
|
REVIEW: Cancer-Associated Antigens and Antigen Arrays in Serological Diagnostics of Malignant TumorsP. V. Belousov1*, D. V. Kuprash1, A. Yu. Sazykin2, S. V. Khlgatian2, D. N. Penkov1, Yu. V. Shebzukhov2, and S. A. Nedospasov1,21Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; fax: (499) 135-1405; E-mail: belous_p@rambler.ru; kuprash@online.ru2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-4716 * To whom correspondence should be addressed. |
Received December 20, 2007; Revision received January 28, 2008
The appearance of antibodies to cancer-associated antigens in biological fluids (particularly, in blood sera) of cancer patients is now a well-established fact, and their detection by immunochemical methods is a promising approach to diagnostics of malignant neoplasms. In this review, we consider some immunobiological aspects of the most extensively studied cancer-associated B-cell antigens, various applications of autoantibodies as cancer biomarkers, and prospects for the use of antigen arrays for improving diagnostic sensitivity.
KEY WORDS: cancer-associated antigens, autoantibodies, serological diagnostics of cancerDOI: 10.1134/S000629790805009X
Abbreviations: DTC) differentiated thyroid cancer; NUC) nonspecific ulcerative colitis; PNS) paraneoplastic neurological syndrome; PSA) prostate-specific antigen; Tg-Ab) anti-thyroglobulin antibodies.
Elaboration of methods for noninvasive diagnostics of malignant tumors
is one of the main challenges to contemporary oncology. It encompasses
a tremendous range of diagnostic tasks including early (screening) and
differential diagnostics of tumors, choice of treatment strategy,
prognosis, monitoring of tumor progression, and estimation of
therapeutic efficiency. Among the immense diversity of biological
markers, serum protein biomarkers are especially attractive by virtue
of ready availability of well-characterized inexpensive detection
procedures, e.g. immunoenzymatic analysis, and ease of isolation of
biological material. To date, clinical oncology has at its disposal
several tens of serum cancer biomarkers; however, their practical
application is limited to clinical trials and/or restricted-range
assays in diagnostically complicated cases. More than 20 cancer
biomarkers are now used in routine oncological practice; about 30% of
these markers are designed for detecting neuroendocrine tumors. It is
noteworthy that for the most important goal in the field, i.e. early
detection of tumors by screening, only one cancer biomarker, the serum
prostate-specific antigen (PSA), was recommended unanimously by several
expert groups in Europe and the USA as a reliable tool for screening
and early detection of prostate cancer in older male patients. For
comprehensive information about clinical applications of serum cancer
biomarkers, see the guidelines developed by the US National Academy of
Clinical Biochemistry
(http://www.aacc.org/AACC/members/nacb/LMPG/OnlineGuide/DraftGuidelines/TumorMarkers).
Serum autoantibodies to cancer-associated antigens represent a relatively new class of serological biomarkers and have a number of advantages over conventional protein oncomarkers. Their main characteristics are as follows:
- high specificity in cancer detection;
- lack of specific requirements for biomaterials and sampling;
- high stability in blood, serum, and plasma samples;
- long half-life;
- minimum concentration variations (e.g. diurnal, food-and-drug intake, physical activity, menstrual cycle phases, checkup procedures, etc.).
The mechanisms underlying T-cell-mediated humoral immune responses to cancer-associated antigens are not completely understood and are the subject of intensive studies. Recent experiments have shown that immunogenicity of the overwhelming majority of tumor-associated antigens is related to their aberrant regulation and/or tumor-specific modifications generated by oncogenic processes in tumor cells [1-4]. From this standpoint, serum autoantibodies to cancer-associated antigens can be regarded as specific “reporters” of carcinogenesis.
The ample body of evidence concerning identification and characterization of novel cancer-associated antigens (for methods see our recent review [5]) prompted the construction of a Cancer Immunome database (http://www2.licr.org/CancerImmunomeDB/SEREX_Intro.php), which comprises over 2300 candidate and validated B-cell antigens. At least several tens of them present substantial interest for serological diagnostics of cancer-associated diseases. However, the majority of currently known cancer-associated B-cell antigens have a serious disadvantage--low detection frequency of respective autoantibodies and, as a consequence, low diagnostic sensitivity in revealing cancer, the most critical parameter from the clinical standpoint.
To overcome this problem, it was suggested to combine candidate antigens into arrays in order to optimize diagnostic sensitivity and specificity parameters [6]. In the subsequent sections, we provide a detailed complete description of the most popular B-cell cancer-associated antigens and outline the prospects for combining individual antigens into arrays with the ultimate goal of improving their diagnostic sensitivity.
UNIVERSAL TUMOR ANTIGENS
Tumor suppressor p53. The tumor suppressor p53, the protein product of the p53 gene, is the most extensively studied cancer-associated B-cell antigen. It was discovered in 1979 by two independent groups of investigators [7-12]. Interestingly, the molecular-biological [7-10, 13] and serological [11, 12] methods used for its identification reflect the main activities of p53 in mammalian cells, i.e. those of a tumor suppressor and a cancer-associated antigen. With few exceptions, the protein product of the p53 gene is not detected in tissues of higher mammals [14], but it can be activated under cell stresses of various origin [15, 16].
Mutations in the p53 gene and amino acid substitutions induced by it are found in >=50% of patients with malignant tumors. These mutations change the conformational structure of p53 in such a way that it loses the ability to transactivate p53-dependent genes concomitantly with inhibition of DNA repair and formation of genetically unstable cells with a “switched-off” p53-induced apoptotic mechanism [17-19]. It is of note that accumulation of the inactive protein of p53 in cells expressing mutant p53 increases its half-life to several hours (compared to 20 min for wild-type p53). Therefore, p53 accumulation in tumor tissues is almost always synonymous to the presence of mutant p53 [20, 21].
For the majority of malignant neoplasms, the detection frequency of class IgG anti-p53 antibodies in appropriate cohorts of patients varies from 15 to 20%, being maximal in patients with squamous cell carcinomas of esophagus, head, and neck (the latter are predominantly localized in the mouth cavity) and minimum in patients with various malignancies of hematopoietic tissue (lymphomas, leukemias, multiple myelomas) and urogenital tract in males (prostate and testicular carcinomas) as well as in patients with melanomas, glial tumors, and differentiated thyroid carcinomas [22, 23]. In healthy donors, the reactivity of p53 is very low (<1%) [22]. Therefore, p53 represents a highly specific cancer-associated antigen.
Interestingly, in patients with the highest (20-35%) reactivity of p53 (e.g. squamous cell head and neck carcinomas; esophageal, urinary bladder, and colorectal tumors) the detection frequency of mutant/accumulated p53 in neoplastic tissues is also the highest (50-60%). In contrast, anti-p53 antibodies are seldom (2-8%) found in patients with leukemias, multiple myelomas, prostate cancer, and differentiated thyroid carcinomas where p53 gene mutations are rare (<10%). Immune responses to p53 are never detected in patients whose tumor histotypes (e.g. melanomas and testicular carcinomas) do not carry mutant p53 [22, 24]. These findings suggest that mutations in the coding region of the p53 gene and intracellular accumulation of the corresponding protein product are the main factors triggering immune responses to p53.
With a few exceptions, mutations/accumulation of mutant p53 in neoplastic tissues correlate with induction of humoral immune responses to p53. At the same time, mutations/accumulation of p53 do not stimulate B-cell-mediated immune responses, since only some patients whose tumors express mutant p53 are p53-seropositive. In-depth analysis of the literature on distribution of p53 mutations in tumors of p53-seropositive and p53-seronegative patients led Soussi to conjecture that these two cohorts do not differ by the presence of characteristic p53 mutations and that the same individual can be p53-seropositive or p53-seronegative [22]. This suggests that prediction of B-cell response to p53 is beyond the capabilities of any existing mutations. Moreover, seroconversion in initially p53-seronegative patients is never observed despite the initial p53 status of the tumor and the clinical course of the disease [25-27]. This suggests that some other intrinsic characteristics of the organism, e.g. MHC haplotype, can play the role of cofactors eliciting humoral responses to p53 in patients whose tumors express mutant p53.
Mapping of human immunogenic p53 epitopes made it possible to establish the localization of dominant B-cell epitopes of p53 in N- and C-terminal regions, i.e. outside the fragment carrying amino acid substitutions in the mutant protein [28-30].
Finally, in immunoenzymatic analysis serum autoantibodies to p53 recognize mutant and wild-type p53 with equal efficiency [31-33]. In this way, mutations in the coding region of the p53 gene, being nonimmunogenic per se, stimulate intracellular accumulation of the protein product of p53, which, in turn, plays the role of an immunogenic stimulus for the system of tumor immunosurveillance. Supporting evidence in favor of this hypothesis can be derived from the presence of anti-p53 autoantibodies in mice with SV40-induced tumors. In these tumors, p53 is intact; its accumulation is a result of its binding and stabilization by the SV40 large T-antigen [7, 13]. The immunogenic activity of mutant p53 can also be induced by conformational changes and exposure of a vast majority of epitopes that are masked in the wild-type p53 tetramer.
From the early diagnostics standpoint, localization of p53 mutations on the time axis of a multistage carcinogenesis model is ideal--it is a rather late “molecular” event in carcinogenesis but rather early “clinical” event. Indeed, the cell clone carrying mutant p53 is precancer by definition, and malignization of cells carrying mutant p53 is a question of time. On the other hand, progression of dysplastic foci carrying mutant p53 to invasive microcarcinoma is a long-term process lasting from several months to several years. Analysis of changes in the p53 status of a tissue or an organ with a high risk of cancer, such as lungs in smokers, esophagus in patients with reflux esophagitis, mammary gland in female patients with a family history of breast cancer, etc., enables cancer detection at the very earliest stage, i.e. long before the appearance of the first clinical manifestations of tumor growth. If, for one reason or another, the use of such markers for screening purposes is inexpedient, they can be used for discrimination between groups of moderately high and very high cancer risk in which aggressive diagnostic/treatment strategy (with respect to precancer foci) is warranted.
Serum antibodies to p53 play the role of a surrogate marker for mutant p53. Seroreactivity of p53 in patients with precancerous lesions and healthy donors with a high risk of cancer were examined for the purpose of screening of these autoantibodies as candidate markers for early detection of cancer [26, 29, 34-39]. Here we shall not go into details of classical analyses of p53 seroreactivity in risk groups for lung cancer (smokers) [26, 29, 36] or liver angiosarcomas (industrial workers with long-term exposure to vinyl chloride) [35], but, rather, will focus our attention on the results of two less known studies of p53 reactivity in patients with nonspecific ulcerative colitis (NUC) as a risk factor for colon cancer [38, 39].
NUC is a severe polyetiological inflammatory disease of the colon. Depending on localization of the inflammation foci and the volume of tissue involved, the relative risk of colorectal cancer in NUC patients varies from 2 to 15 compared to the control level. The cumulative risk of cancer in patients with NUC estimated 10, 20, and 30 years after the onset of the disease is 2, 8, and 18%, respectively, i.e. NUC has every reason to be regarded as a facultative precancer state.
Repeated colonoscopy with multiple biopsies from different regions of the inflamed colon is the main diagnostic procedure for very early detection of colorectal cancer in NUC patients. However, this approach has a number of limitations, such as high cost, compliance, etc. Therefore, a search for novel biomarkers as a reasonable alternative to colonoscopy, which might reduce procedural frequency and identify cohorts of patients with very high cancer risk in which aggressive and costly diagnostic strategies are warranted, is a task of paramount importance.
Low (9% [38] and 15% [39]) frequencies of reactivity of p53 in NUC patients was found by two independent teams of investigators. However, the situation changes dramatically if we take into account colonoscopy findings and degree of tumor progression in this cohort of patients.
The presence of dysplastic foci in NUC patients is regarded not only as a precancer state, but also as a marker of synchronous cancer in other regions of the affected colon. Cioffi et al. [38] failed to detect the presence of dysplastic foci in this category of patients, while in studies by Yoshizawa et al. [39] dysplasia and/or histologically validated tumors were recorded in 13 out of 286 patients. It is noteworthy that in patients of this group p53 seroreactivity (8 of 13, 62%) significantly exceeded that in the combined (noncancer) sampling (44 of 370, 12%, p < 0.001) (combined data from two studies). (Hereinafter, p was calculated using two-sided Fisher exact test.) The specificity and sensitivity of anti-p53 antibodies used as a marker for early diagnostics of NUC-associated colorectal cancer were estimated as 88 and 62%, respectively.
According to Yoshizawa et al. [39], screening of patients' sera for serum antibodies to p53 should not be regarded as an alternative to colonoscopy even at more advanced stages because of very low sensitivity of the assay. At the same time, this approach can be used as a supplementary test, e.g. wherever the results of colonoscopic examination show the minimum risk of inflammation, negative anti-p53 antibody test can be regarded as an indication for increasing the time till the next colonoscopy. This test can also replace colonoscopy in patients withdrawing from serial endoscopic procedures.
Anti-apoptotic proteins of the BIRC family. The ability of cells to evade apoptosis provoked by genotoxic factors, lack of contact inhibition (anoikis), proapoptotic signals, oncogene overexpression, etc., is the main way for malignant tumors to escape from anti-tumor control, while overexpression of anti-apoptotic proteins is a distinguishing feature of many malignant tumors. Inhibitors of apoptosis (IAP) are one of the most extensively studied families of proapoptotic proteins overexpressed in the vast majority of malignant neoplasms and able to induce direct inhibition of caspases and procaspases [40]. The fact that overexpression of some proteins can be accompanied by generation of autoimmune responses to these proteins led Rohayem et al. to suggest that one of representatives of this family, namely, Survivin (BIRC5, baculoviral IAP repeat-containing protein 5), manifests the properties of a cancer-associated B-cell antigen [41]. Antibodies to Survivin were detected in 11 of 51 (22%) patients with lung cancer and in 4 of 49 (8%) patients with colon cancer. The reactivity frequencies of BIRC5 and its homolog, BIRC7 (Livin), were studied in patients with different types of malignant tumors (Table 1) and did not exceed 5% in control groups (healthy donors).
Table 1. Reactivities of BIRC family
antigens in cohorts of patients with various types of malignant
tumors
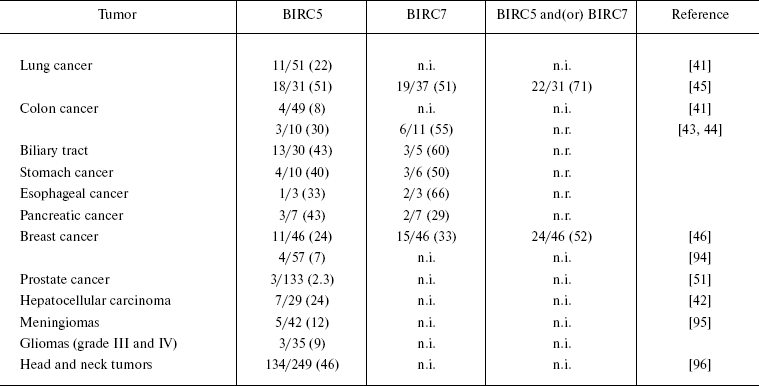
Note: The figures in parentheses designate the percent of
seropositive patients to the total number of patients in a cohort;
n.i., not investigated; n.r., not reported.
It is noteworthy that the reactivity spectra of BIRC5 and BIRC7 do not completely overlap; therefore, these proteins can be used simultaneously for increasing the sensitivity of immunoassays. In experiments by Yagihashi et al. [42-46], the sensitivity of cancer detection simultaneously with both antigens exceeded that of individual antigen assays 1.5-fold.
mRNA-binding proteins of the IMP family. In a search for novel cancer-testis antigens, Chen et al. [47] performed immunoscreening of a cDNA expression library obtained from mRNA of a melanoma cell line using an autologous serum from a patient with melanoma comprising antibodies against two known cancer-testis antigens--MAGE-1 and NY-ESO-1. It was found that more than 50% (33 of 61) of isolated and purified reactive clones corresponded to cDNAs of KOC (KH-domain containing gene overexpressed in cancer) gene (overexpressed in pancreatic carcinoma [48]) and two of its homologs. One of these homologs, p62, identified by Zhang et al. as a hepatocellular carcinoma-specific B-cell antigen, demonstrated high reactivity in patients with hepatocellular carcinoma (20 seropositive patients of 95, i.e. 21%). In healthy donors and in patients with non-cancer liver diseases, seroreactivity was absent (0 of 70 and 0 of 77, respectively, p < 0.001) [49]. Later, autoantibodies to at least one of these antigens (p62 and KOC) were found in 18-30% of patients depending on tumor type [50, 51]. Malignant ovarian and breast tumors were the least immunogenic judging from humoral immune responses to p62 and KOC (9 and 15%, respectively) [50]. In control groups, their reactivity did not exceed 3%. Interestingly, despite localization of immunodominant sites of the proteins in the same domains (N-terminal RRM motifs), virtually all p62 and/or KOC-positive sera contained antibodies to only one of two antigens [50]. Simultaneous application of two antigens increased the diagnostic sensitivity nearly twofold in comparison with median individual sensitivity (20.5 versus 11.6/12.2%).
The amino acid sequences of p62 and KOC contain several RNA-binding motifs--four hnRNP K homology (KH)-domains of the central and C-terminal fragments and two RRM motifs in the N-terminal fragment suggesting involvement of these proteins in mRNA processing. Studies by Nielsen et al. showed that p62 and KOC bind to the 5´-nontranslated region of the third leader mRNA of the embryonic growth factor IGF-II (insulin-like growth factor II) and repress the translation of this transcript [52]. Therefore p62 and KOC were termed as IMP/IGF2BP (Insulin-like Growth Factor 2 mRNA-Binding Proteins)--IMP-2 and IMP-3, respectively.
The mRNA-binding function of p62 and KOC raises the question about mechanisms of immune responses to these proteins. It was found that some RNA or DNA fragments of DNA/RNA-protein complexes demonstrate immunogenic activity in different autoimmune diseases and play a role in generation of autoimmune responses due to activation of specific receptors of the innate immunity [53]. The p62 and KOC homolog, IMP-1, antibodies against which were found in 11 of 133 (8.3%) patients with prostate cancer [51], forms stable ribonucleoprotein particles with IGF-II mRNA [54]. In turn, IGF-II is overexpressed and plays a key role in the pathogenesis of many malignant tumors [55]. These data suggest that overexpression of not only IMP family members (which was validated in experiments with many malignant neoplasms), but also of the “endogenous adjuvant” (IGF-II mRNA), plays a role in immunogenic activity of IMP proteins.
Hence, IMPs, as members of a new family of cancer-associated antigens, present substantial interest for future studies by virtue of their high (on the average, 20%) cancer-associated reactivity, mRNA-binding activity, and a key role of IGF-II in carcinogenesis.
Cyclin family. In a search for targets for humoral autoimmune responses to intracellular antigens in patients with hepatocellular carcinomas, Covini et al. [56] hypothesized that overexpression of the key cell cycle regulators--cyclins--in tumors of different histogenesis may play a role of an immunogenic stimulus triggering the synthesis of anticyclin antibodies. To test this hypothesis, the authors examined the seroreactivity of cyclins A (CCNA), B1 (CCNB1), D1 (CCNB1), and E (CCNE) and also of cyclin-dependent kinase 2 (CDK2) in patients with hepatocellular carcinomas, active chronic hepatitis, and liver cirrhosis as well as in healthy donors (control) [56]. Autoantibodies to cyclin B1 were found in 15 of 100 (15%) patients with hepatocellular carcinomas, in 3 of 70 (4.2%) patients with liver cirrhosis, and in 1 of 70 (1.4%) patients with chronic hepatitis; none of 70 healthy donors were seropositive for cyclin B1. In patients with hepatocellular carcinomas, cyclin A and CDK2 demonstrated very few reactivities, and autoantibodies to other antigens were absent. Later, Suzuki et al. [57] demonstrated high (20-45%) detection frequency of high anti-CCNB1 antibody titers in patients with colon, breast, and pancreatic carcinomas. Lower titers of these antibodies were found in patients with lung cancer and precancerous bronchopulmonary dysplasias. In these cohorts, the frequency of humoral immune responses correlated with CCNB1 overexpression in cancer/precancer foci. Recently Ersvaer et al. [58] found anti-CCNB1 antibodies in 7 of 65 (11%) patients with acute myeloid leukemia.
Overexpression of other members of the cyclin family is characteristic of many neoplasms; some of them also seem to be cancer-associated antigens. In malignant tumors of different histogenesis, cyclin D1 is overexpressed more frequently than other cyclins. Serum antibodies to this protein were found in 7 of 45 (16%) patients with prostate cancer (Gleason score 7-9) and in 2 of 96 (2.2%) healthy donors (p = 0.0049).
These findings suggest that at least two cyclins (B1 and D1) represent highly specific cancer-associated antigens and deserve further investigation from both practical (design of diagnostic antigen arrays) and theoretical (study of regularities of autoimmune responses to these proteins) standpoints.
Catalytic subunit of cAMP-dependent protein kinase A (PKA). Normal mammalian cells contain two intracellular forms (isozymes) of cAMP-dependent protein kinase A--PKA I and PKA II. The latter have a common catalytic subunit C, but differ in regulatory subunits (RI and RII, respectively). During growth of tumor cells of different histogenesis, the catalytic subunit of PKA otherwise termed as ECPKA (extracellular protein kinase A) is secreted into the culture medium. High (in comparison with control group of healthy donors) levels of this protein were found in blood sera of patients with tumors [59, 60].
Since aberrant excretion of the protein, which under normal conditions is localized exclusively inside the cell, is a potent immunological stimulus for B-cells, it was hypothesized that ECPKA may represent a cancer-associated B-cell antigen [61]. Indeed, higher (in comparison with control groups of healthy donors, as well as patients with systemic lupus erythematosus and Carney complex, which is an autosomal-dominant endocrine disease associated with loss-of-function mutation of the regulatory subunit of PKA RIalpha) antibody titers were found in virtually all cohorts of cancer patients [61]. On the whole, the specificity and sensitivity of anti-ECPKA antibodies were estimated as 90 and 87%, respectively. If specificity is typical of virtually all cancer-associated antigens, the sensitivity of this tumor marker significantly exceeds that of other autoantibody oncomarkers, which makes it an indispensable tool in advanced diagnostic studies based on the use of antibodies to cancer-associated antigens. Considering that serum ECPKA levels in melanoma patients decrease after surgical removal of the tumor [62], identification of anti-ECPKA antibodies also may be useful for monitoring tumor responses to implemented therapy.
ANTIGENS WITH RESTRICTED EXPRESSION IN NORMAL TISSUES
Cancer-testis antigens. The family of cancer-testis (cancer/gamete) antigens (CTA) is among the most intensively studied classes of cancer-associated antigens. The first antigen of this group--MAGE-1--was identified by T-cell epitope cloning [63, 64]. Further studies based on the use of this and other techniques (e.g. screening of expression libraries with blood sera from cancer patients, bioinformatic analysis in silico, etc.) enabled identification of more than 40 CTA gene families (containing from 1 to 12 genes) [65]. Their respective proteins are expressed by a vast variety of neoplasms (but not normal tissues), with the exception of testis. The latter belongs to immunologically privileged tissues; therefore, aberrant expression of CTA triggers T-cell and/or humoral immune responses as to a de novo introduced immunogen. High tissue specificity of gene expression, lack of autoimmune injuries associated with aberrant expression of CTA, and ability of the latter to trigger spontaneous antitumor responses culminated in the development of a wide panel of candidate cancer vaccines which now are undergoing different phases of clinical trials [66-68].
However, the use of cancer-testis antigens as tumor biomarkers is limited by low detection frequency of respective antibodies in patients with cancer (as a rule, 5-10%). Classical studies by Stockert et al. [69], who investigated humoral immune responses to four popular cancer-testis antigens (NY-ESO-1, MAGE-1, MAGE-3, and SSX2), demonstrated that only anti-NY-ESO-1 autoantibodies could successfully overcome the 5% reactivity barrier and established significant differences between healthy donors (0 of 70) and patients with melanomas (12 of 127, 9.4%, p = 0.0049) and ovarian cancer (4 of 32, 13%, p = 0.0085). These findings are consistent with the results of more recent studies suggesting that maximum seroreactivity of individual cancer-testis antigens does not exceed the 10% threshold [70-72]. Notwithstanding, autoantibodies to cancer-testis antigens can be used as additional markers in diagnostic antigen arrays.
Monitoring of therapeutic efficiency of a broad range of immunotherapeutic protocols is yet another area of application of autoantibodies to cancer-testis antigens. The results obtained at early stages of clinical testing of antigen-specific protocols of cancer immunotherapy suggest that antigen-specific CD4+ and CD8+ lymphocytes and serum antibodies against antigen used for vaccination can be used as surrogate markers of antitumor immune response [66-68]. As T-cell-mediated and humoral response do not completely overlap, autoantibodies to cancer-testis antigens can be used as valuable diagnostic markers of antitumor immunity during clinical testing of immunotherapeutic protocols based on cancer-testis antigens.
Differentiation antigens. Under the term “differentiation antigens” are understood antigens selectively expressed by certain tissues and respective tumors. An immense variety of differentiation tumor antigens of different histogenesis have been identified, among which ANKRD30A/NY-BR-1, the differentiation antigen of mammary gland glandular epithelium, was studied in especially great detail [73-80]. Although some immunological aspects of B-cell immunity of many of these antigens (including ANKRD30A/NY-BR-1) were not studied in large cohorts of cancer patients, these proteins attract attention as candidate T-cell antigens for immunotherapy. Some differentiation antigens display not so much cancer-associated reactivity as relatedness to autoimmune tissue injuries of different histogenesis. For example, autoantibodies to the differentiation antigen of brain glial cells (GFAP) were found in patients with gliomas [81] and autism [82]; autoantibodies to tyrosinase, the differentiation antigen of melanocytes [83], were identified in patients with melanomas and vitiligo, while autoantibodies to thyroglobulin, the differentiation antigen of the thyroid gland follicular epithelium [84], were found to be associated with differentiated thyroid cancer and autoimmune diseases of the thyroid gland. In some cases, similar autoantibodies can be used as indispensable oncomarkers; one of such cases will be considered below in the example of anti-thyroglobulin autoantibodies.
Thyroglobulin is secreted exclusively by follicular epithelium cells of the thyroid gland and its blood level drops below the detection threshold level after total surgical removal of the gland for differentiated thyroid cancer (DTC). Therefore, at any time any detectable level of thyroglobulin is estimated as relapse or distant metastasis. The situation is further aggravated by high detection frequency of anti-thyroglobulin autoantibodies (Tg-Ab) in both clinically healthy individuals (~10%) and patients with DTC (~25%). In thyroglobulin detection by immunometric assay, Tg-Ab (if they are present) block the thyroglobulin epitopes; therefore, this assay often gives false-negative results. To avoid this, all thyroglobulin assays include simultaneous determination of Tg-Ab. If Tg-Ab are found, zero level of thyroglobulin cannot be interpreted as evidence of remission, and further examinations of these patients are carried out using other diagnostic techniques. In some cases, Tg-Ab itself can be used as a tumor marker. For example, seroconversion in initially Tg-Ab-negative patients points to a relapse or distant metastasis, while stable Tg-Ab titers in patients after thyroidectomy are characteristic of persistence. Similarly, negative conversion in Tg-Ab-positive patients is suggestive of remission [84].
Another interesting example can be derived from the analysis of anti-PSA-antibody titers in patients with prostate cancer. PSA (prostate-specific antigen) secreted by prostate epithelium is a glycoprotein encoded by the KLK3 gene. Other serine proteases belonging to the KLK family also undergo clinical tests as candidate cancer biomarkers [85]. In patients with prostate cancer, serum PSA levels are notably increased; however, this increase is also characteristic of inflammation and benign hyperplasias of the prostate. By analogy with the aforementioned extracellular protein kinase A, significant increases in blood plasma levels of PSA can theoretically be estimated as a potent B-cell-mediated stimulating factor. Since estimation of specificity and sensitivity of anti-PSA antibodies is less efficient than PSA itself in diagnostics of prostate cancer, this test is valuable in that it allows discrimination between patients with androgen-dependent (high antibody titers) and androgen-independent (low antibody titers) tumors. However, serum PSA levels in both cohorts are similar [86]. Considering that surgical and/or therapeutic androgenic blockade is a strategy of choice in the treatment of androgen-dependent tumors, while hormone dependence is an important prognostic factor, serum anti-PSA antibodies appear to be a promising additional marker in the diagnostics of prostate cancer.
Onconeural antigens. The group of onconeural antigens stands apart among other cancer-associated antigens. Under normal conditions, these antigens are expressed in the central and peripheral nervous systems, but can also be expressed in neuroendocrine tumors. If such tumors are localized in immunologically unprivileged tissues, the immune system recognizes onconeural antigens as non-self and triggers humoral and T-cell-mediated immune responses to both the tumor and those divisions in the nervous system in which these antigens are expressed. This phenomenon has the name “paraneoplastic neurological syndromes” (PNS) [87].
Autoantibodies to onconeural antigens (hereinafter referred to as onconeural antibodies) are identified in the majority of patients with PNS. Their presence correlates, unambiguously and independently, with malignant growth [88] enabling their application in diagnostics of PNS and, which is especially important, localization of associated tumor foci.
The majority of modern studies are aimed at the analysis of seroreactivity of onconeural antigens in blood sera of PNS patients, which made this procedure a valuable differential diagnostic test in clinical neurology [89]. At the same time, there is evidence that onconeural antibodies predict: (i) presence and histogenesis of tumors, (ii) its anatomical localization, and (iii) presence and/or type of PNS [88]. Up to 20% of patients with particular types of tumors not associated with PNS are seropositive by some onconeural antigens (Table 2), the specificity of onconeural antibodies to oncopathology reaching 95-98%. High utility of onconeural antibodies in serological diagnostic tests leaves no doubt, since their appearance in the blood simultaneously with associated PNS anticipates clinical manifestation of tumor by several months and even years. Therefore, combination of onconeural antigens with other cancer-associated antigens (e.g. p53, Survivin, etc.) is a promising approach to detecting some types of cancer, particularly lung cancer, e.g. by screening of high-risk cohorts. For detailed description of diagnostic aspects of clinical application of onconeural antigens, see [89].
Table 2. Detection frequency of
autoantibodies to some onconeural antigens in cancer patients without
PNS [89, 97, 98]

*In parentheses, the total number of patients with small-cell lung
cancer, with exception of 44 patients in RCVRN1 raw, indicating the
total number of patients with non-small-cell lung cancer, is given.
DIAGNOSTIC AUTOANTIGENIC ARRAYS
As mentioned above, low sensitivity in cancer detection is the main disadvantage of virtually all (excluding ECPKA) cancer-associated B-cell antigens, while high diagnostic sensitivity is the main requirement for virtually all clinical applications of tumor biomarkers.
The second, no less important issue, is a lack of specificity of the majority of autoantibodies to cancer-associated antigens with respect to localization and histogenesis of malignant tumors. Most of them have different, yet overlapping, redundant autoantigenic repertoires. This means that even positive results of a diagnostic test (which in the case of highly specific cancer-associated antigens is a reliable marker of malignant growth) do not allow unequivocal interpretation and demand more exact localization of the tumor. The situation is further complicated by the fact that autoantibodies to many cancer-associated antigens can be found in patients with benign, inflammatory, autoimmune, and precancer diseases, although less frequently than in cancer patients.
The use of antigen arrays allows identification of so-called “autoantibody signatures” of various diseases; the latter represent combinations of reactive antigens able to discriminate between a pathology and a normal state. Association of antigens into arrays allows one to select a combination of antigens whose respective antibodies are strictly specific for the given pathology (~100%). The sensitivity (i.e. detection frequency of antibodies to at least one antigen) of such arrays also can reach 100%.
The first attempt to establish the reactivity of a cancer-associated antigen array in patients with colorectal cancer was undertaken by Scanlan et al. [90] who examined humoral immune responses to 77 candidate antigens identified by serological expression cloning (SEREX). A panel of 13 antigens (p53, MAGEA3, SSX2, NY-ESO-1, HDAC5, MBD2, TRIP4, NY-CO-45, KNSL6, HIP1R, Seb4D, KIAA1416, and LMNA) was selected for further assays. Analysis of autoantibodies reactive to at least one of these antigens made it possible to discriminate between patients with colon cancer and healthy donors with 46% sensitivity and 100% specificity (p < 10-10). The reactivity of each individual antigen (excluding highly immunogenic p53) varied from 3 to 8%. Only five individually tested antigens (p53, MAGEA3, NY-ESO-1, TRIP4, and HIP1R) displayed significant difference in seroreactivity between patients with large intestinal cancer and healthy donors. These data suggest that the use of an array of 13 cancer-associated antigens allowed 6-9-fold increase in sensitivity of cancer detection with preservation of 100% specificity of each individual antigen.
Studies by Zhang et al. [6] demonstrated a stepwise increase in the sensitivity of diagnostics of different types of cancer (64 cases of breast cancer, 46 cases of colorectal cancer, 91 cases of stomach cancer, 56 cases of lung cancer, 206 cases of prostate cancer, and 65 cases of hepatocellular carcinoma) on increase in the number of antigens included into the array from 5-20% for individual antigens (c-MYC, p53, CCNB1, p62, KOC, IMP1, and BIRC5) to 44-68% when using complete array (ELISA format). It is important to note that along with the drastic increase in sensitivity, the specificity of cancer detection with the antigen array used comprised 89-95% (compared to different control groups), i.e. decreased by only 5-10% in comparison with individual antigen specificity.
Based on the characteristic seroreactivity patterns of different cancer types, the same authors [91] used a statistical algorithm of recursive partitioning for the analysis of the same seven antigens in the same cohorts of patients (in these studies they added additional control groups and in some cases lowered the reactivity threshold level from +3 to +2 standard deviation of absorption in control group). Depending on type of cancer, this strategy achieved 77-92% sensitivity and 85-91% specificity and classified the patients into three cohorts according to the cancer type. In each of these cohorts, the desired levels of sensitivity and specificity could be achieved by analysis of reactivity to only three (of seven) antigens included in the array.
Using developed in our laboratory SMARTA-2 (serological mini-array of recombinant tumor antigens-2) assay, we examined the reactivity of 20 candidate antigens using panels of blood sera from patients with colon cancer and healthy donors. Five antigens (KIAA1864, MO-TES-391, CCND1, RGS5, and MMP-7) able to differentiate between patients with colon cancer and healthy donors were selected and combined in a panel with overall 98% specificity and 50% sensitivity. Similar analysis of sera from patients with DTC and benign nodular lesions in thyroid gland identified a combination of three antigens (ANKRD30A/NY-BR-1, KIAA1864, and RGS5) for discrimination between two cohorts in cancer revealing 95% specificity and 48% sensitivity (p = 0.00062), while in separate use, only ANKRD30A/NY-BR-1 demonstrated significant difference in reactivity between groups.
A comparison of the literature and our experimental data suggests that combination of different tumor-associated antigens in multiobject arrays increases the sensitivity of cancer diagnostics from 5-15 to 50-90% with a rather small (0-15%) loss of specificity. The number of antigens required for attaining optimum sensitivity and specificity does not exceed 10-20 for each cancer type. The number of specific antigens in the final panels depends on a number of factors:
- sensitivity of each individual antigen (the higher the sensitivity, the smaller the number of antigens needed in an array);
- degree of redundancy of reactivity profiles for each individual antigen (the greater the overlapping of reactivity profiles of individual antigens, the larger the number of antigens needed in an array);
- specific clinical task (diagnostic kits for early detection of cancer should include fewer targets than diagnostic kits for establishing the nature of primary foci in metastatic tumors of unknown primary localization).
Identification of “autoantibody fingerprints” of malignant tumors does more than opening up fresh opportunities for serological diagnostics and immunological analysis of malignant tumors. The majority of practitioners in the field agree that reactivity or autoantigenic patterns reflect not only specific interactions of tumors with the immune system, but also profound intracellular processes coupled with malignant transformation, growth and metastasis [1-4, 92]. Indeed, immunogenic activity of tumor antigens is stimulated by their aberrant regulation, which, in turn, directly correlates with the role of these proteins in molecular processes associated with carcinogenesis. Thus, immunogenicity of p53 is a result of its mutations and pathological accumulation inside the cell due to formation and maintenance of a malignant phenotype, escape from apoptotic control, and disturbances in DNA repair conferring high genetic “flexibility” on malignant cells. As far as immunogenicity of cancer-testis antigens is concerned, it is coupled with realization of an embryogenetic program of differentiated cells, e.g. “pregnancy” of somatic cells [93]. Elucidation of the whole autoantigenic repertoire may not only culminate in the development of novel diagnostic procedures, but help identify new molecules involved in carcinogenesis, objects for targeted therapy, etc. Elaboration of advanced serological diagnostic technologies on the basis of antigen arrays for identification of novel cancer autoantigens and autoantigenic repertoires is a task of paramount importance, particularly with regard to its clinical and investigative implications. It is our hope that this review will give a new impetus to such studies particularly in Russia.
The authors thank Dr. M. A. Lagarkova for valuable comments, Drs. V. E. Vanushko, K. V. Lanshchakov and other staff members of the Department of Endocrine Surgery of the Endocrine Research Center for collaboration, counseling, and discussions, Drs. C. Gouttefangeas and H.-G. Rammensee for cooperation and help in cloning CCND1, RGS5, and MMP-7 antigens, and Dr. D. Jager for generous supply of the ANKRD30A/NY-BR-1 clone.
This study has been carried out with the financial support of the Programs for Basic Research “Molecular and Cellular Biology” and “Basic Science to Medicine” of the Presidium of Russian Academy of Sciences, Russian Foundation for Basic Research (grant No. 05-04-49075), and the UICC Yamagiwa-Yoshida Memorial International Cancer Study Grant.
REFERENCES
1.Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J.,
and Schreiber, R. D. (2002) Nat. Immunol., 3,
991-998.
2.Dunn, G. P., Old, L. J., and Schreiber, R. D.
(2004) Immunity, 21, 137-148.
3.Dunn, G. P., Old, L. J., and Schreiber, R. D.
(2004) Annu. Rev. Immunol., 22, 329-360.
4.Tan, E. M. (2001) J. Clin. Invest.,
108, 1411-1415.
5.Shebzukhov, I., Belousov, P. V., Khlgatian, S. V.,
Sazykin, A. I., Kuprash, D. V., and Nedospasov, S. A. (2007) Mol.
Gen. Mikrobiol. Virusol., 3-6.
6.Zhang, J. Y., Casiano, C. A., Peng, X. X., Koziol,
J. A., Chan, E. K., and Tan, E. M. (2003) Cancer Epidemiol.
Biomarkers Prev., 12, 136-143.
7.Melero, J. A., Stitt, D. T., Mangel, W. F., and
Carroll, R. B. (1979) Virology, 93, 466-480.
8.Linzer, D. I., and Levine, A. J. (1979)
Cell, 17, 43-52.
9.Linzer, D. I., Maltzman, W., and Levine, A. J.
(1979) Virology, 98, 308-318.
10.Lane, D. P., and Crawford, L. V. (1979)
Nature, 278, 261-263.
11.Rotter, V., Witte, O. N., Coffman, R., and
Baltimore, D. (1980) J. Virol., 36, 547-555.
12.DeLeo, A. B., Jay, G., Appella, E., DuBois, G.
C., Law, L. W., and Old, L. J. (1979) Proc. Natl. Acad. Sci.
USA, 76, 2420-2424.
13.Kress, M., May, E., Cassingena, R., and May, P.
(1979) J. Virol., 31, 472-483.
14.Benchimol, S., Pim, D., and Crawford, L. (1982)
EMBO J., 1, 1055-1062.
15.Kastan, M. B., Onyekwere, O., Sidransky, D.,
Vogelstein, B., and Craig, R.W. (1991) Cancer Res., 51,
6304-6311.
16.Yonish-Rouach, E., Resnitzky, D., Lotem, J.,
Sachs, L., Kimchi, A., and Oren, M. (1991) Nature, 352,
345-347.
17.Bourdon, J. C., Laurenzi, V. D., Melino, G., and
Lane, D. (2003) Cell Death Differ., 10, 397-399.
18.Bourdon, J. C. (2007) Br. J. Cancer,
97, 277-282.
19.Soussi, T. (2005) Br. J. Surg., 92,
1331-1332.
20.Casey, G., Lopez, M. E., Ramos, J. C., Plummer,
S. J., Arboleda, M. J., Shaughnessy, M., Karlan, B., and Slamon, D. J.
(1996) Oncogene, 13, 1971-1981.
21.Dowell, S. P., Wilson, P. O., Derias, N. W.,
Lane, D. P., and Hall, P. A. (1994) Cancer Res., 54,
2914-2918.
22.Soussi, T. (2000) Cancer Res., 60,
1777-1788.
23.Fenton, C. L., Patel, A., Tuttle, R. M., and
Francis, G. L. (2000) Ann. Clin. Lab. Sci., 30,
179-183.
24.Malaguarnera, R., Vella, V., Vigneri, R., and
Frasca, F. (2007) Endocr. Relat. Cancer, 14, 43-60.
25.Hammel, P., Boissier, B., Chaumette, M. T.,
Piedbois, P., Rotman, N., Kouyoumdjian, J. C., Lubin, R., Delchier, J.
C., and Soussi, T. (1997) Gut, 40, 356-361.
26.Zalcman, G., Schlichtholz, B., Tredaniel, J.,
Urban, T., Lubin, R., Dubois, I., Milleron, B., Hirsch, A., and Soussi,
T. (1998) Clin. Cancer Res., 4, 1359-1366.
27.Buttitta, F., Marchetti, A., Gadducci, A.,
Pellegrini, S., Morganti, M., Carnicelli, V., Cosio, S., Gagetti, O.,
Genazzani, A. R., and Bevilacqua, G. (1997) Br. J. Cancer,
75, 230-235.
28.Vennegoor, C. J., Nijman, H. W., Drijfhout, J.
W., Vernie, L., Verstraeten, R. A., Mensdorff-Pouilly, S., Hilgers, J.,
Verheijen, R. H., Kast, W. M., Melief, C. J., and Kenemans, P. (1997)
Cancer Lett., 116, 93-101.
29.Schlichtholz, B., Tredaniel, J., Lubin, R.,
Zalcman, G., Hirsch, A., and Soussi, T. (1994) Br. J. Cancer,
69, 809-816.
30.Lubin, R., Schlichtholz, B., Bengoufa, D.,
Zalcman, G., Tredaniel, J., Hirsch, A., de Fromentel, C. C.,
Preudhomme, C., Fenaux, P., Fournier, G., Mangin, P., Laurent-Puig, P.,
Pelletier, G., Schlumberger, M., Desgrandchamps, F., le Duc, A.,
Peyrat, J. P., Janin, N., Bressac, B., and Soussi, T. (1993) Cancer
Res., 53, 5872-5876.
31.Winter, S. F., Minna, J. D., Johnson, B. E.,
Takahashi, T., Gazdar, A. F., and Carbone, D. P. (1992) Cancer
Res., 52, 4168-4174.
32.Schlichtholz, B., Legros, Y., Gillet, D.,
Gaillard, C., Marty, M., Lane, D., Calvo, F., and Soussi, T. (1992)
Cancer Res., 52, 6380-6384.
33.Labrecque, S., Naor, N., Thomson, D., and
Matlashewski, G. (1993) Cancer Res., 53, 3468-3471.
34.Cawley, H. M., Meltzer, S. J., de Benedetti, V.
M., Hollstein, M. C., Muehlbauer, K. R., Liang, L., Bennett, W. P.,
Souza, R. F., Greenwald, B. D., Cottrell, J., Salabes, A., Bartsch, H.,
and Trivers, G. E. (1998) Gastroenterology, 115,
19-27.
35.Trivers, G. E., Cawley, H. L., DeBenedetti, V.
M., Hollstein, M., Marion, M. J., Bennett, W. P., Hoover, M. L.,
Prives, C. C., Tamburro, C. C., and Harris, C. C. (1995) J. Natl.
Cancer Inst., 87, 1400-1407.
36.Lubin, R., Zalcman, G., Bouchet, L., Tredanel,
J., Legros, Y., Cazals, D., Hirsch, A., and Soussi, T. (1995) Nat.
Med., 1, 701-702.
37.Ralhan, R., Nath, N., Agarwal, S., Mathur, M.,
Wasylyk, B., and Shukla, N. K. (1998) Clin. Cancer Res.,
4, 2147-2152.
38.Cioffi, M., Riegler, G., Vietri, M. T., Pilla,
P., Caserta, L., Carratu, R., Sica, V., and Molinari, A. M. (2004)
Inflamm. Bowel. Dis., 10, 606-611.
39.Yoshizawa, S., Matsuoka, K., Inoue, N., Takaishi,
H., Ogata, H., Iwao, Y., Mukai, M., Fujita, T., Kawakami, Y., and Hibi,
T. (2007) Inflamm. Bowel. Dis., 13, 865-873.
40.LaCasse, E. C., Baird, S., Korneluk, R. G., and
MacKenzie, A. E. (1998) Oncogene, 17, 3247-3259.
41.Rohayem, J., Diestelkoetter, P., Weigle, B.,
Oehmichen, A., Schmitz, M., Mehlhorn, J., Conrad, K., and Rieber, E. P.
(2000) Cancer Res., 60, 1815-1817.
42.Yagihashi, A., Asanuma, K., Kobayashi, D., Tsuji,
N., Torigoe, T., Sato, N., and Watanabe, N. (2005) Autoimmunity,
38, 445-448.
43.Yagihashi, A., Asanuma, K., Tsuji, N., Torigoe,
T., Sato, N., Hirata, K., and Watanabe, N. (2003) Clin. Chem.,
49, 1206-1208.
44.Yagihashi, A., Asanuma, K., Nakamura, M., Araya,
J., Mano, Y., Torigoe, T., Kobayashi, D., and Watanabe, N. (2001)
Clin. Chem., 47, 1729-1731.
45.Yagihashi, A., Asanuma, K., Kobayashi, D., Tsuji,
N., Shijubo, Y., Abe, S., Hirohashi, Y., Torigoe, T., Sato, N., and
Watanabe, N. (2005) Lung Cancer, 48, 217-221.
46.Yagihashi, A., Ohmura, T., Asanuma, K.,
Kobayashi, D., Tsuji, N., Torigoe, T., Sato, N., Hirata, K., and
Watanabe, N. (2005) Clin. Chim. Acta, 362, 125-130.
47.Chen, Y. T., Gure, A. O., Tsang, S., Stockert,
E., Jager, E., Knuth, A., and Old, L. J. (1998) Proc. Natl. Acad.
Sci. USA, 95, 6919-6923.
48.Mueller-Pillasch, F., Lacher, U., Wallrapp, C.,
Micha, A., Zimmerhackl, F., Hameister, H., Varga, G., Friess, H.,
Buchler, M., Beger, H. G., Vila, M. R., Adler, G., and Gress, T. M.
(1997) Oncogene, 14, 2729-2733.
49.Zhang, J. Y., Chan, E. K., Peng, X. X., and Tan,
E. M. (1999) J. Exp. Med., 189, 1101-1110.
50.Zhang, J. Y., Chan, E. K., Peng, X. X., Lu, M.,
Wang, X., Mueller, F., and Tan, E. M. (2001) Clin. Immunol.,
100, 149-156.
51.Shi, F. D., Zhang, J. Y., Liu, D., Rearden, A.,
Elliot, M., Nachtsheim, D., Daniels, T., Casiano, C. A., Heeb, M. J.,
Chan, E. K., and Tan, E. M. (2005) Prostate, 63,
252-258.
52.Nielsen, J., Christiansen, J., Lykke-Andersen,
J., Johnsen, A. H., Wewer, U. M., and Nielsen, F. C. (1999) Mol.
Cell Biol., 19, 1262-1270.
53.Deane, J. A., and Bolland, S. (2006) J.
Immunol., 177, 6573-6578.
54.Nielsen, J., Kristensen, M. A., Willemoes, M.,
Nielsen, F. C., and Christiansen, J. (2004) Nucleic Acids Res.,
32, 4368-4376.
55.Pavelic, K., Bukovic, D., and Pavelic, J. (2002)
Mol. Med., 8, 771-780.
56.Covini, G., Chan, E. K., Nishioka, M., Morshed,
S. A., Reed, S. I., and Tan, E. M. (1997) Hepatology, 25,
75-80.
57.Suzuki, H., Graziano, D. F., McKolanis, J., and
Finn, O. J. (2005) Clin. Cancer Res., 11, 1521-1526.
58.Ersvaer, E., Zhang, J. Y., McCormack, E., Olsnes,
A., Anensen, N., Tan, E. M., Gjertsen, B. T., and Bruserud, O. (2007)
Eur. J. Haematol., 79, 210-225.
59.Cho, Y. S., Park, Y. G., Lee, Y. N., Kim, M. K.,
Bates, S., Tan, L., and Cho-Chung, Y. S. (2000) Proc. Natl. Acad.
Sci. USA, 97, 835-840.
60.Cvijic, M. E., Kita, T., Shih, W., DiPaola, R.
S., and Chin, K. V. (2000) Clin. Cancer Res., 6,
2309-2317.
61.Nesterova, M. V., Johnson, N., Cheadle, C.,
Bates, S. E., Mani, S., Stratakis, C. A., Khan, I. U., Gupta, R. K.,
and Cho-Chung, Y. S. (2006) Cancer Res., 66,
8971-8974.
62.Kita, T., Goydos, J., Reitman, E., Ravatn, R.,
Lin, Y., Shih, W. C., Kikuchi, Y., and Chin, K. V. (2004) Cancer
Lett., 208, 187-191.
63.Van der Bruggen, P., Traversari, C., Chomez, P.,
Lurquin, C., de Plaen, E., van den Eynde, B., Knuth, A., and Boon, T.
(1991) Science, 254, 1643-1647.
64.Traversari, C., van der Bruggen, P., van den
Eynde, B., Hainaut, P., Lemoine, C., Ohta, N., Old, L., and Boon, T.
(1992) Immunogenetics, 35, 145-152.
65.Simpson, A. J., Caballero, O. L., Jungbluth, A.,
Chen, Y. T., and Old, L. J. (2005) Nat. Rev. Cancer, 5,
615-625.
66.Slingluff, C. L., Jr., Petroni, G. R.,
Chianese-Bullock, K. A., Smolkin, M. E., Hibbitts, S., Murphy, C.,
Johansen, N., Grosh, W. W., Yamshchikov, G. V., Neese, P. Y.,
Patterson, J. W., Fink, R., and Rehm, P. K. (2007) Clin. Cancer
Res., 13, 6386-6395.
67.Jager, E., Karbach, J., Gnjatic, S., Neumann, A.,
Bender, A., Valmori, D., Ayyoub, M., Ritter, E., Ritter, G., Jager, D.,
Panicali, D., Hoffman, E., Pan, L., Oettgen, H., Old, L. J., and Knuth,
A. (2006) Proc. Natl. Acad. Sci. USA, 103,
14453-14458.
68.Davis, I. D., Chen, W., Jackson, H., Parente, P.,
Shackleton, M., Hopkins, W., Chen, Q., Dimopoulos, N., Luke, T.,
Murphy, R., Scott, A. M., Maraskovsky, E., McArthur, G., MacGregor, D.,
Sturrock, S., Tai, T. Y., Green, S., Cuthbertson, A., Maher, D.,
Miloradovic, L., Mitchell, S. V., Ritter, G., Jungbluth, A. A., Chen,
Y. T., Gnjatic, S., Hoffman, E. W., Old, L. J., and Cebon, J. S. (2004)
Proc. Natl. Acad. Sci. USA, 101, 10697-10702.
69.Stockert, E., Jager, E., Chen, Y. T., Scanlan, M.
J., Gout, I., Karbach, J., Arand, M., Knuth, A., and Old, L. J. (1998)
J. Exp. Med., 187, 1349-1354.
70.Jager, E., Stockert, E., Zidianakis, Z., Chen, Y.
T., Karbach, J., Jager, D., Arand, M., Ritter, G., Old, L. J., and
Knuth, A. (1999) Int. J. Cancer, 84, 506-510.
71.Sugita, Y., Wada, H., Fujita, S., Nakata, T.,
Sato, S., Noguchi, Y., Jungbluth, A. A., Yamaguchi, M., Chen, Y. T.,
Stockert, E., Gnjatic, S., Williamson, B., Scanlan, M. J., Ono, T.,
Sakita, I., Yasui, M., Miyoshi, Y., Tamaki, Y., Matsuura, N., Noguchi,
S., Old, L. J., Nakayama, E., and Monden, M. (2004) Cancer Res.,
64, 2199-2204.
72.Fujita, S., Wada, H., Jungbluth, A. A., Sato, S.,
Nakata, T., Noguchi, Y., Doki, Y., Yasui, M., Sugita, Y., Yasuda, T.,
Yano, M., Ono, T., Chen, Y. T., Higashiyama, M., Gnjatic, S., Old, L.
J., Nakayama, E., and Monden, M. (2004) Clin. Cancer Res.,
10, 6551-6558.
73.Jager, D., Stockert, E., Gure, A. O., Scanlan, M.
J., Karbach, J., Jager, E., Knuth, A., Old, L. J., and Chen, Y. T.
(2001) Cancer Res., 61, 2055-2061.
74.Seil, I., Frei, C., Sultmann, H., Knauer, S. K.,
Engels, K., Jager, E., Zatloukal, K., Pfreundschuh, M., Knuth, A.,
Tseng-Chen, Y., Jungbluth, A. A., Stauber, R. H., and Jager, D. (2007)
Int. J. Cancer, 120, 2635-2642.
75.Theurillat, J. P., Zurrer-Hardi, U., Varga, Z.,
Barghorn, A., Saller, E., Frei, C., Storz, M., Behnke, S., Seifert, B.,
Fehr, M., Fink, D., Rageth, C., Linsenmeier, C., Pestalozzi, B., Chen,
Y. T., Knuth, A., Jager, D., and Moch, H. (2008) Int. J. Cancer,
122, 1585-1591.
76.Jager, D., Karbach, J., Pauligk, C., Seil, I.,
Frei, C., Chen, Y. T., Old, L. J., Knuth, A., and Jager, E. (2005)
Cancer Immun., 5, 11.
77.Nissan, A., Jager, D., Roystacher, M., Prus, D.,
Peretz, T., Eisenberg, I., Freund, H. R., Scanlan, M., Ritter, G., Old,
L. J., and Mitrani-Rosenbaum, S. (2006) Br. J. Cancer,
94, 681-685.
78.Jager, D., Filonenko, V., Gout, I., Frosina, D.,
Eastlake-Wade, S., Castelli, S., Varga, Z., Moch, H., Chen, Y. T.,
Busam, K. J., Seil, I., Old, L. J., Nissan, A., Frei, C., Gure, A. O.,
Knuth, A., and Jungbluth, A. A. (2007) Appl. Immunohistochem. Mol.
Morphol., 15, 77-83.
79.Theurillat, J. P., Zurrer-Hardi, U., Varga, Z.,
Storz, M., Probst-Hensch, N. M., Seifert, B., Fehr, M. K., Fink, D.,
Ferrone, S., Pestalozzi, B., Jungbluth, A. A., Chen, Y. T., Jager, D.,
Knuth, A., and Moch, H. (2007) Cancer Immunol. Immunother.,
56, 1723-1731.
80.Varga, Z., Theurillat, J. P., Filonenko, V.,
Sasse, B., Odermatt, B., Jungbluth, A. A., Chen, Y. T., Old, L. J.,
Knuth, A., Jager, D., and Moch, H. (2006) Clin. Cancer Res.,
12, 2745-2751.
81.Schmits, R., Cochlovius, B., Treitz, G., Regitz,
E., Ketter, R., Preuss, K. D., Romeike, B. F., and Pfreundschuh, M.
(2002) Int. J. Cancer, 98, 73-77.
82.Singh, V. K., Warren, R., Averett, R., and
Ghaziuddin, M. (1997) Pediatr. Neurol., 17, 88-90.
83.Fishman, P., Merimski, O., Baharav, E., and
Shoenfeld, Y. (1997) Cancer, 79, 1461-1464.
84.Harish, K. (2006) Endocr. Regul.,
40, 53-67.
85.Borgono, C. A., Michael, I. P., and Diamandis, E.
P. (2004) Mol. Cancer Res., 2, 257-280.
86.Nesterova, M., Johnson, N., Cheadle, C., and
Cho-Chung, Y. S. (2006) Biochim. Biophys. Acta, 1762,
398-403.
87.Albert, M. L., and Darnell, R. B. (2004) Nat.
Rev. Cancer, 4, 36-44.
88.Pittock, S. J., Kryzer, T. J., and Lennon, V. A.
(2004) Ann. Neurol., 56, 715-719.
89.Belousov, P. V., Shezbukhov, I., Nedospasov, S.
A., and Kuprash, D. V. (2007) Mol. Gen. Mikrobiol. Virusol.,
6-13.
90.Scanlan, M. J., Welt, S., Gordon, C. M., Chen, Y.
T., Gure, A. O., Stockert, E., Jungbluth, A. A., Ritter, G., Jager, D.,
Jager, E., Knuth, A., and Old, L. J. (2002) Cancer Res.,
62, 4041-4047.
91.Koziol, J. A., Zhang, J. Y., Casiano, C. A.,
Peng, X. X., Shi, F. D., Feng, A. C., Chan, E. K., and Tan, E. M.
(2003) Clin. Cancer Res., 9, 5120-5126.
92.Casiano, C. A., Mediavilla-Varela, M., and Tan,
E. M. (2006) Mol. Cell Proteomics, 5, 1745-1759.
93.Old, L. J. (2007) Cancer Immun., 7,
19.
94.Al Joudi, F. S., and Iskandar, Z. A. (2006)
Med. J. Malaysia, 61, 302-306.
95.Soling, A., Plugge, E. M., Schmitz, M., Weigle,
B., Jacob, R., Illert, J., Holzhausen, H. J., and Rainov, N. G. (2007)
Int. J. Oncol., 30, 123-128.
96.Chang, J. T., Wong, F. H., Liao, C. T., Chen, I.
H., Wang, H. M., and Cheng, A. J. (2004) Clin. Chem., 50,
1261-1264.
97.Bazhin, A. V., Savchenko, M. S., Shifrina, O. N.,
Demoura, S. A., Chikina, S. Y., Jaques, G., Kogan, E. A., Chuchalin, A.
G., and Philippov, P. P. (2004) Lung Cancer, 44,
193-198.
98.Graus, F., Delattre, J. Y., Antoine, J. C.,
Dalmau, J., Giometto, B., Grisold, W., Honnorat, J., Smitt, P. S.,
Vedeler, C., Verschuuren, J. J., Vincent, A., and Voltz, R. (2004)
J. Neurol. Neurosurg. Psychiatry, 75, 1135-1140.