
|
REVIEW: Human Oncogenic Viruses: Hepatitis B and Hepatitis C Viruses and Their Role in HepatocarcinogenesisV. E. GurtsevitchBlokhin Russian Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia; fax: (495) 324-1205; E-mail: gurtsevitch@crc.umos.ru |
Received November 26, 2007
Chronic infections caused by hepatitis B virus (HBV) and/or hepatitis C virus (HCV) are the main risk factors for the development of hepatocellular carcinoma (HCC) in humans. Both viruses cause a wide spectrum of clinical manifestations ranging from healthy carrier state to acute and chronic hepatitis, liver cirrhosis, and HCC. HBV and HCV belong to different viral families (Hepadnoviridae and Flaviviridae, respectively); they are characterized by different genetic structures. Clinical manifestations of these viral infections result from the interaction between these viruses and host hepatocytes (i.e. between viral and cell genomes). Proteins encoded by both viruses play an important role in processes responsible for immortalization and transformation of these cells. Chronic inflammation determined by host immune response to the viral infection, hepatocyte death and their compensatory proliferation, as well as modulation of expression of some regulatory proteins of the cell (growth factors, cytokines, etc.) are the processes that play the major role in liver cancer induced by HBV and HCV.
KEY WORDS: hepatitis B virus, hepatitis C virus, carcinogenesis, signaling pathwaysDOI: 10.1134/S0006297908050039
Abbreviations: AFB1) aflatoxin B1; ER) endoplasmic reticulum; HBV) hepatitis B virus; HCC) hepatocellular carcinoma; HCV) hepatitis C virus; NF-kappaB and AP-1) transcription factors; ORFs) open reading frames; PKC) protein kinase C.
Oncovirology originated from the wonderful discovery of American
scientist Peyton Rous, who originally demonstrated in 1911 the
possibility of tumor induction in birds by a cell free extract. The
virus present in the filtrate and responsible for the development of
sarcoma in hens was later named Rous sarcoma virus, and Rous became a
Nobel Prize Laureate in 1966. In the middle of twentieth century, a
significant number of viruses inducing tumors in animals were
identified. For example, viruses causing leukemias in mice, cats,
monkeys, and horned cattle were discovered. Later it was demonstrated
that basically all representatives of the animal world suffer from
tumors of viral origin. Humans are not an exception. There is evidence
that about 15-20% of all human tumors are of viral origin. The most
frequent human tumors directly associated with certain viruses include
hepatic cancer, cervical carcinoma, nasopharyngeal cancer, Burkitt's
lymphoma, Hodgkin's lymphoma, and many others. The goal of this study
was to integrate known literature data on the most widespread oncogenic
viruses in humans, particularly hepatitis B virus and hepatitis C virus
(HBV and HCV, respectively) with emphasis on molecular mechanisms of
interactions of these viruses and the host organism.
Chronic infection with HBV and HCV is one of the most important risk factors of the development of hepatic cancer, which is also known as hepatocellular carcinoma (HCC). Although there is certain similarity in clinical manifestations of hepatitis induced by these viruses and creating background for subsequent development for HCC, these are two different viruses. HBV is a DNA-containing virus, which belongs to hepadnaviruses (Hepadnoviridae), whereas HCV is a RNA-containing virus of the flavivirus family (Flaviviridae).
Convincing evidence exists that more than 50% of the registered cases of HCC is associated with HBV infection, 25% is associated with HCV, and only in 22% of patients appearance of this type of cancer has no evident links with viral infection [1, 2]. In the latter case chronic exposure to aflatoxin B1 (AFB1), alcohol, and other factors may be considered as putative etiological factors. Mortality of HCC is in the third place after mortality of lung and stomach cancers in the world, and each fifth diagnosed tumor in the world is HCC [3, 4]. Primary evidence for a close interrelationship between these viruses and HCC has been obtained during accurate epidemiological studies, which have recognized increased risk for HCC in patients infected with HBV and HCV compared with non-infected individuals [5, 6].
HBV
HBV is widely distributed in the world's human population: about 400 million of people are chronically infected with this virus. HBV can be actively transmitted by perinatal, parenteral, and sexual exposures. Studies have shown that after several decades of HBV infection liver cirrhosis appears in 30-40% of infected persons and HCC develops in 1-5% of cirrhotic patients (this is about 500,000 cases per year) [6-8]. Chronic HBV carriers have a 100-fold increase in risk of developing HCC [5]. There is geographical variability in HCC morbidity, which correlates with the level of HBV infection: it is rather low (up to 2%) in the USA and European countries and approaches 10% and sometimes 20% in African and Southwest Asian countries. These differences in HCC morbidity are determined by percent of HBV infection among population and various risk factors including chronic HCV infections, exposure to AFB1, alcohol abuse, obesity, and diabetes mellitus [4]. Four serotypes (adv, ayw, adr, and ayr) and seven genotypes of HBV (from A to G) have been identified; they are characterized by distinct geographic distribution.
The study of HBV as etiological factor of HCC was started in 1970 after cloning of its genome [7, 9]. Comparison of HBV with woodchuck hepatitis virus inducing HCC in 100% of these rodents revealed their genetic relationship. Subsequently, the woodchuck model of viral-induced HCC has been actively used for studies of biological properties of HBV [9, 10].
Studies of virus-associated incidences of HCC in man have shown that most frequently HBV is integrated into the genome of actively proliferating cells, and each individual tumor is monoclonal in the type of integration of viral DNA. However, a specific site of viral integration into the cell genome has not been identified so far. HBV lacks a “classical” oncogene; it is possible that other viral genes, for example, products of gene X and some other genes exhibit the oncogenic functions.
Genomic structure of HBV and gene expression. HBV is a small partially double stranded DNA-containing virus of 3200 bp in length. Its genome has unusual structure, which consists of different size of DNA strands (Fig. 1). The minus-chain is full-sized and at its 5´_end it contains covalently bound protein identified as viral DNA-polymerase. The plus chain is shorter than the full sized minus-chain and its 5´ end contains oligoribonucleotides. At the 5´ ends of both strands there are regions containing short (11 nucleotides) direct repeats [11]. In contrast to replication of all known DNA-containing mammalian viruses, replication of hepadnaviruses occurs via intermediate RNA and use of reverse transcriptase; this is typical for RNA-containing viruses. In HBV the genome for open reading frames (ORFs) has been identified on the minus DNA strand. Viral DNA polymerase exhibiting reverse transcriptase activity is encoded by ORF P (defined as gene P). The nucleocapsid core protein is encoded by ORF preC (pre-core)/C (core), which is also defined as the preC/C gene; this protein undergoes posttranslational modification followed by formation of “e” antigen (HBeAg), the serological marker suggesting high level of viral replication and the main structural capsid protein (HbcAg). ORF preS1/preS2/S, which is also defined as preS/S gene, encodes several surface proteins of viral envelope (HbsAgs). Finally, ORF X (also defined as gene X) encodes viral regulatory protein HBx (HbxAg). It is the best-studied protein, which is characterized as transcription transactivator of cellular and viral genes [10-16].
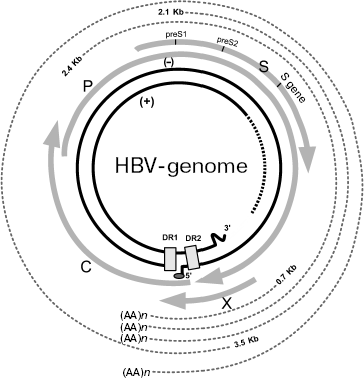
During infection, viral particles are attached to the surface of the infected cells (however, a membrane receptor responsible for interaction with virus remains unknown). At the next step, virions release their nucleocapsids into the cytoplasm. Nucleocapsids are then translocated into the nucleus where their genomic DNA is transformed into covalently closed circular DNA. This DNA is a template for transcription of pre-genomic and sub-genomic RNAs catalyzed by host RNA-polymerase II. Pre-genomic RNA serves as the template for reverse transcription as well as for translation of proteins encoded by genes preC/C and P; these proteins are formed in the endoplasmic reticulum (ER). Assembly of virus also occurs in ER [17].Fig. 1. Structure of HBV genome (modified from [11]). The inner ring shows viral DNA, hatches designate region of single strand breaks. Four gray arrows correspond to transcripts of the viral genes: C (nucleocapsid, core protein), P (viral polymerase), S (surface glycoprotein), and X (protein HBx). Hatched lines of the periphery of this figure designate viral RNAs, which are formed in the infected cells. DR1 and DR2 are two direct repeats of 11 nucleotides each at the 5´ ends of minus and plus strands of DNA. (AA)n is amino acid sequence.
HBx and its properties. HBx is a protein product of gene X. It consists of 154 amino acid residues and functions as a universal transcription transactivator. This protein is produced (although at low level) in periods of acute and chronic hepatitis. It can transactivate gene expression by interacting with a wide spectrum of viral and cellular regulatory elements [18]. For example, it has been demonstrated that HBx transactivates promoters and enhancers of some HBV genes (as well as genes of other viruses, such as HIV) and also cellular genes controlling the cell cycle, cell proliferation, or apoptosis. These include: c-jun, c-fos, c-myc, TP53, AP-1, NF-kappaB, SP1, and others (Fig. 2) [12, 19-21]. During early stages of investigation of HBx, it was suggested that this protein plays a decisive role in carcinogenesis associated with HBV. However, subsequent studies demonstrated that ectopic expression of HBx was not accompanied by transformation of either primary or immortalized rodent cells but induced their apoptosis. Moreover, hepatic tumors did not appear in most transgenic mice carrying HBx. When rodent cells were transformed in vitro, HBx was able to interact with H-ras [22]. It was also found that the effect induced in transfected cells by only HBx protein differed from the effect induced by full sized viral genome in the infected cells. While HBx alone stimulated cell proliferation, expression of the whole protein spectrum by full sized viral genome (including regulatory proteins HBx and PreS2) suppressed cell cycle progression [23].
Although pleiotropic functions of HBx have been well demonstrated in various experiments, it still remains unclear which mechanism and regulatory pathways of the protein product of gene X are especially important for induction of HCC. Some studies have demonstrated that HBx can activate protein kinase C (PKC), which in turn triggers activation of transcription factors NF-kappaB and AP-1 via signaling cascades (Fig. 2) [24]. However, the transcription level of HBx remained at a rather low level, which did not exceed a 15-fold increase compared with HBx level in normal cells. Results of other studies rule out such possibility and suggest that HBx does not influence PKC activity and that PKC does not play an essential role in transcription activation mediated by HBx [25].Fig. 2. Main signaling pathways activated by HBV proteins HBx and PreS2 (modified from [37]). PKC, protein kinase C; Pyk2, cytosolic Ca2+-dependent proline-rich tyrosine kinase 2; Src, proto-oncogene; the encoded protein is thyrosine kinase; Grb2, growth factor receptor-bound protein 2; Ras, proto-oncogene; c-Raf-1, cytoplasmic protein serine/threonine kinase; MEK, mitogen-activated extracellular regulated kinase 2; MAP2, mitogen-activated protein 2; NF-kappaB, nuclear factor kappaB; IkappaB, inhibitor of NF-kappaB; AP-1, activator protein-1.
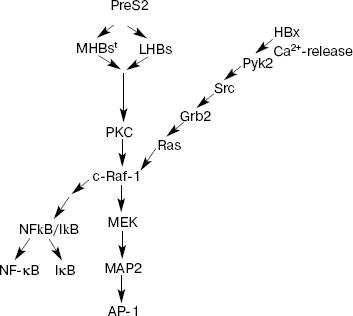
HBV effects on various promoters involve not only activation of PKC, but also other pathways, e.g. activation of kinase cascade c-Raf1-MEK/MAP2. In this case, HBx functions as intracellular cytoplasmic activator of Src family tyrosine kinases [26]. Certain evidence exists that this protein can activate Ca2+-dependent proline-enriched tyrosine kinases (Pyk2) [27]. Activation of these kinases may modulate regulatory effects on the cell resulting in activation of NF-kappaB and modification of activity of other cellular genes including Src activation (Fig. 2). There is evidence that HBx may be involved in activation of focal adhesion kinase (FAK), which acts as an Src kinase regulator. Activation of these signal pathways occurs when HBx is located in the extranuclear space of the cell. However, HBx fraction may also be localized in the nucleus as well. It is possible that various localization of HBx is associated with various functions of this protein: HBx localized in cytoplasm modulates intracellular signaling pathways; some authors suggest that HBx localized in the nucleus may directly interact with transcription factors or indirectly influence functions associated with transcription. For example, direct interaction of HBx with CREB (cAMP responsive element-binding protein) and ATF 2 (activating transcription factor 2) results in the increase of affinity for DNA binding [28].
HBx activating cell transcription factors causes a negative influence on various stages of DNA repair by increasing the number of critical mutations. Experiments on cell cultures demonstrated that HBx expression actually inhibited DNA repairing ability; this confirms that HBx influences mutagenesis [29].
Since HCC usually appears after a long period of chronic infections in a small proportion of infected individuals, there are some concerns whether expression of just one gene X is sufficient for induction of HCC. The level of protein product of this gene X (HBx) is low in infected cells and it is also characterized by a short half-life period. The oncogenic potential of HBx was studied in numerous experiments in vitro and in vivo. In cell cultures, its transforming potential is low (near the background level) and can be detected only when cells have already been immortalized by other oncogenes [30]. In vivo experiments have not revealed any serious pathologies or hepatic tumors in transgenic mice carrying gene X; however, HBx weakly stimulated the development of tumors in certain strains of transgenic mice (CD-1 strain), where tissue content of this protein was high [31]. It was also demonstrated that in transgenic mice HBx increased hepatocarcinogenesis induced by gene c-myc [32]. In other series of experiments hepatic tumors appeared in transgenic mice producing only one viral envelope protein, LHBs [33, 34]. This suggests that even production of viral envelope protein may be hepatotoxic and sufficient for tumor induction. Overproduction of viral envelope proteins, especially LHBs and possibly MHBs results in their accumulation inside cells, and this may promote predisposition of such cells to stress, which may finally result in HCC [35]. Although results obtained by various authors are a bit inconsistent, they suggest that HBx is a nonspecific pleiotropic transcription transactivator, which, however, lacks high transforming activity. Nevertheless, under conditions of increased expression of this protein and existence of certain genetic background, HBx can stimulate tumor formation. Obviously this occurs during multistage transformation employing cellular oncogenes, for example, Ha-ras, responsible for avoidance of apoptosis mediated by HBx.
Family of activators encoded by gene preS/S and their properties. HBV DNA integrated into hepatocyte genome encodes not only HBx but also another family of regulatory proteins known as transcription activators, PreS2. The ORF of gene preS/S is subdivided into three coding domains: preS1, preS2, and S; their alternative initiation may result in synthesis of envelope proteins of different sizes: large LHBs (PreS1 + PreS2 + S), medium MHBs (PreS2 + S), and small SHBs (S). Transcription activators PreS2 include LHBs protein (large HBV surface protein of the envelope) and MHBst (medium size protein truncated at C-terminal region). These proteins exhibit regulatory functions.
MHBst. Regulatory functions of the PreS2 (MHBst and LHBs) transcription activators are linked to cytoplasmic orientation of a PreS2 domain. The full sized variant of MHBs proteins lacks transcriptional activator activity: in this case, the PreS2 domain is oriented into the ER lumen. Functionally active MHBst proteins originating from MHBs proteins appeared as the result of deletion of the 3´ end of preS2/S. This deletion covers at least 70 amino acid residues, belonging to the hydrophobic region III of S-domain. Interestingly, deletion of preS2/S 3´ end sequence resulting in formation of functionally active MHBst activators was found in one third part of all examined cases of HCC associated with HBV. This suggests biological importance of PreS2 activator family for hepatocarcinogenesis [25, 36, 37].
Studies have shown that there are at least two types of MHBst activators. One type of these proteins is localized in ER (in contrast to full sized MHBs); their processing and secretion in Golgi apparatus is impossible in this case. The other type of MHBst transactivators includes proteins lacking three transmembrane sites; these proteins are not linked to ER membranes and they are uniformly distributed in nucleus and cytoplasm [38].
LHBs. The other representatives of PreS2 activators, LHBs proteins, are also characterized by two variants of their positioning versus membrane [39]. In one type of proteins there is the first functionally active transmembrane site located within the initial stretch of S-domain (8-21 amino acid residues): in this case, PreS1-PreS2 domain of LHBs is facing the ER lumen. In the other type of LHBs proteins, this transmembrane site does not function and PreS1/PreS2 domain is facing the cytoplasm. As in the case of MHBst proteins, cytoplasmic orientation of domain PreS1/PreS2 determines regulatory function of LHBs proteins, which are referred to the PreS2 family of activators mainly due to this feature [40].
There is significant difference of PreS2 transactivator family (LHBs and MHBst) from MHBs: LHBs and MHBst can undergo phosphorylation by PKC. As transactivators LHBs and MHBst initiate PKC-dependent activation of signaling pathway c-Raf-1/MEK/ERK (extracellular signal regulated kinase), which is a necessary precondition for activation of transcription factors AP-1 and NF-kappaB (Fig. 2). In contrast to HBx, this activation does not require the presence of Ras oncogene and is accompanied by activation of such cell kinases as c-Raf and MAP2. Importance of these transactivating potentials of MHBst in vivo has been demonstrated using a specially developed model of transgenic mice producing in the liver one of the PreS2 activators, MHBst. In spite of production of insignificant amounts of MHBst, specific activation of signaling cascade c-Raf-1/MEK/ERK and increased hepatocyte proliferation was found in young transgenic mice. At the age of 15 months and above, these mice had high frequency of hepatic tumors [25]. Tumor formation was also observed in LHBs-transgenic mice [33], which were characterized by overproduction of LHBs protein accumulated in cytoplasm followed by subsequent formation of so-called glass-hepatocytes (i.e. hepatocytes characterized by the histological sign of chronic HBV infection and containing viral surface proteins in ER). The authors explain tumor formation in these transgenic mice by chronic inflammation. The effect of LHBs is considered as an additional factor influencing appearance of tumors. Thus, stress related to increased load with corresponding viral protein and inflammation probably result in formation of critical mutations and constant activation of signaling cascade PKC-/c-Raf-1/MEK/ERK. The effect of both factors is obviously similar to the effect of tumor promoter in the two-stage model of carcinogenesis; the function of this promoter consists in activation of a wide spectrum of key transcription factors controlling cell proliferation.
Effect of integration of HBV DNA into the hepatocyte genome. Almost all hepatomas associated with HBV contain viral DNA integrated into chromosomes [41]. Usually multiple integration takes place and the integration site varies [41, 42]. Interestingly, integration of HBV DNA into cellular genome does not play an essential role in viral replication, but causes persistence of the viral genome in cells. In many cases, integration of viral genomes is accompanied by their rearrangement or deletions [43]. HBV integration can cause deletions in chromosomes of infected cells. Taking into consideration these observations, it was suggested that integration of viral genome into cellular DNA caused impairments in functioning of key regulators of the cell cycle [43]. However, site-specific integration of virus or integration of HBV genome near known oncogenes (cis-activation) was considered as a very rare event in cells with HBV-associated HCC. Recently, PCR analysis was used to show that HBV integration into cell genes occurs rather frequently, even at stages of early viral infection after appearance of acute hepatitis terminated by independent recovery [44]. Virus may be integrated into genes regulating cell-signaling pathways related to control of proliferation and differentiation of cells. This could be the gene of human telomerase (hTERT), which is subjected to cis-activation in hepatomas associated with HBV [41, 45]. All these data suggest that integration processes play an important role in HBV associated carcinogenesis.
HCV
According to epidemiological evaluation, about 170 million people in the world are chronically infected with HCV. The infected population varies from a fraction of a percent in European countries and USA up to 20% in countries of Asia and the Near East. Drug addicts, hemophilic patients, and medical personnel are at the highest risk of HCV infection. Parenteral transmission remains the primary mode of spread; however, other pathways (e.g. sexual, perinatal) are also possible. Chronic HCV infection (as well as HBV infection) causes the development of severe chronic hepatitis usually transforming into cirrhosis; in 3-4% of cirrhotic cases HCC appears. Epidemiological studies have shown that HCV infection causes a 50-fold increase in risk of developing HCC [5].
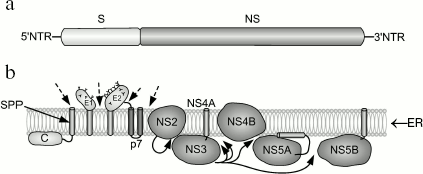
Genomic structure of HCV and gene expression. HCV is an RNA-containing virus. Its genome encodes a single polyprotein consisting of 3100 residues. Cleavage of this polyprotein with cell and viral proteases results in formation of about 10 polypeptides (Fig. 3). Cleavage of protein components NS2-3 and NS-3-4A by viral proteases results in release of three nonstructural proteins from this polyprotein; two structural proteins are cleaved from this polyprotein by cellular ER signal peptidase. Subsequent cleavage involving signal peptide peptidase occurs at the C-terminal region of the capsid protein [46-48]. The following proteins encoded by HCV are known: nucleocapsid protein (core proteins) of 21 kD is involved in nucleocapsid formation, binding to RNA; E1 (of 31-35 kD) is an envelope glycoprotein interacting with E2; E2 (70 kD) is the envelope glycoprotein, a receptor for binding with other molecules; E2 interacts with E1; p7 (7 kD) is an ion channel; NS2 (21 kD) is a component of NS2-3 protease; NS3 (69 kD) is the N-terminal domain of peptidase and C-terminal domain of NTPase/helicase; NS4A (6 kD) is a peptidase cofactor; NS4B (27 kD) is an integral membrane protein; NS5A (56-58 kD) is a phosphoprotein controlling replication of HCV genome by suppressing interferon; NS5B (68 kD) is RNA-dependent RNA polymerase. At both ends of HCV RNA, there are regulatory nontranslated regions (NTR). Thus, the genetic structure of HCV is similar to the structure of retroviral RNA [2].
During hepatocyte infection, virus interacts with surface cell receptor; this results in endocytosis of a viral particle. Subsequent fusion of viral envelope with endosomal membrane releases the HCV genome into the cytosol. The HCV genome is a plus (+) RNA strand, which is directly translated, and all viral proteins are synthesized simultaneously. Synthesis of HCV proteins causes intracellular membrane rearrangements followed by formation of so-called membrane network, where RNA is replicated. Assembly of nonstructural proteins (NS3-NS5B) employs cellular factors; this results in formation of a complex responsible for RNA replication. Accumulation of genomic RNA and structural proteins results in assembly of nucleocapsid, followed by formation of its own envelop within the intracellular space. The resulting viral particle leaves the infected cell and enters the extracellular space. It should be noted that HCV lacks reverse transcriptase activity and it is not integrated into the cell genome. In contrast to HBV (for which a specific cell membrane receptor has not been identified yet), there are some putative cell membrane receptors for HCV; these include CD81, LDL-R, and some other proteins [49].Fig. 3. Structure of the HCV genome (a) and processing of HCV-encoded polyprotein (b) (modified from [49]). a) A single protein encoded by HCV consists of structural (S) proteins occupying two-thirds of the N-terminal regions and nonstructural (NS) proteins located at one-third of the N-terminal region and two-thirds of the C-terminal region; b) results of HCV polyprotein processing and localization of 10 proteins associated with ER membrane. Dashed arrows indicate cleavage sites for host signal peptidase. Curved arrows indicate sites for cleavage of NS2-3 and NS3-4A proteins. The arrow inside the membrane shows cleavage by host signal peptide peptidase (SPP). Transmembrane domains E1 and E2 are shown after cleavage by signal peptidase and change in orientation of their C-terminal region.
Biological properties of HCV. According to the leading hypothesis, the development of HCV (as also HBV)-induced HCC is associated with continuous death of virus-infected hepatocytes, compensatory substitution of dead cells by dividing hepatocytes, followed by appearance of mutations in their genomes. It is also possible that in contrast to carcinogenesis associated with HBV infection, the carcinogenesis associated with HCV may involve other viral and some other factors.
Persistent HCV infection is obviously one of the most important conditions for the development of HCC. HCV employs various mechanisms for the development of persistent infection and escape from the immune response. One of these mechanisms consists of formation of so-called quasi-viral particles, which appear in the result of errors in one of the stages of viral replication, and they are selected by their ability for reproduction under conditions of potent immune response [50]. Proteins encoded by HCV play a major role in impairments in cell metabolism and/or modulation of signaling pathways directly involved in transformation of hepatocytes and appearance of HCC. Nucleocapsid protein is one of them. Modifying intracellular signaling pathways, it prevents immune response-induced death of infected cells. For example, it inhibits apoptosis mediated by TNF-alpha. It is known that TNF-alpha is the main cytokine accompanying inflammation. It is secreted by macrophages and T-cells and plays a major role in acute infections. Nucleocapsid protein interacts with cytoplasmic domains of tumor necrosis factor receptor 1 (TNFR1), lymphotoxin b, and gC1q and blocks the FAS/TNF-alpha signaling pathway [51, 52]. Blockade of the TNF-alpha signaling pathway promotes survival of infected hepatocytes and thus provides persistence of HCV infection. This protein also activates transcription factor NF-kappaB involved in regulation of immune response [53]. It has been shown that NF-kappaB content is higher in hepatocytes of patients with chronic HCV infection, and such hepatocytes are less sensitive to TNF-alpha [47]. Thus, the nucleocapsid protein promotes persistence of infection, suppresses immune system of an infected person, and simultaneously it “inverts” functions of proteins controlling cell growth and differentiation.
E1 and E2, two viral envelope glycoproteins, play an important role in various steps of the HCV life cycle. For example, they are required for integration of virus into cells [54] and also for assembly of viral particles [55]. The heterodimer E1/E2 is a viral component presented on the surface of HCV viral particles; it is a putative candidate ligand for cell receptors.
P7 is a polypeptide localized inside the HCV polyprotein at the site joining structural and nonstructural proteins. Its cleavage from polyprotein involves cell signal peptidase. Polypeptide P7 is a small protein consisting of two transmembrane domains, and its N- and C-terminal segments face the ER lumen [56]. This polypeptide is not involved in RNA replication, and it remains unclear whether it represents a virion component. However, it has been demonstrated that in artificial lipid membranes P7 is responsible for ion channel functioning [57]; P7 contains functionally important and specific for a certain genotype sequences required for manifestation of viral infectivity [58].
NS2 is an integral membrane protein that is not involved in viral replication. The function of the mature form of this protein remains unknown; however, before its cleavage from polyprotein it functions as the protease involved in cleavage of the protein complex NS2/NS3 [59, 60].
NS3 is a multifunctional protein possessing serine protease activity in the N-terminal domain and RNA helicase/NTPase activity in the C-terminal domain. Protease activity of NS3 increases in the presence of NS4A protein as cofactor. NS3 lacks its own transmembrane domain, but it noncovalently interacts with the central domain of NS4A, which is a transmembrane protein. In the case of NS3 coexpression with NS4A, the former is associated with ER or ER-like membranes. In the case of independent expression, NS3 is diffusely distributed in the cytoplasm and nucleus [61]. There is evidence that NS3 interacts with some cellular proteins, and it is probably involved in hepatocellular carcinogenesis [62].
A hydrophobic N-terminal domain of NS4A is required for interaction with ER with NS3. NS4A stabilizes the protease. The protease complex NS3-4A formed during infection is responsible for processing of polyprotein encoded by the HCV genome; this processing occurs in the region below NS3. The protease complex activity is important for formation of components involved in replication of viral RNA [63]. The protease complex NS3-4A is also involved in blockade of activation of natural antiviral response by host cells [64].
NS4B is a highly hydrophobic nonstructural protein. It contains four transmembrane domains, and its N- and C-terminal regions are located in the cytosol. A fraction of the N-terminal domain can also be detected in the ER lumen [65]. One of the known functions of NS4B consists in intracellular rearrangement of membranes, for example, in formation of membrane structures maintaining RNA replication [66, 67].
NS5A is a membrane-associated protein; in its N-terminal region, it has a unique amphipathic alpha-helix that functions as a planar membrane anchor. Like many other HCV proteins, NS5A is detected in association with ER or ER-derived membranes. NS5A plays an important role in replication of the HCV genome. This protein is also involved in blockade of interferon effect and modulation of cell signaling pathways; this not only promotes infection persistence but may also result in cell transformation. In NS5A there is a region determining sensitivity to interferon. Mutagenesis experiments have shown that this region is required for the interaction of NS5A with PKR; this protein kinase suppresses viral translation and mediates antiviral effects of interferon [68]. Activated PKR is also a mediator of apoptosis in response to some types of cell stress. Indeed, administration of double-stranded RNA into certain cell types activates PKR and causes apoptosis [69]. Thus, NS5A (as well as nucleocapsid protein) can suppress immune response and protect cells against apoptosis.
NS5B is a membrane-associated protein; its C-terminal transmembrane domain is essential for RNA replication in cell culture [70]. Like many other HCV proteins, NS5B is detected in association with ER or ER-derived membranes. NS5B is an RNA-dependent RNA polymerase (replicase), which represents the catalytic component of the HCV replication machinery. This enzyme synthesizes RNA using RNA as a template. Under experimental conditions, NS5B can initiate RNA synthesis de novo. This fact is interpreted as an indication that a similar process may take place in vivo. Formation of viral replicase on the cytosolic surface of ER may play an important role in molecular events associated with HCV infection. It is relevant to suggest that nonstructural HCV proteins located in ER and virus-specific ER-like membrane structure (demonstrated by means of electron microscopy) may influence ER, alter its structure, and induce stress [66].
TP53 GENE
Study of genetic and epigenetic mechanisms of hepatocarcinogenesis has shown that the appearance of HCC is a complex process accompanied by impaired functioning of many genes and therefore modification of the whole spectrum of signaling cascades. Activation of many proto-oncogenes and inactivation of suppressor genes due to genetic and epigenetic damages represent biological processes characterizing the development of this neoplasm. Tumor cells of patients with HCC of viral and nonviral origin are characterized by impairments in the signaling pathways RB1, p53, and Wnt. Nevertheless, numerous observations demonstrate doubtful results on the role of tumor suppressors, for example TP53 in the development of HCC. For example, cases of HCC of viral origin are characterized by frequent impairments in the signaling pathway RB1; these include methylation of p16INK4a and RB1 promoters and also amplification of cyclin D1 [71-73]. Impairments in signaling pathways of the TP53 gene are more typical for cases of hepatic tumors associated with aflatoxin B1 treatments and characteristic mutation in the 249th codon (resulting in substitution of Arg for Ser) may be a genetic marker for this type of tumors [74, 75]. Thus, there is evidence suggesting that the existence and frequency of TP53 mutation in tumor tissue from patients with HCC are significantly influenced by environmental factors involved in tumor induction and even by geographic zone. Indeed, cases of HCC in China and certain regions of Africa with high exposure of the population to aflatoxin are characterized by high frequency of TP53 mutations. This suggests that the mechanism underlying appearance of HCC differs from that in Europe and the USA.
Based on incidences of HCC in children infected with HBV, one can suggest existence of genetic predisposition to this type of tumor. Relatively short latent period for its development in children is consistent with this hypothesis. Such predisposition inherited as critical mutations in germinal cells and also somatic mutations accumulated due to HBV infection may probably shorten the latent period of tumor development [75].
Special attention was paid to investigation of mechanism of interaction between HBx and gene suppressor TP53. Results of some studies have shown an indirect inhibitory effect of HBx on TP53; this effect involving a chain of signaling pathways repressed TP53 transcription [76]. Other studies revealed direct interaction between HBx and p53 followed by formation of complexes neutralizing p53 functioning; this resulted in inhibition of apoptosis [77]. However, analysis of intracellular content of these proteins has shown that in infected hepatocytes p53 content is much higher than HBx content, and in a certain proportion of cases p53 does not function [77]. Thus, functional consequences of direct interaction between HBx and p53 still require further investigations.
ROLE OF STRESS IN THE DEVELOPMENT OF HCC
HBV and HCV employing ER for realization of the integral part of their replication strategy antagonize ER response to stress and also consequences of signals from ER subjected to stress. ER stress is a homeostatic mechanism, which regulates cell metabolism and protein synthesis in response to folding impairments and protein biosynthesis. Moderate ER stress modulates initiation of protein synthesis and can attenuate cell growth, whereas strong or long-term ER stress can result in caspase-12 mediated apoptosis.
Noncytopathic flaviviruses of HCV type carry our stress induced and ER signaling at the sublethal level, whereas stress signals induced by other viruses are lethal and cause apoptosis [78]. Long-term consequences of ER sublethal signals remain unclear, but one can suggest that constant stress of this organelle causes certain changes in cell physiology and predisposes cells to transformation [2].
Signaling from ER susceptible to stress is closely related to cell metabolism and intracellular redox status. For example, ER stress results in oxidative stress [79], which exerts a strong effect on cell metabolism. Changes in cell metabolism can cause an increase of mutation processes including stimulation of cell proliferation and (in extreme cases) to apoptosis [80]. Taking into consideration serious consequences of oxidative stress, many researchers believe that it is involved in the development of HCC induced by chronic HBV and HCV infection.
Studies of mechanisms of oxidative stress have shown that oxidative stress activates signaling cascades (including MAP kinase pathway), which can seriously influence regulation of cell growth and transformation processes [80]. Particularly, MAP kinases regulate cell proliferation and apoptosis in response to external and internal stimuli; they may be involved in pathogenesis of some diseases associated with oxidative stress.
Oxidative stress also activates hepatic stellate cells (also known as Ito cells). These cells are the main connective tissue cells in the liver. They are involved in formation of extracellular matrix and required for normal growth and differentiation of cells during liver damage. In this case, the stellate cells divide in response to various cytokines, growth factors, and chemokines (proinflammatory cytokines) produced by the damaged liver. Chronic activation of stellate cells in response to oxidative stress induced by viral replication may contribute to fibrogenesis and increased proliferation of hepatocytes chronically infected with HBV and HCV. Thus, stellate cells are involved in regulation of growth, differentiation, and turnover of hepatocytes; all these events together with activation of MAP kinases may result in appearance of HCC [2].
ROLE OF IMMUNITY IN DEVELOPMENT OF HCC
Cytotoxic T lymphocytes (CTL), also known as cytotoxic T cells or killer T cells, have a major influence on infection and hepatocarcinogenesis as well. These cells selectively interact with polypeptide epitopes of viral polymerase and envelope and also with cells of the natural immune system including dendritic cells, macrophages, and natural killers (NK). Activity of CTL cells is primarily important for “liberation” of the human body from virus and therefore for recovery of patients from acute hepatitis [81, 82]. Persons chronically infected with HBV are obviously characterized by attenuated T cell response to viral antigens [83]. It was suggested (and then confirmed in experiments with syngeneic mice) that immunological control of infection employed a special population of T cells, CD8+. These cells lack a cytolytic effect, but they suppress viral replication and do not act on viable noninfected hepatocytes [84]. Death of infected hepatocytes may cause liver damage accompanied by hepatic insufficiency in extreme cases. Death of these cells also results in constant regeneration of hepatocytes; this increases probability of appearance of mutant cells. The microenvironment in which hepatocytes divide may be enriched by oxidant-type mutagens appearing in response to liver cell damage (including immunological treatments) [85]. These pathological processes are stimulated by increased hepatic fibrosis; this creates conditions for the development of HCC. Summarizing all these data, one can conclude that in humans at least three branches of the immune system are involved in HBV associated hepatic pathology: T-cell and B-cell immunity and also the NK/NKT system [86].
PROPHYLAXIS OF HBV- AND HCV-ASSOCIATED CASES OF HCC
It has already been mentioned that about 400 and 170 million people in the world are chronic carriers of HBV and HCV, respectively. These viral infections may cause acute and chronic liver damage--cirrhosis and HCC. HBV-induced liver damage is a process preferentially control by the immune system. Taking into consideration modes of host-virus interrelationships, the time course of chronic infection can be subdivided into three stages: immunological tolerance, immunological clearance, and the phase of viral integration. Safe and effective vaccines have been developed for treatment of HBV infections and its severe consequences. According to WHO recommendations, such vaccines need to be introduced into a routine program of immunization of children and teenagers in all countries. Effectiveness of universal immunization has already been proved in some countries, and vaccinated children are characterized by a sharp decrease in number of HBV carriers. Moreover, it has been proved that such vaccination protects children not only against viral infection, but also against acute and chronic forms of hepatitis and HCC [87, 88].
In accordance with WHO recommendations, obligatory vaccination against HBV was introduced in Russia. It involves newborn babies, young children, teenagers, and persons of risk groups. For prophylaxis of HBV and HCC spread all pregnant women and blood donors are tested for HbsAg, and the use of aflatoxins, potential carcinogens, is forbidden in agriculture. Although modern antiviral therapy is safe and rather effective, it is not ideal. However, there is hope that it will be optimized in the nearest future.
Prophylaxis of HCV spread also involves testing of pregnant women and blood donors for antibodies against this virus, and certain efforts are undertaken for development and trial of vaccines against HCV.
HBV- and HCV-associated carcinogenesis is now considered as a multifactorial process. Integration of both viruses in the hepatocyte genome may occur at early stages of viral infection. This influences many genes regulating cell proliferation, differentiation, and apoptosis. Chronic inflammation induced by viral infection results in a complex of interrelated degenerative and regenerative processes, promoting accumulation of critical mutations in hepatic cells. Regulatory proteins of HBs and PreS2 transactivators may exhibit functions similar to functions of tumor promoters. This results in selection of cells characterized by activation of genes responsible for cell proliferation, cytokine (IL-6) synthesis, etc. Cytokines secreted by cells promote microenvironment formation from neighboring fibroblasts, endothelial, and other cells. These cells secrete growth factors causing the paracrine type stimulation of hepatocyte proliferation. Increased hepatocyte proliferation can result in genetic defects, which will promote selection of cells with accelerated proliferation and acquired malignant transformation. Attenuated T-cell immune response of patients to viral antigens plays an important role in promotion of the tumor process, because virus-transformed hepatocytes may escape from immunological control.
Considering the perspectives of HCC studies, one can suggest that use of methods of proteomic and genomic analysis would clarify the molecular mechanisms responsible for the development of hepatocarcinomas associated with HBV and HCV. Modern technologies can probably reveal new diagnostic markers and develop more effective medical and prophylactic preparations.
The author is deeply grateful to A. V. Liechtenstein for critical discussion of this manuscript and valuable advice and to S. V. Diduk for help in preparation of figures.
REFERENCES
1.Anthony, P. P. (2001) Histopathology,
39, 109-118.
2.Block, T. M., Mehta, A. S., Fimmel, C. J., and
Jordan, R. (2003) Oncogene, 22, 5093-5107.
3.Montalto, G., Cervello, M., Giannitrapani, L.,
Dantona, F., Terranova, A., and Castagnetta, L. A. (2002) Ann. N. Y.
Acad. Sci., 963, 13-20.
4.Parkin, D. M., Bray, F., Ferlay, J., and Pisani, P.
(2001) Int. J. Cancer, 94, 153-156.
5.Cohen, J. (1999) Science, 285,
26-30.
6.Conjeevaram, H. S., and Lok, A. S. (2003) J.
Hepatol., 38 (Suppl. 1), 90-103.
7.Dane, D. S., Cameron, C. H., and Briggs, M. (1970)
Lancet, 1, 695-698.
8.Perrillo, R. P. (2003) J. Hepatol., 39
(Suppl. 1), S177-S180.
9.Buendia, M. A. (1992) Adv. Cancer Res.,
59, 167-226.
10.Baumert, T. F., Thimme, R., and von Weizsacker,
F. (2007) World J. Gastroenterol., 3, 82-90.
11.Ryu, W. S. (2003) J. Biochem. Mol. Biol.,
36, 138-143.
12.Feitelson, M. A. (1999) J. Cell Physiol.,
181, 188-202.
13.Fujiyama, A., Miyanohara, A., Nozaki, C.,
Yoneyama, T., Ohtomo, N., and Matsubara, K. (1983) Nucleic Acids
Res., 11, 4601-4610.
14.Galibert, F., Mandart, E., Fitoussi, F.,
Tiollais, P., and Charnay, P. (1979) Nature, 281,
646-650.
15.Ono, Y., Onda, H., Sasada, R., Igarashi, K.,
Sugino, Y., and Nishioka, K. (1983) Nucleic Acids Res.,
11, 1747-1757.
16.Pasek, M., Goto, T., Gilbert, W., Zink, B.,
Schaller, H., MacKay, P., Leadbetter, G., and Murray, K. (1979)
Nature, 282, 575-579.
17.Szabo, E., Paska, C., Kaposi, N. P., Schaff, Z.,
and Kiss, A. (2004) Pathol. Oncol. Res., 10, 5-11.
18.Yen, T. S. (1996) J. Biomed. Sci.,
3, 20-30.
19.Su, F., and Schneider, R. J. (1996) J.
Virol., 70, 4558-4566.
20.Yeh, C. T. (2000) J. Gastroenterol.
Hepatol., 15, 339-341.
21.Waris, G., and Siddiqui, A. (2003) J.
Biosci., 28, 311-321.
22.Kim, Y. C., Song, K. S., Yoon, G., Nam, M. J.,
and Ryu, W. S. (2001) Oncogene, 20, 16-23.
23.Friedrich, B., Wollersheim, M., Brandenburg, B.,
Foerste, R., Will, H., and Hildt, E. (2005) J. Hepatol.,
43, 696-703.
24.Luber, B., Lauer, U., Weiss, L., Hohne, M.,
Hofschneider, P. H., and Kekule, A. S. (1993) Res. Virol.,
144, 311-321.
25.Hildt, E., Munz, B., Saher, G., Reifenberg, K.,
and Hofschneider, P. H. (2002) EMBO J., 21, 525-535.
26.Klein, N. P., and Schneider, R. J. (1997) Mol.
Cell Biol., 17, 6427-6436.
27.Bouchard, M. J., Wang, L. H., and Schneider, R.
J. (2001) Science, 294, 2376-2378.
28.Haviv, I., Shamay, M., Doitsh, G., and Shaul, Y.
(1998) Mol. Cell Biol., 18, 1562-1569.
29.Becker, S. A., Lee, T. H., Butel, J. S., and
Slagle, B. L. (1998) J. Virol., 72, 266-272.
30.Hohne, M., Schaefer, S., Seifer, M., Feitelson,
M. A., Paul, D., and Gerlich, W. H. (1990) EMBO J., 9,
1137-1145.
31.Kim, C. M., Koike, K., Saito, I., Miyamura, T.,
and Jay, G. (1991) Nature, 351, 317-320.
32.Terradillos, O., Billet, O., Renard, C. A., Levy,
R., Molina, T., Briand, P., and Buendia, M. A. (1997) Oncogene,
14, 395-404.
33.Chisari, F. V., Klopchin, K., Moriyama, T.,
Pasquinelli, C., Dunsford, H. A., Sell, S., Pinkert, C. A., Brinster,
R. L., and Palmiter, R. D. (1989) Cell, 59,
1145-1156.
34.Chisari, F. V. (1995) Hepatology,
22, 1316-1325.
35.Xu, Z., Jensen, G., and Yen, T. S. (1997) J.
Virol., 71, 7387-7392.
36.Murakami, S. (1999) Intervirology,
42, 81-99.
37.Lupberger, J., and Hildt, E. (2007) World J.
Gastroenterol., 13, 74-81.
38.Hildt, E., and Hofschneider, P. H. (1998)
Recent Results Cancer Res., 154, 315-329.
39.Prange, R., and Streeck, R. E. (1995) EMBO
J., 14, 247-256.
40.Hildt, E., Urban, S., and Hofschneider, P. H.
(1995) Oncogene, 11, 2055-2066.
41.Paterlini-Brechot, P., Saigo, K., Murakami, Y.,
Chami, M., Gozuacik, D., Mugnier, C., Lagorce, D., and Brechot, C.
(2003) Oncogene, 22, 3911-3916.
42.Beasley, R. P., Hwang, L. Y., Lin, C. C., and
Chien, C. S. (1981) Lancet, 2, 1129-1133.
43.Thorgeirsson, S. S., and Grisham, J. W. (2002)
Nat. Genet., 31, 339-346.
44.Murakami, Y., Saigo, K., Takashima, H., Minami,
M., Okanoue, T., Brechot, C., and Paterlini-Brechot, P. (2005)
Gut, 54, 1162-1168.
45.Hytiroglou, P., and Theise, N. D. (2006) Am.
J. Gastroenterol., 101, 839-841.
46.McLauchlan, J., Lemberg, M. K., Hope, G., and
Martoglio, B. (2002) EMBO J., 21, 3980-3988.
47.Penin, F., Dubuisson, J., Rey, F. A., Moradpour,
D., and Pawlotsky, J. M. (2004) Hepatology, 39, 5-19.
48.Reed, K. E., and Rice, C. M. (2000) Curr. Top.
Microbiol. Immunol., 242, 55-84.
49.Dubuisson, J. (2007) World J.
Gastroenterol., 13, 2406-2415.
50.Forns, X., and Bukh, J. (1999) Clin. Liver
Dis., 3, 693-716.
51.Tai, D. I., Tsai, S. L., Chen, Y. M., Chuang, Y.
L., Peng, C. Y., Sheen, I. S., Yeh, C. T., Chang, K. S., Huang, S. N.,
Kuo, G. C., and Liaw, Y. F. (2000) Hepatology, 31,
656-664.
52.Kittlesen, D. J., Chianese-Bullock, K. A., Yao,
Z. Q., Braciale, T. J., and Hahn, Y. S. (2000) J. Clin. Invest.,
106, 1239-1249.
53.Zhu, N., Ware, C. F., and Lai, M. M. (2001)
Virology, 283, 178-187.
54.Cocquerel, L., Voisset, C., and Dubuisson, J.
(2006) J. Gen. Virol., 87, 1075-1084.
55.Wakita, T., Pietschmann, T., Kato, T., Date, T.,
Miyamoto, M., Zhao, Z., Murthy, K., Habermann, A., Krausslich, H. G.,
Mizokami, M., Bartenschlager, R., and Liang, T. J. (2005) Nat.
Med., 11, 791-796.
56.Carrere-Kremer, S., Montpellier-Pala, C.,
Cocquerel, L., Wychowski, C., Penin, F., and Dubuisson, J. (2002) J.
Virol., 76, 3720-3730.
57.Griffin, S. D., Harvey, R., Clarke, D. S.,
Barclay, W. S., Harris, M., and Rowlands, D. J. (2004) J. Gen.
Virol., 85, 451-461.
58.Sakai, A., Claire, M. S., Faulk, K.,
Govindarajan, S., Emerson, S. U., Purcell, R. H., and Bukh, J. (2003)
Proc. Natl. Acad. Sci. USA, 100, 11646-11651.
59.Pallaoro, M., Lahm, A., Biasiol, G., Brunetti,
M., Nardella, C., Orsatti, L., Bonelli, F., Orru, S., Narjes, F., and
Steinkuhler, C. (2001) J. Virol., 75, 9939-9946.
60.Schmidt-Mende, J., Bieck, E., Hugle, T., Penin,
F., Rice, C. M., Blum, H. E., and Moradpour, D. (2001) J. Biol.
Chem., 276, 44052-44063.
61.Wolk, B., Sansonno, D., Krausslich, H. G.,
Dammacco, F., Rice, C. M., Blum, H. E., and Moradpour, D. (2000) J.
Virol., 74, 2293-2304.
62.Tellinghuisen, T. L., and Rice, C. M. (2002)
Curr. Opin. Microbiol., 5, 419-427.
63.Lindenbach, B. D., and Rice, C. M. (2005)
Nature, 436, 933-938.
64.Foy, E., Li, K., Wang, C., Sumpter, R., Jr.,
Ikeda, M., Lemon, S. M., and Gale, M., Jr. (2003) Science,
300, 1145-1148.
65.Lundin, M., Monne, M., Widell, A., von Heijne,
G., and Persson, M. A. (2003) J. Virol., 77,
5428-5438.
66.Egger, D., Wolk, B., Gosert, R., Bianchi, L.,
Blum, H. E., Moradpour, D., and Bienz, K. (2002) J. Virol.,
76, 5974-5984.
67.Elazar, M., Cheong, K. H., Liu, P., Greenberg, H.
B., Rice, C. M., and Glenn, J. S. (2003) J. Virol., 77,
6055-6061.
68.Gale, M. J., Jr., Korth, M. J., and Katze, M. G.
(1998) Clin. Diagn. Virol., 10, 157-162.
69.Kaufman, R. J. (1999) Proc. Natl. Acad. Sci.
USA, 96, 11693-11695.
70.Moradpour, D., Brass, V., Bieck, E., Friebe, P.,
Gosert, R., Blum, H. E., Bartenschlager, R., Penin, F., and Lohmann, V.
(2004) J. Virol., 78, 13278-13284.
71.Edamoto, Y., Hara, A., Biernat, W., Terracciano,
L., Cathomas, G., Riehle, H. M., Matsuda, M., Fujii, H., Scoazec, J.
Y., and Ohgaki, H. (2003) Int. J. Cancer, 106,
334-341.
72.Chen, Y. W., Klimstra, D. S., Mongeau, M. E.,
Tatem, J. L., Boyartchuk, V., and Lewis, B. C. (2007) Cancer
Res., 67, 7589-7596.
73.Lai, P. B., Chi, T. Y., and Chen, G. G. (2007)
Apoptosis, 12, 387-393.
74.Hussain, S. P., Schwank, J., Staib, F., Wang, X.
W., and Harris, C. C. (2007) Oncogene, 26, 2166-2176.
75.Mu, L. N., Cao, W., Zhang, Z. F., Cai, L., Jiang,
Q. W., You, N. C., Goldstein, B. Y., Wei, G. R., Chen, C. W., Lu, Q.
Y., Zhou, X. F., Ding, B. G., Chang, J., and Yu, S. Z. (2007) Cancer
Causes Control, 18, 665-675.
76.Lee, S. G., and Rho, H. M. (2000)
Oncogene, 19, 468-471.
77.Ueda, H., Ullrich, S. J., Gangemi, J. D., Kappel,
C. A., Ngo, L., Feitelson, M. A., and Jay, G. (1995) Nat.
Genet., 9, 41-47.
78.Jordan, R., Wang, L., Graczyk, T. M., Block, T.
M., and Romano, P. R. (2002) J. Virol., 76,
9588-9599.
79.Wang, X., McCullough, K. D., Franke, T. F., and
Holbrook, N. J. (2000) J. Biol. Chem., 275,
14624-14631.
80.Finkel, T., and Holbrook, N. J. (2000)
Nature, 408, 239-247.
81.Liang, T. J., Rehermann, B., Seeff, L. B., and
Hoofnagle, J. H. (2000) Ann. Intern. Med., 132,
296-305.
82.Chisari, F. V., and Ferrari, C. (1995)
Springer Semin. Immunopathol., 17, 261-281.
83.Webster, G., and Bertoletti, A. (2001) Mol.
Immunol., 38, 467-473.
84.Guidotti, L. G., Rochford, R., Chung, J.,
Shapiro, M., Purcell, R., and Chisari, F. V. (1999) Science,
284, 825-829.
85.Friedman, S. L. (2000) J. Biol. Chem.,
275, 2247-2250.
86.Kakimi, K., Lane, T. E., Chisari, F. V., and
Guidotti, L. G. (2001) J. Immunol., 167, 6701-6705.
87.Kao, J. H., and Chen, D. S. (2002) Lancet
Infect. Dis., 2, 395-403.
88.Colombo, M., and Donato, M. F. (2005) Semin.
Liver Dis., 25, 155-161.