Cooperation of Photosystem I with the Plastoquinone Pool in Oxygen Reduction in Higher Plant Chloroplasts
B. N. Ivanov
Institute of Basic Biological Problems, Russian Academy of Sciences, ul. Institutskaya 2, 142290 Pushchino, Moscow Region, Russia; fax: (496) 733-0532; E-mail: ivabor@issp.serpukhov.su
Received April 27, 2007; Revision received June 1, 2007
The possible functions of a light-induced electron transfer to oxygen in the photosynthetic electron transport chain of higher plant chloroplasts are considered. The thermodynamic preconditions, as well as the experimental data about the participations of ferredoxin, the components of photosystems I and II, and plastoquinone in oxygen reduction are examined. It is concluded that, even in the presence of ferredoxin and ferredoxin + NADP+, oxygen reduction is carried out mainly by the membrane-bound carriers of the photosynthetic electron transport chain. The hypothesis is put forward that most superoxides, which are produced by reduction of O2 molecules by the intramembrane components of the acceptor side of photosystem I, are reduced within the membrane by the plastohydroquinone molecules to the hydrogen peroxide. It is assumed that the H2O2 molecules that originate as the result of this process serve for signaling about the redox state of the plastoquinone pool.
KEY WORDS: chloroplast, thylakoid membrane, electron transport chain, plastoquinone pool, oxygen, superoxide radicalDOI: 10.1134/S0006297908010173
Abbreviations: Chl) chlorophyll; DNP-INT) dinitrophenyl-2-iodo-4-nitrothymol; Fd) ferredoxin; FNR) ferredoxin-NADP-reductase; PETC) photosynthetic electron transport chain; PQ) plastoquinone; PQH*) plastosemiquinone; PQH2) plastohydroquinone, plastoquinol; PSI) photosystem I; PSII) photosystem II; QA and QB) primary and secondary electron acceptors of PSII, specialized membrane-bound forms of PQ.
POSSIBLE FUNCTIONS OF OXYGEN REDUCTION IN THE PHOTOSYNTHETIC
ELECTRON TRANSPORT CHAIN
Demonstration of the formation of hydrogen peroxide (H2O2) during illumination of isolated chloroplasts [1] was the first evidence of the reduction of O2 molecules in the course of photosynthetic electron transport. Later it was shown that, even under conditions optimal for photosynthesis, the rate of the concurrent oxygen reduction can be up to 30% of the rate of CO2 reduction [2, 3]. Many possible physiological functions of the transfer of the electrons coming into a photosynthetic electron transport chain (PETC) from water oxidation to oxygen are discussed in the literature [4-8]. The drain of electrons to oxygen, decreasing the reduction level of PETC components, protects them from photoinhibition, which is stimulated under anaerobic conditions [4]. This drain also promotes operation of photosystem I (PSI) cyclic electron flow, which requires a certain degree of plastoquinone (PQ) pool oxidation [5]. Proton accumulation in the intrathylakoid space due to electron transport to oxygen under conditions of CO2 deficiency can provide protection against photoinhibition owing to the increase in energy dissipation in the pigment matrix; this increase results from both the structural changes of light-harvesting complexes mediated by exchange of magnesium ions for protons, and the increase in the zeaxanthin content in these complexes on the activation of violaxanthin-deepoxidase [6]. The proton gradient occurring across the thylakoid membrane in the course of electron flow to O2 was widely considered as the source of ATP synthesis supplementing that initiated by electron flow to NADP+ [7]. The current values of both H+/e- ratio and H+/ATP ratio, 3 and 4, respectively, provide, through electron flow to NADP+, an ATP/NADPH ratio equal to 1.5, which is required for the Calvin cycle to operate. However, stationary photosynthesis requires an amount of ATP exceeding that used in the Calvin cycle, since ATP is consumed also in the processes of metabolite transport, of biosynthesis of polysaccharides and proteins, etc.; so additional ATP synthesis? at the expense of either electron flow to oxygen, or PSI cyclic flow ?seems necessary. H2O2 is the main signaling molecule in plant cells [8], and its formation in the PETC is an additional and now widely studied function of oxygen reduction in the course of photosynthetic electron transport.
In spite of the important functions attributed to oxygen reduction in the PETC, the mechanisms of this process remain unclear. There is no consensus as to what chloroplast component serves as the basic immediate reductant of O2 molecules; either the membrane-bound carriers of the PETC, or ferredoxin (Fd), or the stromal components reduced by ferredoxin.
OXYGEN REDUCTION IN CHLOROPLASTS OUTSIDE THYLAKOID MEMBRANES. THE
ROLE OF FERREDOXIN
Robinson [9] supposed that the enzymes participating in stromal metabolism might be the basic direct reductants of O2 molecules; this role was suggested for the enzymes (reduced by Fd) in nitrogen metabolism in the absence of the specific substrate. The assumption about participation of the stromal components in O2 reduction has been put forward again [10, 11] to explain the differences in both the rates and the characteristics (Km, etc.) of oxygen reduction in isolated thylakoids versus intact chloroplasts. From experiments with isolated thylakoids supplied with various flavoenzymes, which stimulated oxygen reduction, it was assumed that the direct reductant of O2 molecules by the electrons from the PETC is monodehydroascorbate-reductase [11]. However, this enzyme when reduced was oxidized by oxygen rather slowly in the absence of thylakoids [11], and additional studies on its possible participation in electron transfer from the PETC to oxygen are required.
The water-soluble component of PETC, Fd, which has a low redox-potential (figure), when added to the isolated thylakoids also stimulated oxygen reduction, and the level of the stimulation depended on the concentration of this protein [12-14]. This fact implied that in vivo Fd also plays a significant role in oxygen reduction. However, in these studies, in order to reach oxygen reduction rates close to the maximum rates of electron transport along the PETC, high Fd concentrations were required, corresponding to a Fd/Chl ratio of 5-10 in the experimental vessel [14], which is more than three orders greater than in vivo. The requirement for such high concentrations was explained by the necessity to accumulate high levels of reduced Fd, if the rate of its oxidation by oxygen is low. Actually, the pseudo-first order rate constant of the direct oxidation of reduced Fd by oxygen was found to be low, namely, 0.1-0.2 sec-1 [15, 16]. In a number of studies, the participation of Fd in oxygen reduction in vivo was not counted at all, and this process was considered to be mediated only by membrane-bound PETC carriers (see [10]). However, there were no experimental data in the literature about the ratio of rates of two possible electron flows to oxygen, namely, through Fd and immediately from membrane-bound PETC carriers situated “before” Fd. Recently, in experiments with isolated thylakoids supplied with Fd (added at concentrations corresponding to Fd/Chl ratio of ~1 in the vessel when NADP+ reduction rates were close to the rates observed in vivo), we have found that only 30-50% of electrons participating in oxygen reduction are transferred through Fd, both in the absence and in the presence of NADP+ [17]. In vivo, Fd is oxidized not only by ferredoxin-NADP-reductase (FNR) transferring the electrons to NADP+, but also by other stromal enzymes; so, its participation in oxygen reduction may be even lower. Thus, the main electron flow to oxygen takes a course from the membrane-bound carriers of the PETC. This flow in experiments with isolated thylakoids was either 5 or 60% of the total linear electron flow along the PETC in the presence and in the absence of NADP+, respectively [17].
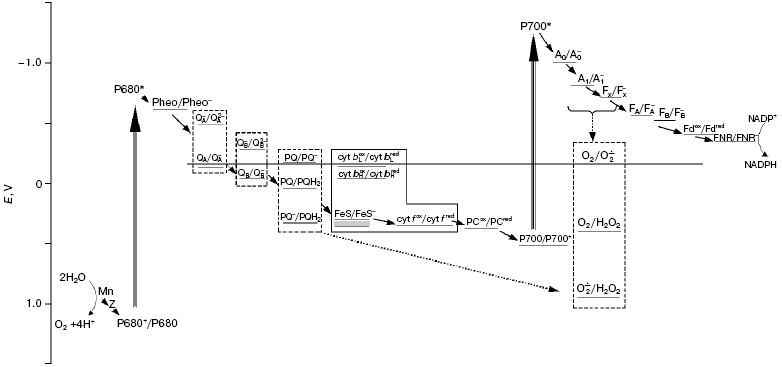
Scheme of the photosynthetic electron transport chain including the oxygen pool. The dashed line shows the redox potential of the O2/O2*- pair. The solid line outlines components of a b6/f-complex; a dashed line outlines the components having several redox forms. The dotted arrows show both the reduction of O2 molecules by the carriers of PSI within the membrane, and the reduction of superoxide radicals formed by plastohydroquinone molecules. The redox potentials are shown at pH 7.0. Mn, manganese cluster of the water-oxidizing complex; Z, redox-active tyrosine of D1 polypeptide; P680, primary electron donor in PSII; Pheo, pheophytin; QA and QB, primary and secondary quinone acceptors in PSII; PQ, PQH2, and PQH* are plastoquinone, plastoquinol, and plastosemiquinone, respectively; cyt bL and cyt bH, low- and high-potential hemes of cytochrome b6; FeS, the Rieske iron-sulfur center; cyt f, cytochrome f; PC, plastocyanin; P700, primary electron donor in PSI; A0 and A1, primary and secondary electron acceptors in PSI; FX, FA, and FB are tertiary iron-sulfur acceptors in PSI; Fd, ferredoxin; FNR, ferredoxin-NADP reductase
OXYGEN REDUCTION BY MEMBRANE-BOUND CARRIERS OF THE PHOTOSYNTHETIC CHAIN
Fd-NADP-reductase (FNR) and PSI complex. In earlier work [13], two reactions of the Fd-dependent reduction of O2 were found, and the value of Km(Fd) of one of them (the smaller) was close to the value of Km(Fd) of the reaction of NADP+ reduction under the same experimental conditions [18]. The analysis of data suggested that FNR participated in the electron transfer to O2. Thermodynamically possible (figure), the participation of FNR in the process of O2 reduction was proposed also by other researchers [19], but until now there has not been enough experimental data in favor of this pathway.
It is commonly accepted that reduction of oxygen by the membrane carriers of the PETC is almost entirely carried out by the components of the acceptor side of PSI [10]. They have sufficiently low redox potentials (figure) to effectively reduce an O2 molecule to a superoxide radical, O2*-, which is considered to be the main reduced oxygen species initially generated in thylakoids [20]. Asada et al. [20], from experiments with membrane fragments enriched with PSI, for the first time assumed that O2*- is generated within the thylakoid membrane. Further studies [21, 22] demonstrated additional evidence in favor of intramembrane reduction of O2 in PSI complex; in particular, the kinetics of cytochrome c reduction after a short (10 µsec) flash was interpreted as reflecting the diffusion of superoxide from the place of its generation to the membrane surface [21]. The immediate reductant of O2 in PSI has not been established precisely. The iron-sulfur clusters FA and FB, located in the PSI subunit exposed into stroma [23], produce superoxide, most likely outside of a lipid membrane phase. The reduction of oxygen by the PSI acceptors buried in the membrane, namely, by A1 (phyloquinone) and FeS cluster Fx, having Em equal to -820 and -730 mV, respectively [24], is very favorable thermodynamically (figure). It is necessary to take also into account the higher concentration of O2 in the hydrophobic membrane phase than in the aqueous solution. The fast oxidation of these carriers by their oxidizers, Fx and FA/B, respectively, within the PSI complex (?t1/2 are equal to 200 and 250 nsec, respectively) can decrease the probability of their reactions with O2 molecules. However, these times have been measured when electron transfer was initiated by rare flashes, i.e. under conditions when all the components of the PSI acceptor side were oxidized. The maximal rates of the reduced Fd usage in the metabolic reactions in stroma (Calvin cycle, activation of enzymes, reductions of nitrogen, oxaloacetate, and glutathione) in saturating light correspond to a transfer of 1e- from FA/B to Fd per 4-8 msec [9]. If the rate of electron input to Fx and/or A1 is higher, then the accumulation of these carriers in the reduced states and, accordingly, their reactions with O2 become very probable.
PSII complex. The inhibition of oxygen reduction by diuron in intact thylakoids is regarded as evidence that QA, the primary electron acceptor in PSII does not participate in this process [20]. Actually, the redox potential of the QA/QA- pair is slightly more positive than that of the O2/O2·- pair (figure), and doubly reduced QA2- does not accumulate under physiological conditions [25]. E'0 of the QB/QB- pair, the secondary electron acceptor in PSII, -45 mV [26], is even more positive. However, diuron-sensitive and having low rate (0.5-1.5 µmol/mg Chl per h), H2O2 formation was found in the thylakoid fragments enriched with PSII [27]; this was interpreted as being the result of oxygen reduction by QB. Possibly, reduction of O2 molecules at the acceptor side of PSII was induced just by the membrane fragmentation, which could lead, for instance, to an increase in the lifetime of the reduced QB-. Oxygen consumption in PSII membrane fragments is considerably stimulated after damage to the water-oxidizing complex [28]. We concluded that the fundamental contribution of this stimulation is the embedding of O2 molecules in the peroxides of organic molecules, a process which is promoted by oxidation of these molecules by the long-lived cation-radical P680+. Experiments, however, implied that this stimulation of oxygen consumption is partially due to an increase in oxygen reduction [28]; but what component of PSII transfers the electrons to oxygen is not yet clear (see review [29]).
Cytochrome b6f complex. The low potential components of the b6f-complex are not usually considered as participants in the reduction of O2 in higher plant thylakoids. The high potential heme of cytochrome b6, which is reduced after the first oxidation of plastohydroquinone (PQH2), has Em of only -45 mV [30]. However, the potential of the low potential heme was estimated to be -150 mV [30] or even -170 mV [31]; and if electron transfer to high potential heme is hampered, the electron from the low potential heme can be accepted by molecular O2. This process, with low effectiveness due to small difference in the values of the redox potentials of the corresponding pairs (figure), would compete with the Q-cycle, and this could have a regulating function as was earlier assumed [32]. The fact that inhibition by antimycin of the cytochrome b oxidation in bc1-complexes of mitochondria and yeast provoked stimulation of oxygen reduction, together with analysis of the action of inhibitors of other reactions in this complex, led to the conclusion that ubisemiquinone arising in a quinol-oxidizing site of both complexes serves as the reductant of O2 [33, 34]. Such data are absent for the cytochrome complex of the thylakoid membranes, but this process cannot be excluded.
Plastoquinone pool. The possibility of functioning of the so-called plastid terminal oxidase in the thylakoid membrane, capable of oxidizing the quinols by oxygen with formation of water, is considered in the literature [35]. Available data fail to confirm, however, any significant participation of this enzyme, which is present there in very small amounts in the oxidation of PQ pool in the thylakoids of higher plants [36].
Components of the PQ pool are the only intramembrane carriers, which are not included in the molecular complexes where electron transfer pathways are structurally determined. This circumstance provides them with an opportunity to react with carriers outside the PETC. The light-induced oxygen consumption in isolated thylakoids in the presence of dinitrophenyl-2-iodo-4-nitrothymol (DNP-INT), a non-autooxidizable inhibitor of PQH2 oxidation by the b6f-complex, has been revealed [37]. Diuron as well as catalase suppressed this consumption almost to zero, indicating respectively both that the reduction of O2 occurs “after” PSII and that it is accomplished by electrons coming into the PETC from water oxidation. Since an appreciable contribution of the PSII acceptor side components into the oxygen reduction in intact thylakoids is absent (see above), conspicuous difference in the pH dependence of the oxygen consumption rate between the untreated thylakoids and those treated with DNP-INT demonstrated the existence of oxygen reduction in the PQ pool.
It was concluded [37] that an immediate reductant of O2 molecules in the PQ pool is plastosemiquinone (PQ*-):
PQ*- + O2 = PQ + O2*-. (1)
Thermodynamic properties of both pairs, PQ/PQ*- (pK = 6.0, E'0 = -170 mV [31]) and O2/O2*- (pK = 4.8, E'0 = -160 mV), satisfactorily explained the increase in the oxygen consumption rate in the presence of DNP-INT, from close to zero at pH 5.0, up to stationary level starting at pH 6.2-6.5 [37]. In this pH range, the difference in the redox potentials of these pairs changed from +50 mV (unfavorable for O2 reduction) to -10 mV (favorable). One-electron reduction of O2 was supported by the fact that ascorbate as a superoxide trap increased the rate of oxygen consumption, and this increase was completely suppressed by superoxide dismutase [37]. Generation of the superoxide radical in the light in the presence of DNP-INT was observed also with the use of a special electrode [38].
Calculated from the rate of oxygen consumption, the rate of superoxide generation in the PQ pool turned out to be higher than the rate of its appearance in the medium; and it was assumed [37] that superoxide formed in the PQ pool was “up-reduced” to H2O2 by PQH2 within the membrane:
PQH2 + O2*- = PQ*- + H2O2. (2)
Reaction (2) is thermodynamically favorable owing to the appreciable difference in the values of E'0 of the pairs PQ*-/PQH2, 370 mV [31], and O2*-/H2O2, 940 mV; the calculated equilibrium constant is higher than 109. Thus, the reduction of oxygen in the PQ pool occurs within the membrane as an autocatalytic process, namely, PQ*- produced in reaction (2) reduces a new molecule of O2 in reaction (1), and so forth.
If the dismutation reaction is the sole route of semiquinone production, its concentration in the PQ pool depends on the presence of protons. The equilibrium constant of the reaction
QH2 + Q = 2QH* (3)
for ubiquinone has been given as 10-10 [39]. However, for the reaction
QH2 + Q = 2Q*- + 2H+, (4)
which, for plastoquinone, is possible in the pH range from 6.0 (pK of PQH*) to 10.8 (pK1 of PQH2 [31]) the thermodynamic calculation of the equilibrium constant from the values of the redox potentials of the pairs PQ/PQ*- and PQ*-/PQH2 gives a value of 10-3 for pH 10. The membrane phase is considered as “aprotic”, and the volumetric molar concentration of protons in this phase can be even less than in aqueous solution at pH 10; however, transient occurrence of the semiquinone charged head in the water-bearing area of the membrane is possible. An additional generation of semiquinone in reaction (2) can increase the stationary concentration of this species and, accordingly, the rate of oxygen reduction.
With increasing light intensity, the rate of oxygen reduction in thylakoids in the presence of DNP-INT was quickly saturated and turned out to be estimably less than the rate in the total PETC in the absence of inhibitor, with dioxygen as a sole electron acceptor in both cases [40]. However, the participation of the PQ pool in oxygen reduction in the total PETC did increase and reached 60-70% at pH 6.5 and 7.8 [41]. At pH 5.0, when PQH* for thermodynamic reasons cannot reduce O2, as has been observed experimentally [37], participation of PQ pool in oxygen reduction at high light intensity was, nevertheless, 50% [41]. These unexpected results led to the assumption that molecules of PQH2 could reduce the superoxide produced not only in the PQ pool but also in the acceptor side of PSI, the main site of reduction of O2 molecules in the PETC. Thus, PQ pool participation in oxygen reduction at pH 5.0 is determined by the fact that PQH2 molecules do participate in this process; that this participation is close to 50% indicates that, at high light intensity, virtually all the superoxides originating from PSI are reduced by PQH2. The higher contribution of the PQ pool to oxygen reduction at higher pH values, obviously, is the result of participation of PQ*- in the reduction of O2 molecules at these pH values.
The concentration of plastoquinone molecules in the thylakoid membrane, 20 per 1000 Chl, is an order of magnitude greater than that of the reaction centers of PSII [42]; the quinone and quinol forms quickly diffuse in the membrane [43]; and they spread along the whole membrane interior, with a slightly higher concentration in the stromal lamella [44]. The microdomain PQ pool organization assumed on the basis of measurements under infrequent flashes [45], obviously, does not influence its reduction under continuous illumination: an average time of domain reorganization was estimated to be 60 msec, and a full reduction of the entire PQ pool occurred for less than 2 sec [45].
The data obtained earlier [21, 46] show that superoxide generated in the thylakoid membrane via oxygen reduction is preserved there over the seconds time range. In studies by Asada [10], it was repeatedly stressed that the superoxide radical is rather stable in the aprotic phase of the thylakoid membrane. It was recently shown that even in aqueous solution at pH 11, the half-life of superoxide was close to 15 sec [47]. The permeability of the membrane for superoxide was estimated to be 20 nm/sec [21], i.e. during its lifetime it is capable of diffusing inside the membrane for a long distance from the place of its generation, and the negative surface charges at the membrane boundaries impede its escape from the membrane. Since, under continuous illumination, the PQ pool is reduced along the whole membrane, the probability of reaction of the superoxide generated in PSI with PQH2 molecules is very high.
Characteristics of the PQ pool oxidation after illumination can serve as confirmation of the reaction of PQH2 with superoxide. It was found that in the thylakoids [48, 49], as well as in leaves [36], this oxidation, estimated from an increase of the area above the fluorescence induction curve, consisted of two phases, the first fast and the second slow. In our work [50], the same kinetics was found at the post-illumination appearance of oxidized plastoquinone. In thylakoids, the PQ pool reduced in the light was not oxidized in the dark under anaerobic conditions [50, 51], and under aerobic conditions, the apparent first order rate “constant” for the first phase of oxidation [50] was approximately three orders higher than the constant of oxidation of PQH2 molecules placed in liposomes [51]. This distinction can be the consequence of oxidation of PQH2 only in the thylakoids, by superoxide being accumulated in the thylakoid membrane in the light. The “constants” of the first phases and a percentage ratio of two phases depended on light intensity during illumination: “the constant”, as well as the contribution of the first fast phase, were higher after illumination with stronger light [50]. It was found [50] that the effect of the light intensity on the characteristics of PQ pool oxidation only correlated with an increase in the rate of oxygen reduction in PSI under increasing light intensity. This correlation confirmed the coupling of the initial rate of PQ pool oxidation with superoxide accumulation in the membrane in the light.
INCLUDING THE POOL OF OXYGEN INTO ELECTRON TRANSPORT IN THE
THYLAKOID MEMBRANE
The above review leads to the suggestion that, in the thylakoid membrane, the components of the acceptor side of PSI function in concert with plastoquinone pool to reduce O2 to H2O2, namely, superoxide produced from O2 in PSI is reduced to H2O2 by PQH2. This process seems to be an integral part of the electron transport system in the chloroplasts of higher plants. The PETC scheme taking into account the values of the redox potentials of the participants of this cooperative oxygen reduction is presented in the figure. Oxygen content in the thylakoid membrane (2-10 per 1000 Chl assuming the membrane volume as 4.6 ml per gram of Chl [52]) exceeds that of other PETC carriers excepting plastoquinone; the content is higher in the hydrophobic areas where the solubility of the gas is higher. The population of oxygen molecules residing in the membrane under stationary conditions can be labeled as oxygen pool. Similar to the PQ pool, the oxygen pool includes oxidized (O2), singly reduced (O2*-) and doubly reduced (H2O2) molecules (figure). These species are the reactants and the products of the above two consecutive reactions of O2 reduction to H2O2. However, in contrast to the PQ pool, the intramembrane oxygen pool serves as the final electron acceptor and has dynamic steady-state composition: O2 molecules including those formed due to water oxidation constantly come to the membrane, as they are converted there into more reduced species, i.e. superoxide and peroxide.
It is known that in the thylakoids of higher plants the amount of the reaction centers of PSII is 2-3 times higher than the amount of the reaction centers of PSI [42, 53]. Some sophisticated mechanisms capable of providing the ratio of 1 for the reaction centers of the photosystems, which participate in stationary linear electron transport, were proposed [54]. It follows from the PETC model proposed in this work that possibly there is no requirement of this “diminution” of the reaction center ratio.
Under the majority of stress conditions, the stomata are closed and CO2 influx to the chloroplasts is limited. This, in turn, results in deficiency of NADP+ in the stroma. This not only stimulates O2 reduction at the acceptor side of PSI, but also increases the level of PQ pool reduction, since PQH2 oxidation by a b6f-complex is slowed down due to the lowered ATP consumption. Under such conditions, the significance of the proposed cooperation of the oxygen reduction reactions obviously increases.
The cooperation in oxygen reduction of the PETC segments separated by photoreaction is capable of increasing adjustability of the electron transport system to constantly changing environmental conditions. Connection of these segments through the oxygen pool can coordinate regulation of changes in the electron transport. In particular, reduction by the components of the PQ pool of the superoxide generated in PSI within the membrane promotes effective drain of electrons from the acceptor side of PSII, decreasing the danger of the photoinhibition of this photosystem. Superoxide reduction by the electrons from the PQ pool can also be considered as one way to protect the membrane components from these destructive molecules. It was found that sustaining the PQ pool in the reduced state significantly decreased lipid peroxidation and pigment bleaching in the thylakoid membranes [55].
The proposed scheme implies that a significant share of the H2O2 molecules generated due to operation of the PETC should be formed within the thylakoid membrane, where reaction of PQH2 with superoxide occurs. In fact, it was shown experimentally that, under conditions when the participation of the PQ pool in oxygen reduction exceeds 50%, approximately the same share of electrons coming into the PETC at water splitting participates in H2O2 formation within the membrane [56, 57]. Both values were less at lower light intensities. Participation of the PQ pool in oxygen reduction has one more important consequence. It is known that the change of the redox state of the PQ pool in the chloroplasts initiates many of the adaptive reactions in a plant cell (and even in surrounding cells where this state does not change), in particular, control of transcription of chloroplast and nuclear genes [58]. The mechanism of this communication is still unclear. However, a logical candidate for the signaling agent is H2O2 molecules formed in the thylakoid membrane with participation of PQH2 as described above.
The author expresses deep gratitude to S. A. Khorobrykh, M. M. Mubarakshina, and M. A. Kozuleva for the participation in the work allowing the presented hypothesis to be put forward and to Prof. Edwards for valuable comments and the help in the course of preparation of the manuscript.
This work was supported by grant 05-04-48629 from the Russian Foundation for Basic Research.
REFERENCES
1.Mehler, A. H. (1951) Arch. Biochem.
Biophys., 33, 65-77.
2.Osmond, C. B., and Grace, C. E. (1995) J. Exp.
Bot., 46, 1351-1362.
3.Badger, M. R., von Caemmerer, S., Ruuska, S., and
Nakano, H. (2000) Phil. Trans. R. Soc. Lond. B, 355,
1433-1446.
4.Park, Y.-I., Chow, W. S., Osmond, C. B., and
Anderson, J. M. (1996) Photosynth. Res., 50, 23-32.
5.Ziem-Hanck, U., and Heber, U. (1980) Biochim.
Biophys. Acta, 591, 266-274.
6.Horton, P., Ruban, A. V., and Walters, R. G. (1996)
Annu. Rev. Plant Physiol. Plant Mol. Biol., 47,
655-684.
7.Heber, U. (1973) Biochim. Biophys. Acta,
305, 140-152.
8.Foyer, C. H., and Noctor, G. (2003) Physiol.
Plant., 119, 355-364.
9.Robinson, J. M. (1988) Physiol. Plant.,
72, 666-680.
10.Asada, K. (1999) Ann. Rev. Plant Physiol.
Plant Mol. Biol., 50, 601-639.
11.Miyake, C., Schreiber, U., Hormann, H., Sano, S.,
and Asada, K. (1998) Plant Cell Physiol., 39,
821-829.
12.Allen, J. (1975) Nature, 256,
599-600.
13.Ivanov, B. N., Red'ko, T. P., Shmeleva, V. L.,
and Mukhin, E. N. (1980) Biokhimiya, 45, 1425-1432.
14.Furbank, R. T., and Badger, M. R. (1983)
Biochim. Biophys. Acta, 723, 400-409.
15.Golbeck, J. H., and Radmer, R. (1984) in
Advances in Photosynthesis Research, Vol. I (Sybesma, C., ed.),
Martinus Nijhoff/Dr. W. Junk Publishers, The Hague, pp. 561-564.
16.Hosein, B., and Palmer, G. (1983) Biochim.
Biophys. Acta, 723, 383-390.
17.Kozuleva, M. A., Naidov, I. A., Mubarakshina, M.
M., and Ivanov, B. N. (2007) Biofizika, 52, 650-655.
18.Red'ko, T. P., Shmeleva, V. L., Ivanov, B. N.,
and Mukhin, E. N. (1982) Biokhimiya, 47, 1695-1699.
19.Goetze, D. C., and Carpenter, R. (1994) Can.
J. Bot., 72, 256-260.
20.Asada, K., Kiso, K., and Yoshikawa, K. (1974)
J. Biol. Chem., 249, 2175-2181.
21.Takahashi, M., and Asada, K. (1988) Arch.
Biochem. Biophys., 267, 714-722.
22.Hormann, H., Neubauer, C., Asada, K., and
Schreiber, U. (1993) Photosynth. Res., 37, 69-80.
23.Fromme, P., Jordan, P., and Kraub, N. (2001)
Biochim. Biophys. Acta, 1507, 5-31.
24.Hoshina, S., and Itoh, S. (1993) in
Photosynthesis. Photoreactions to Plant Productivity (Abrol, Y.
P., Mohanty, P., and Govindjee, eds.) Oxford & IBH Publishing, New
Delhi, pp. 51-82.
25.Hideg, E., Kalai, T., Hideg, K., and Vass, I.
(1998) Biochemistry, 37, 11405-11411.
26.Diner, B. A., Petrouleas, V., and Wendoloski, J.
J. (1991) Physiol. Plant., 81, 423-436.
27.Zastrizhnaya, O. M., Khorobrykh, A. A., Khristin,
M. S., and Klimov, V. V. (1997) Biochemistry (Moscow),
62, 357-361.
28.Khorobrykh, S. A., Khorobrykh, A. A., Klimov, V.
V., and Ivanov, B. N. (2002) Biochemistry (Moscow), 67,
683-688.
29.Ivanov, B., and Khorobrykh, S. (2003) Antiox.
Redox. Signal., 5, 43-53.
30.Kramer, D. M., and Crofts, A. R. (1994)
Biochim. Biophys. Acta, 1184, 193-201.
31.Hauska, G., Hurt, E., Gabellini, N., and Lockau,
W. (1983) Biochim. Biophys. Acta, 726, 97-133.
32.Ivanov, B. N. (1993) in Photosynthesis.
Photoreactions to Plant Productivity (Abrol, Y. P., Mohanty, P.,
and Govindjee, eds.) Oxford & IBH Publishing, New Delhi, pp.
109-128.
33.Ksezenko, M., Konstantinov, A. A., Khomutov, G.
B., Tikhonov, A. N., and Ruuge, E. K. (1983) FEBS Lett.,
155, 19-24.
34.Muller, F., Crofts, A. R., and Kramer, D. M.
(2002) Biochemistry, 41, 7866-7874.
35.Aluru, M., and Rodermel, S. R. (2004) Physiol.
Plant., 120, 4-11.
36.Nixon, P. J., and Rich, P. R. (2006) in The
Structure and Function of Plastids (Wise, R. R., and Hoober, J. K.,
eds.) Springer, The Netherlands, pp. 237-251.
37.Khorobrykh, S. A., and Ivanov, B. N. (2002)
Photosynth. Res., 71, 209-219.
38.Cleland, R. E., and Grace, S. C. (1999) FEBS
Lett., 457, 348-352.
39.Mitchell, P. (1976) J. Theor. Biol.,
62, 327-367.
40.Mubarakshina, M. M. (2006) PhD Thesis, Institute
of Basic Biological Problems, Russian Academy of Sciences,
Pushchino.
41.Khorobrykh, S. A., Mubarakshina, M. M., and
Ivanov, B. N. (2004) Biochim. Biophys. Acta, 1657,
164-167.
42.Melis, A., and Brown, J. S. (1980) Proc. Natl.
Acad. Sci. USA, 77, 4712-4719.
43.Rich, P. R. (1984) Biochim. Biophys. Acta,
768, 53-79.
44.Jennings, R. C., Garlaschi, F. M., and Gerola, P.
D. (1983) Biochim. Biophys. Acta, 722, 144-149.
45.Kirchhoff, H., Horstmann, S., and Weis, E. (2000)
Biochim. Biophys. Acta, 1459, 148-168.
46.Shuvalov, V. A., and Krasnovskii, A. A. (1975)
Biokhimiya, 40, 358-366.
47.Fujiwara, K., Kumata, H., Kando, N., Sakuma, E.,
Aihara, M., Morita, Y., and Miyakawa, T. (2006) Int. J. Environm.
Anal. Chem., 86, 337-346.
48.Golbeck, J. H., and Kok, B. (1979) Biochim.
Biophys. Acta, 547, 347-360.
49.McCauley, S. W., and Melis, A. (1986)
Photosynth. Res., 8, 3-16.
50.Ivanov, B. N., Mubarakshina, M. M., and
Khorobrykh, S. A. (2007) FEBS Lett., 581, 1342-1346.
51.Kruk, J., and Strzalka, K. (1999) Photosynth.
Res., 62, 273-279.
52.Lawlor, D. W. (1987) Photosynthesis:
Metabolism, Control, and Physiology, Longman
Scientific & Technical, Singapore.
53.Chow, W. S., Hope, A. B., and Anderson, J. M.
(1989) Biochim. Biophys. Acta, 973, 105-108.
54.Jursinic, P., and Dennenberg, R. (1989)
Photosynth. Res., 21, 197-200.
55.Hundal, T., Forsmark-Andree, P., Ernster, L., and
Andersson, B. (1995) Arch. Biochem. Biophys., 324,
117-122.
56.Mubarakshina, M. M., Khorobrykh, S. A., Kozuleva,
M. A., and Ivanov, B. N. (2006) Dokl. Biokhim. Biofiz.,
408, 113-116.
57.Mubarakshina, M. M., Khorobrykh, S. A., and
Ivanov, B. N. (2006) Biochim. Biophys. Acta, 1757,
1496-1503.
58.Allen, J. F., and Pfannschmidt, T. (2000)
Phil. Trans. R. Soc. Lond. B, 355, 1351-1359.