaperons and, as a result of this, their fragmentation and propagation [136]. On the other hand, the results point to the significance of amino acid repeats of the NR region for the process of Sup35 polymerization [137]. The shortened Sup35 from which four repeats were removed (from second to fifth) formed polymers in vitro at a considerably lower rate compared with the full-size Sup35. The recombinant Sup35, containing two additional copies of the second repeat, polymerized in vitro at a rate exceeding that of the wild-type Sup35 due to reduction of the prion conversion lag phase, i.e. to a more rapid formation of “seeds”.
According to the proposed model, the curing of [PSI+] by the mutant Sup35 protein in the case of its co-expression with normal Sup35 can be mediated either by a defect in Sup35 polymerization or by disturbance in fragmentation of formed polymers [136].
VARIANTS OF [PSI+]
As noted above, strain variability is one of main features of prions. The phenomenon of prion variability is also characteristic of yeast prions. Strains of yeast prions are called variants. [PSI+] variants differ in mitotic stability and the strength of suppressor phenotype [91]. [PSI+] variants characterized by high mitotic stability and strong suppression of nonsense mutations are called strong. The [PSI+] variants with low mitotic stability and weak suppression level are called weak. The strength of suppressor phenotype is inversely proportional to the level of soluble Sup35 [138-140]. The amount of soluble Sup35 may differ several times in cells with different [PSI+] variants. [PSI+] variants also differ by the size of prion polymers [121]. The stronger is a [PSI+] variant, the smaller are Sup35 polymers. The difference in the Sup35 polymer size between [PSI+] variants most likely points to their different susceptibility to fragmentation. Sup35 polymers of strong [PSI+] are fragmented more intensively compared with polymers of weak [PSI+]; therefore, polymers of strong [PSI+] are smaller than polymers of weak [PSI+]. In fact, the efficiency of [PSI+] elimination upon Hsp104 overproduction depends on the variant of the prion being lost. In addition, there was observed variant-specific loss of [PSI+], maintained by chimeric protein in which the amino-terminal region of S. cerevisiae Sup35 was replaced by an analogous region of Sup35 of P. methanolica, upon overproduction of Ssb1, Ssa1, and Ydj1 chaperons [120]. [PSI+] variants also differ by the ability of prion “seeds” to catalyze conversion of soluble Sup35 to the polymer state: Sup35 aggregates from cells with strong [PSI+] induce prion conversion of soluble Sup35 more efficiently than aggregates from cells with weak [PSI+] [138, 139].
[PSI+] variants are reliably maintained in vivo. This means that the strength of suppressor phenotype and the level of [PSI+] variant mitotic stability do not change in cell generations. The M region of Sup35 protein plays an important role in ensuring stable inheritance of [PSI+] variants, because the removal of the beginning of this region resulted in appearance of undifferentiated [PSI+], i.e. of such [PSI+] in which suppression strength and mitotic stability varied from one cell to another [81]. There is only a single described case concerning structural instability of [PSI+] maintained by the full-length Sup35: the generation of a strong variant from a weak one [138]. Such structural instability is characteristic of very weak [PSI+] variants.
It has been recently shown that [PSI+] variants can be stably maintained in vitro [100]. Polymers of chimeric protein containing a shortened PrD of Sup35 (1-61 aa) and GFP (Sup35-1-61-GFP), isolated from yeast cells containing three different [PSI+] variants ([VH], [VK], [VL]) were used as a “seed” for prion conversion of purified recombinant Sup35-1-61-GFP produced in E. coli. With the use of prion [VH], [VK], and [VL] “seeds”, in vitro fibrils were obtained which were then used for transformation (infection) of [psi-] yeast cells. This resulted in appearance of [PSI+] colonies with three phenotypes corresponding to the original variants. The experiment also showed that the region of the Sup35 prion domain from 1 to 61 aa was sufficient to maintain in vitro differences in properties of Sup35 prion.
Most likely, the [PSI+] variants are different stably maintained prion conformations of Sup35. The following experimental data are in favor of this statement.
Amyloid fibrils spontaneously formed in vitro by the NM fragment of Sup35 protein may differ in growth rate and polarity. Amyloid fibrils formed by Sup35NM in vitro are able to grow in two directions, but the rate of the fibril growth in different directions is different [141]. Besides, amyloid fibrils formed at different temperatures (4 and 37°C) are characterized by different thermostability in the presence of SDS and resistance to proteolysis [101]. Data on different structure of Sup35NM fibrils formed at different temperatures were obtained using EPR spectroscopy [101]. Analysis of such Sup35NM fibrils using a fluorescent label has shown that Sup35NM molecules forming fibrils at 25 or 37°C have a more extended region of the prion domain involved in prion fold as compared with Sup35NM molecules within fibrils formed at 4°C [134].
The recently developed method of transformation of [psi-] cell to the [PSI+] state with the in vitro obtained amyloid fibrils [100, 101] made it possible to show that infection with amyloids formed at 4°C results in appearance of mainly strong [PSI+] variants, whereas cell infection with amyloids formed at 25 or 37°C usually results in weak [PSI+] variants [101]. Thus, it was shown that the variability of [PSI+] properties
is caused by conformational differences of corresponding prion polymers.
STRUCTURE OF AMYLOID FIBRILS
Fibrils formed in vitro by Sup35, Ure2, and HET-s proteins or their prion domains are structurally similar to amyloids involved in pathogenesis of human diseases. To date, about 20 human proteins have been described which are able to form amyloids in vivo [142], like insulin, fragments of light immunoglobulin chains, alpha-sinuclein, Abeta-peptide, and PrP protein. Amyloid fibrils interact with thioflavin T and Congo Red [143] and are resistant to treatment with SDS detergent at room temperature [39]. In addition, regardless of the protein that formed amyloid fibrils, they are structurally similar: they are formed by beta-sheets perpendicular to the fibril axis, and hydrogen bonds, binding polypeptide chains, are oriented along the fibril axis [144].
The ability of proteins to form amyloids depends on their charge and hydrophobicity [145] and is evidently an inherent feature of polypeptide chains, because in vitro under conditions of partial denaturation many peptides and proteins form similar structures. However, such conditions significantly differ from physiological ones [146, 147].
Atomic structure of amyloid fibrils including those formed by Sup35 is unknown. The X-ray diffraction pattern of fibrils formed by the glutamine-asparagine-rich peptide of the Sup35 prion domain, poly-L-glutamine, as well as by the huntingtin fragment [148], made possible a model of the amyloid fibril structure [149]. According to this model, amyloid fibril consists of several protofibrils intertwined with each other. An amyloid protofibril is a cylindrical beta-sheet, a hollow nanotube of 3 nm in diameter, in which the polypeptide chain is “wound” (coiled) around the fibril axis and forms a helix. One turn of such helix includes 20 amino acids, and the turns interact with each other by hydrogen bonds between amides of the main and side chains (each CO of a previous turn interacts with NH of the following one). The orientation of side chains of amino acids of each turn alternate: neighboring side chains are positioned at different sides from the main chain (they are directed inside the helix and outside). In the case of a nanotube formed by Sup35, its C-domain is not involved in this structure, but overhangs from it. One turn of such helix is unstable, because it is stabilized due to interaction with the next turn. Thus, the minimal number of turns forming a stable structure is equal to two, i.e. 40 amino acids comprise a minimal amyloid-forming fragment. The recently obtained X-ray diffraction pattern of fibrils formed by fragments of Sup35 protein (Sup35NM and Sup35N) favors this model [150].
It has been recently shown that the Sup35NM monomers forming a protofibril are oriented relative to each other in such a way that intermolecular hydrogen bonds are formed between identical regions of these molecules [134]. Thus, Sup35NM are positioned relative each other in the orientation “head” (from 25 to 38 aa) to “head” and “tail” (from 91 to 106 aa) to “tail”.
Another model of amyloid protofibril was suggested for the Ure2 protein [151], but it is probably also applicable for Sup35. The structure considered in this model was called beta-serpentine. According to this model, the prion domain of one Ure2 (or Sup35) molecule forms a superpleated beta-structure--serpentine. Serpentines of Ure2 or Sup35 are oriented in register with a slight shift (of approximately 1°) and stabilized by inter- and intramolecular hydrogen bonds. C-Terminal domains of these proteins are not involved in superpleated structure and form a helix around the serpentine.
NATURAL OCCURRENCE OF PRIONS
A general feature of sequences of all S. cerevisiae prion proteins known to date is a high content of asparagine and glutamine residues. This feature is also characteristic of amyloidogenic sequence of human proteins such as huntingtin. It was shown that glutamine-rich sequences tend to form amyloid fibrils in vitro [152], that suggested the possibility of prediction of prion properties of proteins on the basis of their enrichment with glutamine and asparagine residues. The presence of 30 glutamine or asparagine residues over a continuous sequence of 80 aa residues was used as a criterion for identification of such proteins [153]. Analysis of frequency of glutamine-asparagine-rich sequences in different species has shown that in the S. cerevisiae genome 1.69% of ORF (Open Reading Frames) encode such proteins (107 proteins). In Drosophila melanogaster, 472 such proteins were found, which corresponds to 3.47% of all ORF. Taking into account the broad natural occurrence of glutamine-asparagine-rich sequences, one can suppose that prions are not rare in nature. However, it should be noted that not all prion-forming sequences are enriched with asparagine and glutamine residues. For example, they do not include prion domains of PrP and HET-s proteins. Thus, there is still no reliable criterion for evaluation of prion-forming ability of polypeptide sequences, which significantly hinders the prediction of proteins with prion properties.
Recently there appeared data on cytoplasmically inherited phenotypes that may depend on the presence of the yeast prion determinants. At the present time prion proteins responsible for these traits are being intensively searched for. Among them there are the S. cerevisiae determinants [ISP+] and [GAR+] and determinant [cif] of Schizosaccharomyces pombe.
The [ISP+] determinant of S. cerevisiae was discovered in two sup35 mutants with mutations in the 3´ terminal region of the gene [45]. [ISP+] exhibits antisuppressor activity towards these mutations ([ISP+] - Inversion of Suppressor Phenotype). However, [ISP+] is independent of the Sup35 prion domain and may be a prion form of an unknown protein interacting with Sup35. [ISP+] is characterized by dominant manifestation, non-Mendelian type of inheritance, and is reversibly curable using GuHCl, but unlike [PSI+] its maintenance is independent of the Hsp104 chaperon.
S. cerevisiae cells are capable of spontaneous acquiring of the [GAR+] phenotype resistance to the non-hydrolyzed glucose analog D-(+)-glucosamine [46]. The [GAR+] trait exhibits genetic properties of yeast prions. It is inherited as a cytoplasmic determinant, transmitted via cytoduction, and disappears upon deletion of the SSA1 and SSA2 genes encoding chaperons of the Hsp70 family. The protein whose prion properties may define the [GAR+] phenotype is not yet identified.
The prion-like determinant [cif] (calnexin independence factor) of the yeast S. pombe provides the cell viability in the absence of calnexin (Cnx1 protein), a vitally important chaperon of the endoplasmic reticulum. Phenotype Cin (Calnexin independence) is dominantly inherited and upon sporulation is transmitted to the majority of meiotic segregants. It is also transmissible upon transformation of cells with cell extracts free of nucleic acids [48]. At the same time, treatment of extracts of cells with Cin phenotype with proteinase K significantly decreased the efficiency of transmission of the Cin state, which indicated the protein nature of the calnexin independence factor [cif]. The cif1 gene was identified, whose expression from the multicopy plasmid induces calnexin independence. This gene product was called calnexin independence factor Cif1 [47]. The Cif1 protein forms fibrillar aggregates in vitro, which in the case of infection of calnexin-independent cells induces the Cin phenotype. Thus, facts were obtained which show that Cif1 protein, the intracellular function of which is still unknown, is a prion.
PROSPECTS FOR THERAPY OF PRION DISEASES
Prion diseases are presently considered as incurable, although approaches to their therapy are in progress. Spongiform encephalopathies are characterized by the absence of immune response to prion infection. This is due to the fact that the normal PrP form is always present in the organism, including in T and B lymphocytes. However, it was shown in vitro that antibodies to several PrP epitopes inhibit the propagation of PrPSc [154, 155]. Vaccination by recombinant PrP before or immediately after infection and passive immunization by antibodies to some PrP epitopes resulted in inhibition of prion replication and in delay of the disease onset [156, 157]. These experiments confirmed the efficiency of intervention into the immune system for therapy of prion diseases.
The propagation of PrPSc can be stopped using “beta-structure blockers”--peptides enriched with proline and having homology to PrPC [158]. There is also another approach based on the Prnp gene polymorphism. It is known that substitutions Q171R in sheep PrP and E219K in human PrP are inconsistent with formation of the PrPSc prion form. Mutations resulting in such amino acid substitutions in sheep and human PrP were inserted into the mouse Prnp gene [159]. Corresponding recombinant mutant mouse proteins were not converted into the pathological PrP isoform and also inhibited PrPSc formation in the wild-type cell cultures [159]. These mutations had the dominant-negative manifestation, because they hindered conversion of the normal mouse PrP protein to the prion state. To use the dominant-negative PrP mutants in gene therapy of prion diseases, lentivirus vectors for in vivo delivery of DNA encoding them were constructed. It was shown that cell transduction by lentivirus virions, containing the above-described mutant Prnp alleles, results in a significant decrease of PrPSc level in mouse neuroblastoma cultures [160].
Yeast is a convenient model for search and study of factors able to cure cells of prions. Upon expression in yeast cells, mammalian PrP most likely has a tendency to be converted into the prion form, because it is accumulated in the aggregated and protease-resistant form [161]. It would be possible to use yeast containing prion PrPSc for searching drugs with anti-prion properties, but such works either have not been carried out or are not yet published. At the same time, yeast has been used for searching for chemical compounds curing [PSI+] and [URE3] prions. The ability to eliminate [PSI+] was checked for 2500 chemical compounds. Mammalian cells do not contain the Hsp104 chaperon. Therefore, to reveal compounds active towards mammalian prions, the search for anti-prion compounds in yeasts was carried out under conditions of decreased Hsp104 activity. As a result, six compounds eliminating [PSI+] were discovered. Five of them belonged to a new class of molecules, kastellpaolitines, and the sixth one was phenanthridine. All these compounds also cured [URE3] and inhibited formation of PrPSc in mouse neuroblastoma cell culture [162]. It is interesting that two other compounds, anti-prion activity of which was shown previously in mammalian cell culture, namely quinacrine (used as a drug against malaria) and chlorpromazine (used as an antipsychotic drug) cured yeast cells of [PSI+] and [URE3]. It should be noted that all these compounds are active only in mitotically dividing cells and the presence of these agents during several cell generations was necessary for prion curing. Phenanthridine, kastellpaolitines, quinacrine, and chlorpromazine do not act on prion aggregates. They rather affect mechanisms maintaining prion state of proteins. Thus the mechanisms controlling the prion maintenance in mammals and yeasts most likely have features in common. The experiments have shown the efficiency and validity of using the yeast model for searching for agents that cure mammalian prions.
The discovery of prions in yeast and P. anserina has extended our views of prions. It turned out that prions are not only infectious agents, but a universal biological phenomenon as well. In lower eukaryotes, the prion phenomenon is the basis of epigenetic heredity and regulation of gene expression at the posttranslational level. The presence or absence of the [PSI+] determinant in S. cerevisiae can provide adaptive advantages in changing environmental conditions, for example, the presence of [PSI+] inhibits growth in the presence of some sources of nitrogen in some strains, but allows cells of other strains to grow on galactose [41]. Besides, the presence of [PSI+] prion enhances the cell resistance to heat shock [163]. The prion mechanism has potentially useful distinctions compared to the mechanisms of genetic variability. Conversion of proteins to the prion state may occur more frequently than mutations, and manifestation of the prion phenotype may depend on the prion variant. The reverse conversion from prion state to the non-prion one also may be more frequent than reversion of mutations, because cells with the prion phenotype retain information concerning the original state of the protein. This is especially important for adaptive lability of the population, because the temporary phenotype correction in response to the environment variations is often more important that stable phenotype change.
Taking into account the wide occurrence and significance of prions in lower eukaryotes, one can suppose that in higher organisms as well prions can be not only pathogens, but be involved in carrying out physiological functions. Quite recently it has been found that PrP is probably not the only protein of higher eukaryotes with prion properties. The prion-like properties of neuronal isoform of CPEB protein (cytoplasmic polyadenylation element binding protein) of the sea slug Aplysia californica have been demonstrated [164]. CPEB is a translation regulator stimulating polyadenylation of cytoplasmic mRNA and in this way activating their translation. The process of mRNA polyadenylation is switched on by the CPEB binding to CPE (cytoplasmic polyadenylation element) located in the 3´-UTR (3´-non-translated region) of activated mRNA. Prion-like properties of CPEB were discovered during heterologous expression of the gene encoding this protein in yeast cells, since S. cerevisiae is a convenient genetic system for verification of the prion properties. The amino-terminal region of CPEB is extremely rich in glutamine and asparagines (48%), which suggests its prionization ability. It was shown that CPEB could exist in two states--native and aggregated, and just in the aggregated state it was able to bind the CPE signal sequence and thus activate translation of “silent” mRNA. The aggregated active state of CPEB is inherited in yeast as a dominant non-Mendelian determinant and is transmitted from cell to cell via the cytoplasm.
The level of CPEB increases several fold in the presence of serotonin, a mediator of synaptic plasticity involved in the processes of learning and memory [165]. A model was proposed according to which the increase in the expression level of CPEB neuronal form caused by serotonin causes its conversion to the prion state [164]. Thus, synaptic stimulation, accompanied by CPEB multimerization, can cause local activation of translation of “silent” mRNA. The mechanism of signal transduction, including prion conversion, has an advantage, because as the prion state is achieved, it is self-maintained without additional stimulation, providing the possibility of “storage” of information coming from the original impulse. However, up to now no prion properties of CPEB in A. californica neurons were demonstrated.
Four homologs of the CPEB gene were found in the mouse genome [166]. The product of one of these genes, CPEB-3, has an amino-terminal glutamine-rich domain, a product of another CPEB-4 carries a proline-rich domain at the N-terminus, and both proteins are able to aggregate upon expression in yeast cells and neuronal cell culture [167]. It is possible that the existence of mammalian prions involved in the processes of learning and memory soon will be proved.
Originally prions were discovered as infectious agents of new type. However now there is no doubt that significance of prions is much wider and most likely prions are carries of biological information of a specific type--information stored in protein conformation.
This work was supported by grants from the Russian Foundation for Basic Research and the International Science and Technology Center.
REFERENCES
1.Chandler, R. L. (1961) Lancet, 1,
1378-1379.
2.Gajdusek, D. C., Gibbs, C. J., and Alpers, M.
(1966) Nature, 209, 794-796.
3.Alper, T., Cramp, W. A., Haig, D. A., and Clarke,
M. C. (1967) Nature, 209, 764-766.
4.Griffith, J. S. (1967) Nature, 215,
1043-1044.
5.Prusiner, S. B., Garfin, D. E., Cochran, S. P.,
Baringer, J. R., Hadlow, W. J., Eklund, C. M., and Race, R. E. (1978)
Trans. Am. Neurol. Assoc., 103, 62-64.
6.Chesebro, B., Race, R., Wehrly, K., Nishio, J.,
Bloom, M., Lechner, D., Bergstrom, S., Robbins, K., Mayer, L., Keith,
J. M., et al. (1985) Nature, 315, 331-333.
7.Oesch, B., Westaway, D., Walchli, M., McKinley, M.
P., Kent, S. B., Aebersold, R., Barry, R. A., Tempst, P., Teplow, D.
B., Hood, L. E., et al. (1985) Cell, 40, 735-746.
8.Gabriel, J. M., Oesch, B., Kretzschmar, H., Scott,
M., and Prusiner, S. B. (1992) Proc. Natl. Acad. Sci. USA,
89, 9097-9101.
9.Rivera-Milla, E., Stuermer, C. A., and
Malaga-Trillo, E. (2003) Trends Genet., 19, 72-75.
10.Stahl, N., Baldwin, M. A., Teplow, D. B., Hood,
L., Gibson, B. W., Burlingame, A. L., and Prusiner, S. B. (1993)
Biochemistry, 32, 1991-2002.
11.Riek, R., Hornemann, S., Wider, G., Billeter, M.,
Glockshuber, R., and Wuthrich, K. (1996) Nature, 382,
180-182.
12.Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M.,
Serban, A., Groth, D., Mehlhorn, I., Huang, Z., Fletterick, R. J.,
Cohen, F. E., et al. (1993) Proc. Natl. Acad. Sci. USA,
90, 10962-10966.
13.McKinley, M. P., Bolton, D. C., and Prusiner, S.
B. (1983) Cell, 35, 57-62.
14.Prusiner, S. B. (1998) Proc. Natl. Acad. Sci.
USA, 95, 13363-13383.
15.Bueler, H., Fischer, M., Lang, Y., Bluethmann,
H., Lipp, H. P., DeArmond, S. J., Prusiner, S. B., Aguet, M., and
Weissmann, C. (1992) Nature, 356, 577-582.
16.Brandner, S., Isenmann, S., Raeber, A., Fischer,
M., Sailer, A., Kobayashi, Y., Marino, S., Weissmann, C., and Aguzzi,
A. (1996) Nature, 379, 339-343.
17.Blattler, T., Brandner, S., Raeber, A. J., Klein,
M. A., Voigtlander, T., Weissmann, C., and Aguzzi, A. (1997)
Nature, 389, 69-73.
18.Glatzel, M., and Aguzzi, A. (2000) J. Gen.
Virol., 81, 2813-2821.
19.Legname, G., Baskakov, I. V., Nguyen, H. O.,
Riesner, D., Cohen, F. E., DeArmond, S. J., and Prusiner, S. B. (2004)
Science, 305, 673-676.
20.Saborio, G. P., Permanne, B., and Soto, C. (2001)
Nature, 411, 810-813.
21.Castilla, J., Saa, P., Hetz, C., and Soto, C.
(2005) Cell, 121, 195-206.
22.Pattison, I. H. (1965) in Slow, Latent
and Temperate Virus Infections, NINDB Monograph 2
(Gajdusek, D. C., Gibbs, G. J., and Alpers, M. P., eds.) US
Government Printing, Washington DC, pp. 249-257.
23.Prusiner, S. B. (1982) Science,
216, 136-144.
24.Clarke, A. R., Jackson, G. S., and Collinge, J.
(2001) Philos. Trans. R. Soc. Lond. B Biol. Sci., 356,
185-195.
25.Prusiner, S. B., Scott, M., Foster, D., Pan, K.
M., Groth, D., Mirenda, C., Torchia, M., Yang, S. L., Serban, D.,
Carlson, G. A., et al. (1990) Cell, 63, 673-686.
26.Collinge, J., Palmer, M. S., Sidle, K. C.,
Gowland, I., Medori, R., Ironside, J., and Lantos, P. (1995)
Lancet, 346, 569-570.
27.Bruce, M., Chree, A., McConnell, I., Foster, J.,
Pearson, G., and Fraser, H. (1994) Philos. Trans. R. Soc. Lond. B
Biol. Sci., 343, 405-411.
28.Hill, A. F., Desbruslais, M., Joiner, S., Sidle,
K. C., Gowland, I., Collinge, J., Doey, L. J., and Lantos, P. (1997)
Nature, 389, 448-450, 526.
29.Bessen, R. A., and Marsh, R. F. (1994) J.
Virol., 68, 7859-7868.
30.Bessen, R. A., Kocisko, D. A., Raymond, G. J.,
Nandan, S., Lansbury, P. T., and Caughey, B. (1995) Nature,
375, 698-700.
31.Caughey, B., Raymond, G. J., and Bessen, R. A.
(1998) J. Biol. Chem., 273, 32230-32235.
32.Collinge, J., Sidle, K. C., Meads, J., Ironside,
J., and Hill, A. F. (1996) Nature, 383, 685-690.
33.Parchi, P., Castellani, R., Capellari, S.,
Ghetti, B., Young, K., Chen, S. G., Farlow, M., Dickson, D. W., Sima,
A. A., Trojanowski, J. Q., Petersen, R. B., and Gambetti, P. (1996)
Ann. Neurol., 39, 767-778.
34.Telling, G. C., Parchi, P., DeArmond, S. J.,
Cortelli, P., Montagna, P., Gabizon, R., Mastrianni, J., Lugaresi, E.,
Gambetti, P., and Prusiner, S. B. (1996) Science, 274,
2079-2082.
35.Prusiner, S. B. (1991) Science,
252, 1515-1522.
36.Bellinger-Kawahara, C. G., Kempner, E., Groth,
D., Gabizon, R., and Prusiner, S. B. (1988) Virology,
164, 537-541.
37.Jarrett, J. T., and Lansbury, P. T., Jr. (1993)
Cell, 73, 1055-1058.
38.Caughey, B., Kocisko, D. A., Raymond, G. J., and
Lansbury, P. T., Jr. (1995) Chem. Biol., 2, 807-817.
39.Serio, T. R., Cashikar, A. G., Kowal, A. S.,
Sawicki, G. J., Moslehi, J. J., Serpell, L., Arnsdorf, M. F., and
Lindquist, S. L. (2000) Science, 289, 1317-1321.
40.Wickner, R. B. (1994) Science, 264,
566-569.
41.True, H. L., and Lindquist, S. L. (2000)
Nature, 407, 477-483.
42.Coustou, V., Deleu, C., Saupe, S., and Begueret,
J. (1997) Proc. Natl. Acad. Sci. USA, 94, 9773-9778.
43.Derkatch, I. L., Bradley, M. E., Hong, J. Y., and
Liebman, S. W. (2001) Cell, 106, 171-182.
44.Derkatch, I. L., Bradley, M. E., Zhou, P.,
Chernoff, Y. O., and Liebman, S. W. (1997) Genetics, 147,
507-519.
45.Volkov, K. V., Aksenova, A. Y., Soom, M. J.,
Osipov, K. V., Svitin, A. V., Kurischko, C., Shkundina, I. S.,
Ter-Avanesyan, M. D., Inge-Vechtomov, S. G., and Mironova, L. N. (2002)
Genetics, 160, 25-36.
46.Brown, J. C., and Lindquist, S. L. (2005) in
Abstracts of Prion Biology (Joint Cold Spring Harbor
Laboratory/Wellcome Trust Conference).
47.Beauregard, P. B., Guerin, R., Senechal, P.,
Collin, P., and Rokeach, L. A. (2005) in Abstracts of Prion Biology
(Joint Cold Spring Harbor Laboratory/Wellcome Trust
Conference).
48.Collin, P., Beauregard, P. B., Elagoz, A., and
Rokeach, L. A. (2004) J. Cell Sci., 117, 907-918.
49.Lacroute, F. (1971) J. Bacteriol.,
106, 519-522.
50.Tuite, M. F., Mundy, C. R., and Cox, B. S. (1981)
Genetics, 98, 691-711.
51.Aigle, M., and Lacroute, F. (1975) Mol. Gen.
Genet., 136, 327-335.
52.Cooper, T. G. (1982) Basic Life Sci.,
19, 143-161.
53.Magasanik, B. (1992) in The Molecular and
Cellular Biology of the Yeast Saccharomyces, 2nd Edn. (Jones, E.
W., Pringle, J. R., and Broach, J. R., eds.) Cold Spring Harbor
Laboratory Press, Cold Spring Harbor, NY, pp. 283-314.
54.Chisholm, V. T., Lea, H. Z., Rai, R., and Cooper,
T. G. (1987) J. Bacteriol., 169, 1684-1690.
55.Cooper, T. G., Ferguson, D., Rai, R., and Bysani,
N. (1990) J. Bacteriol., 172, 1014-1018.
56.Courchesne, W. E., and Magasanik, B. (1988) J.
Bacteriol., 170, 708-713.
57.Turoscy, V., and Cooper, T. G. (1987) J.
Bacteriol., 169, 2598-2600.
58.Masison, D. C., and Wickner, R. B. (1995)
Science, 270, 93-95.
59.Edskes, H. K., Gray, V. T., and Wickner, R. B.
(1999) Proc. Natl. Acad. Sci. USA, 96, 1498-1503.
60.Komar, A. A., Lesnik, T., Cullin, C., Merrick, W.
C., Trachsel, H., and Altmann, M. (2003) EMBO J., 22,
1199-1209.
61.Coschigano, P. W., and Magasanik, B. (1991)
Mol. Cell Biol., 11, 822-832.
62.Maddelein, M. L., and Wickner, R. B. (1999)
Mol. Cell Biol., 19, 4516-4524.
63.Brachmann, A., Baxa, U., and Wickner, R. B.
(2005) EMBO J., 24, 3082-3092.
64.Saupe, S. J. (2000) Microbiol. Mol. Biol.
Rev., 64, 489-502.
65.Coustou-Linares, V., Maddelein, M. L., Begueret,
J., and Saupe, S. J. (2001) Mol. Microbiol., 42,
1325-1335.
66.Balguerie, A., Dos, R. S., Ritter, C.,
Chaignepain, S., Coulary-Salin, B., Forge, V., Bathany, K., Lascu, I.,
Schmitter, J. M., Riek, R., and Saupe, S. J. (2003) EMBO J.,
22, 2071-2081.
67.Dos Reis, S., Coulary-Salin, B., Forge, V.,
Lascu, I., Begueret, J., and Saupe, S. J. (2002) J. Biol. Chem.,
277, 5703-5706.
68.Maddelein, M. L., Dos Reis, S., Duvezin-Caubet,
S., Coulary-Salin, B., and Saupe, S. J. (2002) Proc. Natl. Acad.
Sci. USA, 99, 7402-7407.
69.Cox, B. (1965) Heredity, 20,
505-521.
70.Liebman, S. W., and Sherman, F. (1979) J.
Bacteriol., 139, 1068-1071.
71.Hawthorne, D. C., and Leupold, U. (1974) Curr.
Top. Microbiol. Immunol., 64, 1-47.
72.Inge-Vechtomov, S. G., and Adrianova, V. M.
(1970) Genetika, 6, 103-115.
73.Cox, B. S., Tuite, M. F., and McLaughlin, C. S.
(1988) Yeast, 4, 159-178.
74.Kushnirov, V. V., Ter-Avanesyan, M. D., Telckov,
M. V., Surguchov, A. P., Smirnov, V. N., and Inge-Vechtomov, S. G.
(1988) Gene, 66, 45-54.
75.Ter-Avanesyan, M. D., Dagkesamanskaya, A. R.,
Kushnirov, V. V., and Smirnov, V. N. (1994) Genetics,
137, 671-676.
76.DePace, A. H., Santoso, A., Hillner, P., and
Weissman, J. S. (1998) Cell, 93, 1241-1252.
77.Doel, S. M., McCready, S. J., Nierras, C. R., and
Cox, B. S. (1994) Genetics, 137, 659-670.
78.Cosson, B., Couturier, A., Chabelskaya, S.,
Kiktev, D., Inge-Vechtomov, S., Philippe, M., and Zhouravleva, G.
(2002) Mol. Cell Biol., 22, 3301-3315.
79.Hoshino, S., Hosoda, N., Araki, Y., Kobayashi,
T., Uchida, N., Funakoshi, Y., and Katada, T. (1999) Biochemistry
(Moscow), 64, 1367-1372.
80.Hosoda, N., Kobayashi, T., Uchida, N., Funakoshi,
Y., Kikuchi, Y., Hoshino, S., and Katada, T. (2003) J. Biol.
Chem., 278, 38287-38291.
81.Bradley, M. E., and Liebman, S. W. (2004) Mol.
Microbiol., 51, 1649-1659.
82.Liu, J. J., Sondheimer, N., and Lindquist, S. L.
(2002) Proc. Natl. Acad. Sci. USA, 99, 16446-16453.
83.Stansfield, I., Jones, K. M., Kushnirov, V. V.,
Dagkesamanskaya, A. R., Poznyakovski, A. I., Paushkin, S. V., Nierras,
C. R., Cox, B. S., Ter-Avanesyan, M. D., and Tuite, M. F. (1995)
EMBO J., 14, 4365-4373.
84.Zhouravleva, G., Frolova, L., Le, G. X., Le
Guellec, R., Inge-Vechtomov, S., Kisselev, L., and Philippe, M. (1995)
EMBO J., 14, 4065-4072.
85.Ter-Avanesyan, M. D., Kushnirov, V. V.,
Dagkesamanskaya, A. R., Didichenko, S. A., Chernoff, Y. O.,
Inge-Vechtomov, S. G., and Smirnov, V. N. (1993) Mol.
Microbiol., 7, 683-692.
86.Frolova, L., Le, G. X., Rasmussen, H. H.,
Cheperegin, S., Drugeon, G., Kress, M., Arman, I., Haenni, A. L.,
Celis, J. E., Philippe, M., et al. (1994) Nature, 372,
701-703.
87.Bailleul, P. A., Newnam, G. P., Steenbergen, J.
N., and Chernoff, Y. O. (1999) Genetics, 153, 81-94.
88.Valouev, I. A., Kushnirov, V. V., and
Ter-Avanesyan, M. D. (2002) Cell Motil. Cytoskeleton, 52,
161-173.
89.Lund, P. M., and Cox, B. S. (1981) Genet.
Res., 37, 173-182.
90.Chernoff, Y. O., Derkach, I. L., and
Inge-Vechtomov, S. G. (1993) Curr. Genet., 24,
268-270.
91.Derkatch, I. L., Chernoff, Y. O., Kushnirov, V.
V., Inge-Vechtomov, S. G., and Liebman, S. W. (1996) Genetics,
144, 1375-1386.
92.Kochneva-Pervukhova, N. V., Poznyakovski, A. I.,
Smirnov, V. N., and Ter-Avanesyan, M. D. (1998) Curr. Genet.,
34, 146-151.
93.Patino, M. M., Liu, J. J., Glover, J. R., and
Lindquist, S. (1996) Science, 273, 622-626.
94.Paushkin, S. V., Kushnirov, V. V., Smirnov, V.
N., and Ter-Avanesyan, M. D. (1996) EMBO J., 15,
3127-3134.
95.Glover, J. R., Kowal, A. S., Schirmer, E. C.,
Patino, M. M., Liu, J. J., and Lindquist, S. (1997) Cell,
89, 811-819.
96.King, C. Y., Tittmann, P., Gross, H., Gebert, R.,
Aebi, M., and Wuthrich, K. (1997) Proc. Natl. Acad. Sci. USA,
94, 6618-6622.
97.Sipe, J. D., and Cohen, A. S. (2000) J.
Struct. Biol., 130, 88-98.
98.Nelson, R., Sawaya, M. R., Balbirnie, M., Madsen,
A. O., Riekel, C., Grothe, R., and Eisenberg, D. (2005) Nature,
435, 773-778.
99.Paushkin, S. V., Kushnirov, V. V., Smirnov, V.
N., and Ter-Avanesyan, M. D. (1997) Science, 277,
381-383.
100.King, C. Y., and Diaz-Avalos, R. (2004)
Nature, 428, 319-323.
101.Tanaka, M., Chien, P., Naber, N., Cooke, R.,
and Weissman, J. S. (2004) Nature, 428, 323-328.
102.Derkatch, I. L., Bradley, M. E., Masse, S. V.,
Zadorsky, S. P., Polozkov, G. V., Inge-Vechtomov, S. G., and Liebman,
S. W. (2000) EMBO J., 19, 1942-1952.
103.Sondheimer, N., and Lindquist, S. (2000)
Mol. Cell, 5, 163-172.
104.Bradley, M. E., Edskes, H. K., Hong, J. Y.,
Wickner, R. B., and Liebman, S. W. (2002) Proc. Natl. Acad. Sci.
USA, 99, 16392-16399.
105.Derkatch, I. L., Uptain, S. M., Outeiro, T. F.,
Krishnan, R., Lindquist, S. L., and Liebman, S. W. (2004) Proc.
Natl. Acad. Sci. USA, 101, 12934-12939.
106.Salnikova, A. B., Kryndushkin, D. S., Smirnov,
V. N., Kushnirov, V. V., and Ter-Avanesyan, M. D. (2005) J. Biol.
Chem., 280, 8808-8812.
107.Chernoff, Y. O., Lindquist, S. L., Ono, B.,
Inge-Vechtomov, S. G., and Liebman, S. W. (1995) Science,
268, 880-884.
108.Parsell, D. A., Kowal, A. S., Singer, M. A.,
and Lindquist, S. (1994) Nature, 372, 475-478.
109.Glover, J. R., and Lindquist, S. (1998)
Cell, 94, 73-82.
110.Lindquist, S. (1997) Cell, 89,
495-498.
111.Shorter, J., and Lindquist, S. (2004)
Science, 304, 1793-1797.
112.Kushnirov, V. V., and Ter-Avanesyan, M. D.
(1998) Cell, 94, 13-16.
113.Eaglestone, S. S., Ruddock, L. W., Cox, B. S.,
and Tuite, M. F. (2000) Proc. Natl. Acad. Sci. USA, 97,
240-244.
114.Cox, B., Ness, F., and Tuite, M. (2003)
Genetics, 165, 23-33.
115.Ness, F., Ferreira, P., Cox, B. S., and Tuite,
M. F. (2002) Mol. Cell Biol., 22, 5593-5605.
116.Ferreira, P. C., Ness, F., Edwards, S. R., Cox,
B. S., and Tuite, M. F. (2001) Mol. Microbiol., 40,
1357-1369.
117.Jung, G., Jones, G., and Masison, D. C. (2002)
Proc. Natl. Acad. Sci. USA, 99, 9936-9941.
118.Grimminger, V., Richter, K., Imhof, A.,
Buchner, J., and Walter, S. (2004) J. Biol. Chem., 279,
7378-7383.
119.Wegrzyn, R. D., Bapat, K., Newnam, G. P., Zink,
A. D., and Chernoff, Y. O. (2001) Mol. Cell Biol., 21,
4656-4669.
120.Kushnirov, V. V., Kryndushkin, D. S., Boguta,
M., Smirnov, V. N., and Ter-Avanesyan, M. D. (2000) Curr. Biol.,
10, 1443-1446.
121.Kryndushkin, D. S., Alexandrov, I. M.,
Ter-Avanesyan, M. D., and Kushnirov, V. V. (2003) J. Biol.
Chem., 278, 49636-49643.
122.Becker, J., Walter, W., Yan, W., and Craig, E.
A. (1996) Mol. Cell Biol., 16, 4378-4386.
123.Oka, M., Nakai, M., Endo, T., Lim, C. R.,
Kimata, Y., and Kohno, K. (1998) J. Biol. Chem., 273,
29727-29737.
124.Cyr, D. M., Lu, X., and Douglas, M. G. (1992)
J. Biol. Chem., 267, 20927-20931.
125.Newnam, G. P., Wegrzyn, R. D., Lindquist, S.
L., and Chernoff, Y. O. (1999) Mol. Cell Biol., 19,
1325-1333.
126.Allen, K. D., Wegrzyn, R. D., Chernova, T. A.,
Muller, S., Newnam, G. P., Winslett, P. A., Wittich, K. B., Wilkinson,
K. D., and Chernoff, Y. O. (2005) Genetics, 169,
1227-1242.
127.Chernoff, Y. O., Newnam, G. P., Kumar, J.,
Allen, K., and Zink, A. D. (1999) Mol. Cell Biol., 19,
8103-8112.
128.Ohba, M. (1997) FEBS Lett., 409,
307-311.
129.Lopez, N., Aron, R., and Craig, E. A. (2003)
Mol. Biol. Cell, 14, 1172-1181.
130.Sondheimer, N., Lopez, N., Craig, E. A., and
Lindquist, S. (2001) EMBO J., 20, 2435-2442.
131.Schwimmer, C., and Masison, D. C. (2002)
Mol. Cell Biol., 22, 3590-3598.
132.Moriyama, H., Edskes, H. K., and Wickner, R. B.
(2000) Mol. Cell Biol., 20, 8916-8922.
133.Santoso, A., Chien, P., Osherovich, L. Z., and
Weissman, J. S. (2000) Cell, 100, 277-288.
134.Krishnan, R., and Lindquist, S. L. (2005)
Nature, 435, 765-772.
135.Parham, S. N., Resende, C. G., and Tuite, M. F.
(2001) EMBO J., 20, 2111-2119.
136.Osherovich, L. Z., Cox, B. S., Tuite, M. F.,
and Weissman, J. S. (2004) PLoS. Biol., 2, E86.
137.Liu, J. J., and Lindquist, S. (1999)
Nature, 400, 573-576.
138.Kochneva-Pervukhova, N. V., Chechenova, M. B.,
Valouev, I. A., Kushnirov, V. V., Smirnov, V. N., and Ter-Avanesyan, M.
D. (2001) Yeast, 18, 489-497.
139.Uptain, S. M., Sawicki, G. J., Caughey, B., and
Lindquist, S. (2001) EMBO J., 20, 6236-6245.
140.Zhou, P., Derkatch, I. L., Uptain, S. M.,
Patino, M. M., Lindquist, S., and Liebman, S. W. (1999) EMBO J.,
18, 1182-1191.
141.DePace, A. H., and Weissman, J. S. (2002)
Nat. Struct. Biol., 9, 389-396.
142.Selkoe, D. J. (2003) Nature, 426,
900-904.
143.Elghetany, M. T., and Saleem, A. (1988)
Stain Technol., 63, 201-212.
144.Sunde, M., and Blake, C. (1997) Adv. Protein
Chem., 50, 123-159.
145.Chiti, F., Stefani, M., Taddei, N., Ramponi,
G., and Dobson, C. M. (2003) Nature, 424, 805-808.
146.Dobson, C. M. (1999) Trends Biochem.
Sci., 24, 329-332.
147.Dobson, C. M. (2001) Philos. Trans. R. Soc.
Lond. B Biol. Sci., 356, 133-145.
148.Perutz, M. F., Pope, B. J., Owen, D., Wanker,
E. E., and Scherzinger, E. (2002) Proc. Natl. Acad. Sci. USA,
99, 5596-5600.
149.Perutz, M. F., Finch, J. T., Berriman, J., and
Lesk, A. (2002) Proc. Natl. Acad. Sci. USA, 99,
5591-5595.
150.Kishimoto, A., Hasegawa, K., Suzuki, H.,
Taguchi, H., Namba, K., and Yoshida, M. (2004) Biochem. Biophys.
Res. Commun., 315, 739-745.
151.Kajava, A. V., Baxa, U., Wickner, R. B., and
Steven, A. C. (2004) Proc. Natl. Acad. Sci. USA, 101,
7885-7890.
152.Perutz, M. F., Johnson, T., Suzuki, M., and
Finch, J. T. (1994) Proc. Natl. Acad. Sci. USA, 91,
5355-5358.
153.Michelitsch, M. D., and Weissman, J. S. (2000)
Proc. Natl. Acad. Sci. USA, 97, 11910-11915.
154.Enari, M., Flechsig, E., and Weissmann, C.
(2001) Proc. Natl. Acad. Sci. USA, 98, 9295-9299.
155.Peretz, D., Williamson, R. A., Kaneko, K.,
Vergara, J., Leclerc, E., Schmitt-Ulms, G., Mehlhorn, I. R., Legname,
G., Wormald, M. R., Rudd, P. M., Dwek, R. A., Burton, D. R., and
Prusiner, S. B. (2001) Nature, 412, 739-743.
156.Sigurdsson, E. M., Sy, M. S., Li, R.,
Scholtzova, H., Kascsak, R. J., Kascsak, R., Carp, R., Meeker, H. C.,
Frangione, B., and Wisniewski, T. (2003) Neurosci. Lett.,
336, 185-187.
157.White, A. R., Enever, P., Tayebi, M., Mushens,
R., Linehan, J., Brandner, S., Anstee, D., Collinge, J., and Hawke, S.
(2003) Nature, 422, 80-83.
158.Soto, C., Kascsak, R. J., Saborio, G. P.,
Aucouturier, P., Wisniewski, T., Prelli, F., Kascsak, R., Mendez, E.,
Harris, D. A., Ironside, J., Tagliavini, F., Carp, R. I., and
Frangione, B. (2000) Lancet, 355, 192-197.
159.Kaneko, K., Zulianello, L., Scott, M., Cooper,
C. M., Wallace, A. C., James, T. L., Cohen, F. E., and Prusiner, S. B.
(1997) Proc. Natl. Acad. Sci. USA, 94, 10069-10074.
160.Crozet, C., Lin, Y. L., Mettling, C.,
Mourton-Gilles, C., Corbeau, P., Lehmann, S., and Perrier, V. (2004)
J. Cell Sci., 117, 5591-5597.
161.Ma, J., and Lindquist, S. (1999) Nat. Cell
Biol., 1, 358-361.
162.Bach, S., Talarek, N., Andrieu, T., Vierfond,
J. M., Mettey, Y., Galons, H., Dormont, D., Meijer, L., Cullin, C., and
Blondel, M. (2003) Nat. Biotechnol., 21, 1075-1081.
163.Eaglestone, S. S., Cox, B. S., and Tuite, M. F.
(1999) EMBO J., 18, 1974-1981.
164.Si, K., Lindquist, S., and Kandel, E. R. (2003)
Cell, 115, 879-891.
165.Si, K., Giustetto, M., Etkin, A., Hsu, R.,
Janisiewicz, A. M., Miniaci, M. C., Kim, J. H., Zhu, H., and Kandel, E.
R. (2003) Cell, 115, 893-904.
166.Theis, M., Si, K., and Kandel, E. R. (2003)
Proc. Natl. Acad. Sci. USA, 100, 9602-9607.
167.Allen, K. D., Si, K., Theis, M., and Kandel, E.
R. (2005) in Abstracts of Prion Biology (Joint Cold Spring Harbor
Laboratory/Wellcome Trust Conference).