
|
REVIEW: Targeted Therapy in the Treatment of Solid Tumors: Practice Contradicts TheoryN. V. Zhukov1* and S. A. Tjulandin21Federal Research-Clinical Center for Pediatric Hematology, Oncology, and Immunology, Ministry of Health Care and Social Security of the Russian Federation, Leninsky pr. 117, Bldg. 2, 117997 Moscow, Russia; E-mail: zhukov.nikolay@rambler.ru2Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia; fax: (495) 324-9834; E-mail: stjulandin@mail.ru * To whom correspondence should be addressed. |
Received December 5, 2007
The basic principle of targeted therapy formulated about ten years ago consists in the design and application of drugs specifically directed against well-defined targets that are critical for tumor survival and not compromising for normal organs and tissues. The past decade has been marked by the appearance of an immense diversity of novel antitumor agents with claimed targeted action. Unfortunately, despite indisputable progress in clinical settings, some popular drugs against solid tumors (e.g. bevacizumab, trastuzumab, erlotinib, gefitinib) nominally assigned to targeted-action drugs, cannot actually be classified with this group being nonconforming to a priori stated goals of targeted therapy. The state-of-the-art and current problems in targeted therapy of solid tumors are reviewed.
KEY WORDS: targeted therapy, breast cancer, colorectal cancer, VEGF, VEGFR, EGFR, HER-2DOI: 10.1134/S000629790805012X
Abbreviations: CEP) circulating endothelial precursors; EGFR) epidermal growth factor receptor; HSC) hemopoietic stem cells; NSCLC) non-small cell lung cancer; VEGF) vasoendothelial growth factor.
Only six decades separate us from the moment of the first clinical
application of an antitumor drug. During this period, antitumor therapy
has gone through several critical phases. Curiously enough, nearly all
major achievements in cytostatic therapy were made in early stages of
its development when practitioners in oncology had at their disposal
only a very small range of antitumor drugs, namely, cisplatin,
doxorubicin, cyclophosphamide, 5-fluorouracil, tamoxifen, etc.; most of
these were developed 20-30 years ago. Today a vast variety of
registered cytostatic drugs are used in the treatment of malignant
tumors, but their quantity has not been transformed into quality, since
novel drugs fail to conform to the main goals of anticancer
therapy--prolongation of the patient's life and, in the ideal case,
increase of cure rate. Even if these goals were realized, the
“gain” from anticancer therapy consisted in a small increase
in overall survival (on average, by 3-5%, less frequently, by 10%).
More often “advantages” of novel drugs included such criteria
as increase of time to tumor progression, convenience in drug use
without influencing survival percentage, lack of toxic side effects of
definite types, etc. Attempts to improve drug efficiency through
many-fold increase in therapeutic dose and the use of inhibitors of
tumor resistance mechanisms or tissue protectors were unsuccessful. The
situation in the chemotherapy of solid tumors on the eve of the XXI
century can be characterized as stagnant. By that time the efficiency
of “classical” chemotherapy directed against tumor genome
and/or the cell proliferation apparatus reached an absolute ceiling.
The main unresolved problems of modern chemotherapy include lack of selectivity, i.e. unwarranted damage normal cells, and empirical choice of anticancer drugs. By virtue of their genomic instability, tumor cells rapidly develop resistance to anticancer agents; therefore, design of new cytostatic drugs with an “old” mechanism of action seldom brings positive results. Moreover, drug toxicity due to same (with tumor) targets in the patient's body (bone marrow, mucous membrane epithelium, etc.) limits the use of many cytostatics in adequate doses, which accelerates development of drug resistance. In contrast to antimicrobial therapy, where such failures result from activation of new resistance mechanisms “under the pressure” of the widely used antibiotics, the newly designed cytostatic drugs failed to improve the efficiency of antitumor treatment, since the mechanisms of resistance are the same as 60 years ago. This suggests that the approach directed to damage of tumor genome and cell-kill make all as possible, and a breakthrough in this field can hardly be expected. The same targets for cytostatic therapy in the tumor itself and in the normal organs and tissues are the main factors responsible for these failures. Undeniable progress has been achieved only in the therapy of tumors whose sensitivity threshold is lower than that of normal organs and tissues. In other situations all additional approaches, such as modification of structure of cytostatic drugs, dose increase with subsequent transplantation of hemopoiesis precursor cells, use of inhibitors of resistance mechanisms, etc., failed to increase the therapeutic index (tumor eradication without fatal damage of patient's normal tissues) despite the use of the most advanced technologies.
Targeted therapy is a radically new approach to drug treatment of malignant neoplasms that appeared during the last decade. Although its goal has not been finally formulated yet, it is implied that its main difference from conventional chemotherapy is in the targeted action on tumor cells not compromising for normal cells and tissues. It was anticipated that the novel approach, independently or in combination with conventional chemotherapy, would be a radical departure from “classical” techniques and a breakthrough in the pharmacotherapy of malignant tumors. Considering that basic principles of targeted therapy were declared about ten years ago, it seems to us expedient to overview its main milestones and to summarize some intermediate results in their practical application.
DEFINITION OF TARGETED THERAPY
What is the purpose of targeted therapy and what do we call targeted-action drugs? No explicit descriptions exists distinguishing targeted therapy from other approaches to anticancer therapy. The first formal statement of its primary goals made by the US Food and Drug Administration (FDA) suggests that a targeted-action drug is understood to be a drug whose prescription has to be preceded by a formally registered diagnostic test proving the existence of a target for its effect. It is required that this diagnostic test be an integral part of an implicit combination, of which a targeted-action drug is a component, and, though supplied under different covers, must always be used as a single whole in order to provide the maximum therapeutic effect [1]. Although this definition provides a comprehensive description of one of the basic principles of targeted therapy, i.e. identification of a target prior to its prescription to a patient (or, more correctly, to patients whose tumors express this target), we do not think it to be complete. The presence of DNA, the main target for alkylating cytostatic agents, in any tumor cell can easily be established with the help of registered diagnostic tests, but it is not the reason for assigning, e.g. cyclophosphamide, to targeted-action drugs. In our opinion, the main requirements for targeted therapy are as follows:
- validated effect on the target (receptor, growth factor, etc.) critical for tumor survival and not compromising for normal organs and tissues;
- validation and estimation of a target(s) by registered diagnostic tests; prediction of efficiency of anticancer therapy (e.g. high efficiency in the presence of a target and zero efficiency in its absence);
- lack of toxicity related to the basic mechanism of the drug action and low or zero level of nonspecific toxicity (allergic reactions, coagulation effects, etc.).
The use of this set of criteria helps overcome the main disadvantages of conventional chemotherapy--empirical prescription of drugs (the same chemotherapy is usually prescribed to all patients with a definite type and stage of tumor without their preliminary allocation into cohorts with the highest prognostic beneficial therapeutic effect) and their nonspecific effect (the use of drugs in maximally effective doses is often limited by their general toxicity).
At present, a vast array of antitumor agents “claimed” as targeted-action drugs by manufacturers and “accepted” for use by practitioners in the field have undergone registration and/or are undergoing clinical trials. The most popular of them are as follows:
- drugs acting on receptors (and ligands thereof), enzymes, etc., mediating signal transmission to tumor cells (antibodies, small molecules, etc.);
- drugs inhibiting tumor microenvironment critical for tumor survival;
- antibodies eliciting immune responses and/or delivering toxic substances (radioactive materials, cytostatic drugs, etc.) to tumor cells.
Most of these agents are devoid of cytotoxic activity and ability to kill or damage tumor cells, but exert pronounced cytostatic effects by inhibiting proliferation and/or stimulating differentiation of tumor cells through inhibition of mechanisms responsible for the formation of the malignant phenotype. In other words, the effects of these drugs are not curative and consist in the retardation of tumor growth or, in the best case, reduction of tumor mass, which does not detract from their merits, the switchover from malignancy to a sluggish chronic process being only somewhat less attractive goal than cure. It is therefore desirable to prolong the drug effect through its long-term administration (lack of cumulative toxicity) and suppression of drug resistance (lack of alternative non-blockading pathways to stimulation of tumor growth and activation of mechanisms responsible for inactivation or elimination of drugs).
In this review, the state-of-the-art in targeted therapy of solid tumors will be considered on the example of some of the most popular targeted-action drugs--monoclonal antibodies (bevacizumab and trastuzumab) and tyrosine kinase inhibitors (erlotinib and gefitinib).
ANTIANGIOGENIC THERAPY AND BEVACIZUMAB
Effects on VEGF (vasoendothelial growth factor) critical for tumor survival and not compromising for normal organs and tissues. Under normal conditions, activation of angiogenesis, i.e. formation of new vasculature, takes place during embryogenesis or, if we deal with adult organisms, in definite physiological states, e.g. pregnancy, wound healing, etc. In healthy adults, only 0.01% of endothelial cells undergo mitosis (once every 10 years) [2]. However, the situation changes radically if tumorigenesis is turned on. Of course, neither tumorigenesis nor the continuous demand of tumors for oxygen and nutrients are “programmed” by the organism in advance. At the moment of their origin, malignant tumors are surrounded by blood vessels supplying normal tissues with oxygen and nutrients; therefore, useful substances can be delivered to tumor cells only by diffusion and only if the tumor is small (1-2 mm3) [3]. Initially, the tumor is devoid of stroma whose cells (fibroblasts, etc.) able to produce special proangiogenic substances initiating the growth of new vessels in post-embryonal period. Therefore, in early steps of tumorigenesis tumor cells must to regain this ability characteristic of their embryonal stage. It is this circumstance rather than enhanced proliferation of tumor cells that increases the time interval till the moment when the tumor mass reaches a certain “clinically significant” level. Owing to heterogeneity of tumor cell populations, proangiogenic substances can be produced only by primary (but not metastatic) tumors. This is manifested in retardation (by 10-15 years) of micrometastasis growth following surgical treatment of breast cancer (clinically detectable metastasis appeared only after production of proangiogenic substances switch-on) or spontaneous regression of distant metastases after surgical removal of primary tumors in patients with renal cancer (if metastases do not produce proangiogenic substances, they regress).
Hypoxia due to continued inadequate blood supply is the main factor stimulating tumor angiogenesis. The ability to stimulate angiogenesis is characteristic of many active substances: VEGF; platelet-derived growth factor (PDGF or thymidine phosphorylase); fibroblast growth factor (FGF-1, FGF-2); angiopoietin-1 (ang-1), etc. These are produced by tumor, stromal and endothelial cells, extracellular matrix, and blood cells [4]. Under normal conditions and during tumor growth, the activity of proangiogenic substances is counterbalanced by antiangiogenic substances. The relative contributions of proangiogenic and antiangiogenic molecules to the formation of tumor vasculature depend on the histological type and origin of tumor, dynamics of their expression in tumorigenesis, as well as tumor regression and relapse. Their effects on the formation of new tumor vasculature are realized through different mechanisms, such as increased permeability of the vascular wall, attraction of circulating endotheliocyte precursors, stimulation of migration and proliferation of endotheliocytes, etc.
In adult organisms, vascularization processes are initiated by circulating endothelial precursors (CEP) and hemopoietic stem cells (HSC) (probably, derived from a common precursor (hemangioblast)) originating from bone marrow. These cells express on their surface special receptors for the main angiogenesis-promoting factor, VEGF [5]. It is CEP and HSC but not mature endotheliocytes that are responsible for the onset of tumor angiogenesis [6]. The ability of these cells residing in bone marrow and peripheral blood to stimulate angiogenesis in adult organisms is a prerequisite to the beneficial effect of “autotransplantation” of bone marrow (or stimulated HSC) in patients with ischemic heart disease.
Tumor angiogenesis never takes place “in the right place and at the right time”, and newly formed vessels are usually deficient in both structure and function. The formation of “immature” vessels with impaired wall structure and irregular architectonics is a result of disbalance between proangiogenic and antiangiogenic effects on tumor angiogenesis. These defects are compensated by increased permeability and high density of tumor microvessels. Some tumor types (e.g. breast and colon cancers, melanomas, etc.) manifest vascular mimicry or a mosaic structure where some parts of their microvascular wall consist of tumor cells instead of endothelial cells [7, 8]. By reason of such mosaic structure, every day up to 106 tumor cells (the whole tumor mass does not usually exceed 1 g) go out into the circulating blood [8]. These factors increase, directly or indirectly, the malignant potential of the tumor and diminish its sensitivity to anticancer therapy. Increased permeability and high density of tumor microvessels as well as vascular mimicry provide adequate supply of tumor tissues with nutrients and stimulate metastasis despite vascular immaturity, while impaired architectonics and structural deficiency of tumor vessels create a high pressure gradient, which impedes circulation and drug delivery to tumor cells.
These events make angiogenesis and de novo formed tumor vasculature attractive targets for anticancer therapy. Compounds stimulating the growth of new vessels and functional activity of their specific receptors and de novo formed vascular endothelium are potential targets for antiangiogenic therapy.
As follows from its definition, antiangiogenic therapy is not curative, being not directed for tumor cells. However, inhibition of tumor angiogenesis and regression of de novo formed tumor vessels can transfer the tumor into a “dormant” state associated with retardation of tumor growth and reduction of its size due to hypoxia. Ideally the tumor and metastasis should become clinically undetectable because nutrients can penetrate them exclusively by diffusion. Inhibition of angiogenesis can lead to “normalization” of intratumoral vasculature [9]. This phenomenon is used by many authors as an example of paradoxical synergism in the effects of conventional cytostatic and antiangiogenic drugs, i.e. “devascularization” theoretically should be associated with suppressed delivery of cytostatic drugs to target cells, but “normalization” of tumor vasculature improves drug delivery even if the number of vessels is reduced.
The most popular cytostatic drugs (taxanes, topoisomerase inhibitors, purine antimetabolites, interferon, etc.) have toxic (direct and indirect) effects on tumor vasculature. This circumstance stimulated the development of a new trend in cancer treatment--metronomic chemotherapy. It entails indication of cytostatic drugs in much lower doses than is necessary for producing “direct” antitumor effect, but high enough to destroy tumor vascular endothelium. These doses are usually nontoxic; therefore, cytostatic drugs can be administered over sufficiently long periods of time without the risk of repair of the damaged endothelium [10]. The use of endothelial cells as targets for anticancer therapy, supposedly, will make it possible to avoid or significantly delay resistance onset, since in contrast to tumor cells the endothelial cell genome is less affected by mutations. Preliminary studies in solid tumors gave encouraging results, but did not go beyond the pilot trial protocol [11, 12]. It has to be admitted that some of the approaches developed long before the advent of metronomic therapy have similar antiangiogenic mechanisms of action. The use of maintenance therapy in patients with acute lymphoblastic leukemias is an example. In these patients, intensive induction therapy and remission consolidation were supported by long-term administration of low doses of cytostatic drugs, which diminished the risk of relapse. Although this therapy found wide acceptance, its contribution to the overall therapeutic effect is rather small and cannot be regarded as full-value targeted therapy, being not directed against specific targets.
VEGF is one of the most potent stimulators of neoangiogenesis. This peptide or, more exactly, one of the representatives of the VEGF family, VEGF-A, was isolated and described in the 1980s under the name “vascular permeability factor” (VPF) [13], which reflects its ability to increase vascular permeability with a ~1000 times greater efficiency than histamine. Several isoforms of VEGF protein (VEGF-A - VEGF-E), placental growth factor (PLGF), and structurally close to them glycoproteins (PDGF) are presently known. Enhanced expression of VEGF isoforms is characteristic of some malignant tumors (breast, lung, pancreas, kidney, ovary, bladder, brain; multiple myelomas, lymphomas, etc.); very often it predicts unfavorable outcome. Hypoxia is the main factor stimulating VEGF synthesis, especially in paranecrotic zones of the tumor. VEGF synthesis under hypoxia is a result of enhanced expression of the hypoxia-induced factor (HIF-1). The gene product of the von Hippel-Lindau (VHL) tumor suppressor is responsible for negative regulation of VEGF [14]. Low activity of this gene is associated with a hereditary syndrome manifested in activation of neoangiogenesis and high risk of malignant tumors (kidney, brain, adrenal glands, pancreas, etc.).
The effect of VEGF is directed at domains adjacent to the site of its synthesis, but it can also affect more distant targets. VEGF exerts its action on endothelial cells through binding to their specific receptors. Different representatives of the VEGF family (VEGF-A, -B, -C, -D and PLGF) with different affinity bind to three major tyrosine kinase receptors--VEGFR-1 (fms-like tyrosine kinase-1, Flt-1), VEGFR-2 (KDR; Flk-1), and VEGFR-3. The effect of the most active isomer (VEGF-A) on endothelial cells is mediated by VEGFR-1 and VEGFR-2; VEGFR-3 is responsible for formation and functioning of lymphatic vessels [15].
These data suggest that VEGF is an attractive target for anticancer therapy, since it allows realization of all the primary goals of antiangiogenic therapy. In modern studies, the activity of VEGF is “neutralized” by specific humanized antibodies, e.g. bevacizumab (Avastin). Their binding to VEGF blocks its ability to act on specific receptors.
Clinical randomized studies established that the combination of bevacizumab with chemotherapy (irinotecan plus 5-fluorouracil) significantly (from 15.6 to 20.3 months) increases median survival in patients with metastatic colorectal cancer receiving no cytostatic therapy before [16]. In patients with colorectal cancer progressed after chemotherapy with irinotecan, bevacizumab added to second line therapy (oxaliplatin plus 5-fluorouracil) significantly increased survival in comparison with the control group (only chemotherapy by the same scheme) from 10.8 to 13 months [17]. In this study, the selection of patients for bevacizumab monotherapy (third arm of the study) had to be stopped because of the worse survival rates.
The therapeutic efficiency of paclitaxel administered as monotherapy or in combination with bevacizumab was studied in patients with chemotherapy-naïve metastatic breast cancer. A statistically significant (p < 0.0001) improvement of progression-free survival was found in the cohort receiving combined therapy (median survival was 13.3 months vs. 6.7 months in patients receiving paclitaxel monotherapy). However, the lifespan (overall survival) in the group receiving combined therapy did not increase [18]. In another protocol, the addition of bevacizumab to chemotherapy (capecitabin) failed to improve the therapeutic effect in patients with early pretreated by chemotherapy metastatic breast cancer [19]. Based on this finding, bevacizumab was not recommended for inclusion in the second and further lines of treatment of metastatic breast cancer.
In patients with metastatic non-small cell lung cancer (NSCLC), bevacizumab in combination with chemotherapy (carboplatin plus paclitaxel) caused a statistically significant increase in overall survival up to 12.3 months vs. 10.3 months in patients receiving only chemotherapy (control) [20].
These data suggest that combined treatment (bevacizumab plus chemotherapy) is a strategy of choice in patients with breast cancer, NSCLC, and colorectal tumors. However, bevacizumab monotherapy was the least efficient in this category of patients. Ovarian cancer is the only tumor where the therapeutic effect of bevacizumab is commensurate with effects of conventional drugs. However, indication of bevacizumab to such patients always entails a high risk of fatal complications, e.g. bowel perforation and peritonitis [21, 22].
The presence of VEGF can be proved and determined using registered diagnostic tests and has prognostic significance for therapy. Depending on its size, any clinically detectable tumor requires new vasculature for its growth. This process is universally known as VEGF-mediated angiogenesis. However, clinical effects of antiangiogenic drugs are observed in particular categories of patients, i.e. despite its a priori universality, anti-VEGF therapy is effective against a limited range of malignant tumors.
Attempts to establish the threshold level of VEGF (proangiogenic substances, or their specific receptors) expression, beyond which the therapeutic effect from bevacizumab is the most possible, were unsuccessful. The targets for these assays (blood, tissues, endothelial or tumor cells) and methods for their analysis were also obscure. In contrast to experimental models used to study bevacizumab effects on angiogenesis in reinoculated tumors localized in a convenient for the investigator place (e.g. subcutaneous fat, abdominal cavity, etc.), in clinical studies of antiangiogenic drugs have to be carried out in different microenvironments depended of primary and metastatic tumor localizations (lung, liver, bones, brain, metastases, etc.). Therefore, the outcome of antiangiogenic therapy depends on many factors, such as specific localization, concentration of proangiogenic and antiangiogenic substances, receptors, etc. We think that prediction of efficiency of anti-VEGF therapy including bevacizumab or any other drug is hardly possible, since their therapeutic effects are diverse and go far beyond stimulation of angiogenesis.
As mentioned above, VEGF effects are mediated via two receptors, VEGFR-1 and VEGFR-2. Even in endotheliocytes and their precursors, these receptors fulfill different functions that are not unambiguous from the antitumor treatment standpoint. In adults, VEGFR-1 and VEGFR-2 are expressed by virtually all vascular endothelial cells (with the exception of brain blood vessels). Effects of VEGFR-2 on vascular endothelium were studied in detail and consist in stimulation of angiogenesis through proliferation, migration, and differentiation of endothelial cells and inhibition of their apoptosis. In addition, this receptor is responsible for increased vascular permeability and formation of vascular islands [23]. The functional role of VEGFR-1 is significantly less understood and more vaguely described as being dependent on the developmental stage and cell type [24]. The role of circulating (non cell-bound) VEGF receptors is not completely understood yet; supposedly, they act as VEGF traps in the systemic circulation. The expression of VEGF receptors and, correspondingly, their putative roles are not restricted to endotheliocytes. VEGFR-1 expression was found in other (including nonendothelial) cells, such as HSC, monocytes, trophoblasts, choriocarcinomas, multiple myelomas, and leukemia cells. VEGFR-2 is expressed in CEP, pancreatic epithelial cells, retinal precursors, megakaryocytes, etc. Coexpression of VEGFR-1 and VEGFR-2 occurs in intact testicular and myometrial cells. High levels of both receptors (co-hyperexpression) are characteristic of renal, bladder, ovarian, breast, and some brain tumors.
In 2003, we revealed expression of VEGF receptors on tumor cells of breast cancer and showed for the first time that different receptors have oppositely directed prognostic significance [25]. In patients with locally-advanced breast cancer, VEGFR-1 expression following cessation of neoadjuvant chemotherapy predict significant increases in survival, whereas VEGFR-2 expression is implicit of significant deterioration of prognosis. The functional role of these receptors in tumor cells are still unclear, but the results of this study is citable and validated with the help of more advanced techniques than those used in our study [26-29]. We proceeded from the assumption that VEGF secreted by tumor cells under hypoxia can inhibit their proliferation by acting on tumor cell expressed VEGFR-1. Similar effects on tumor cell expressed VEGFR-2 provoke metastasis, since all of these mechanisms, be it inhibition of growth or metastasis, allow tumor cells to escape from hypoxia via different routes. It was conjectured that VEGF effects on tumor can realize not only via proangiogenic fashion, but also can be direct, i.e. stimulation or inhibition. This hypothesis did not gain wide acceptance at the time of its formulation in 2003, since antiangiogenic drugs were unavailable at that time. Experimental verification of this hypothesis will make it possible to single out a population of patients with VEGFR-1-expressing tumor cells to whom antiangiogenic therapy based on VEGF inhibition is contraindicated. Otherwise in patients with VEGFR-2 overexpression in tumor cells, VEGF neutralization based on “direct” inhibition of proliferation and metastasis can produce additional antitumor effect.
Lack of toxicity related to the basic mechanism of bevacizumab action and low or zero level of nonspecific toxicity. As expected, bevacizumab is devoid of deleterious side effects (cytopenia, stomatitis, etc.) characteristic of other cytostatic agents, but slightly increases the risk of myelosuppression when used in combination with chemotherapy (apparently, due to its effect on HSC). However, this drug has a number of adverse effects, some of which can be very serious. (These effects are detailed in the instructions for use of the drug in USA, which summarized data (both published and unpublished) of cited studies [17-22]). In patients with colorectal cancer, 2.4% of bevacizumab-treated patients had bowel perforation with fatal outcome in ~ 30%. Despite high clinical efficiency of the bevacizumab plus chemotherapy schedule in patients with ovarian cancer, this treatment had to be abandoned due to high incidence of bowel perforations. In 4-15% of patients who had undergone emergency surgery in the course of bevacizumab treatment, wound healing was complicated by disjunction of external and internal sutures, deficiency of anastomoses, wound hemorrhages, etc.
Thirty-one percent of patients with squamous cell carcinomas and 4% of patients with other histological subtypes of non-small cell lung cancer receiving combined treatment (bevacizumab plus chemotherapy) had severe or fatal hemorrhages. It is noteworthy that in the cohort receiving only chemotherapy such complications were absent. Arterial thrombosis and thromboembolism (insults, transient ischemia, myocardial infarction, pulmonary artery occlusion, etc.), sometimes with fatal outcome, were also characteristic of this group. These complications were found in 4.4% of patients receiving bevacizumab plus chemotherapy (cf. 1.9% in the chemotherapy group). Severe (grades III and IV) arterial hypertension in the bevacizumab-treated group was more frequently occurring (8-18%). In <=3% of patients, bevacizumab induced pronounced proteinuria (>3.5 g protein per day); nephrotic syndrome was found in 0.5% of bevacizumab-treated patients. It is noteworthy that in patients of this group urine protein levels never returned to the normal level after cessation of bevacizumab therapy.
ANTI-HER-2-THERAPY AND TRASTUZUMAB
Effect on HER-2 critical for tumor survival and not compromising for normal tissues and organs. HER-2 (HER-2/neu or ErbB2) is a transmembrane tyrosine kinase receptor related to the family of epidermal growth factor receptors (EGFR). In addition to HER-2, this family includes other receptors--EGFR (ErbB1), ErbB3, and ErbB4. The functional role of these receptors, which are expressed by the overwhelming majority of normal and malignantly transformed epithelial cells, consists in transmission of transmembrane signals. It is conjectured, however, that malignant cells whose activity is stimulated by this pathway manifest qualitative (mutations, structural changes, etc.) or quantitative (overexpression, amplification) transformations of receptors and/or their coding genes. All EGFR receptors have similar structural organization and consist of an extracellular domain responsible for ligand binding, a transmembrane domain responsible for intermediate signal transmission, and a tyrosine kinase domain triggering cascades of intracellular receptor-stimulating reactions [30].
Interactions of EGFR, ErbB3, and ErbB4 ligands with the extracellular domain result in hetero- or homodimerization of respective receptors, i.e. binding of two different or identical receptors. A salient feature of EGF receptors is dimerization and subsequent phosphorylation of the tyrosine kinase fragment and activation of intracellular cascades responsible triggering and transmitting intracellular signals. The role of ligands is played by numerous EGF-related peptides--epidermal growth factor (EGF), amphiregulin, betacellulin, epiregulin, etc. The majority of ErbB receptor ligands are released and act locally (in autocrine or paracrine mode) [31].
HER-2 has no ligands of its own; its hypothetical function is a “partnership” during heterodimerization with other members of this family [32]. The ability for heterodimerization is a prerequisite to effective functioning of “defective” family members. Thus, the heterodimerization of HER-2 devoid of the ligand and ErbB3 devoid of active tyrosine kinase creates a “pair”, which efficiently transmits external signals inside the cell. Heterodimerization also affects the “amplitude” and duration of signal transmission after ligand binding. HER-2-containing heterodimers exert more potent and long-lasting effects owing to the ability of HER-2 to strengthen the binding of the receptor-partner to the ligand, to suppress the internalization of the ligand-receptor complex, and to increase probability of the re-use of receptors which have been internalized [33].
The phosphorylation of tyrosine kinase results in activation of signaling pathways triggering such important for tumor progression processes as cell growth, proliferation, migration, differentiation, and inhibition of apoptosis. In addition to signal transmission, these interactions can induce amplification or separation of signals by changing cell responses to the same stimuli [30].
It is believed that quantitative or qualitative changes in specific receptors (or coding genes) of some representatives of the ErbB family are responsible for the formation and growth of many malignant epithelial tumors (breast, colon, pancreas, lung, bladder, kidney, etc.). Their presence in many tumors is associated with unfavorable prognosis and low efficiency of antitumor therapy [31]. Hyperexpression of HER-2 and/or amplification of HER-2-coding gene (HER-2-positive tumors), found in 25-30% of breast cancer patients, are associated (to be more precise, were associated before the onset of the clinical use of trastuzumab) with significant decrease in survival compared to the population of patients which is similar to the first by the other characteristics. Unfavorable remote clinical prognosis for such patients can be attributed to both higher aggressiveness of HER-2-positive tumors and their lower sensitivity to chemotherapy and endocrine therapy [34, 35]. These clinical findings stimulated the development of a broad range of ErbB-inhibiting agents. Curiously enough, the targeted action on the extracellular domain of HER-2 devoid of the activating ligand gave the most clinically significant results.
Trastuzumab (Herceptin) comprises humanized monoclonal antibodies to the extracellular domain of the HER-2 receptor. Due to its introduction into clinical practice, a number of patients with breast tumors possessing overexpression of HER-2 and/or amplification of the HER-2-coding gene have got a real chance for prolongation of life in metastatic cancer or additional chance for cure at the early stages of the disease. In addition, the use of anti-HER-2-therapy for the first time in oncological practice changed the vector of prognostic significance of the marker: the survival rate of patients with HER-2-positive tumors (earlier considered as the most unfavorable) now exceeds that of patients with HER-2-negative tumors [36]. Whereas median overall survival in patients with metastatic breast cancer increased from 438 days in 1991-1992 to 667 days in 1999-2000 (i.e. within a decade the “gain” comprised 229 days (7.6 months) despite ready availability of many cytostatic drugs) [37], just the addition of trastuzumab to chemotherapy with taxanes increased this parameter in HER-2-positive patients by 7-9 months!
The mechanism of antitumor effect of trastuzumab is still poorly understood. Attempts to attribute this effect to enhanced internalization and degradation of HER-2 failed, while numerous clinical studies demonstrated that the level of receptor expression did not change during trastuzumab therapy (even with clear clinical benefit) [38-40]. Although preclinical studies in vivo revealed that antitumor effect of trastuzumab can be partially related to the trastuzumab-induced antibody-dependent cytotoxicity [41, 42], the reason for lacking of antitumor effects of trastuzumab on other HER-2-overexpressing tumors by universal immune mechanisms is unclear. Nothing is known about the ability of trastuzumab to inhibit HER-2 activity during the formation of functionally active heterodimers. More or less unambiguous is the fact that trastuzumab effects are realized though its interaction with the extracellular domain of HER-2. It was found that ErbB-2 can possess a truncated form devoid of the extracellular domain (p95ErbB2), the expression of this form being accompanied by the resistance to trastuzumab both in experimental and clinical studies. At the same time, according to some data this receptor isoform contains functionally active tyrosine kinase capable of functioning independently of the presence of the extracellular domain [43]. Preclinical studies demonstrated that trastuzumab is rather cytostatic than cytotoxic. After cessation of trastuzumab effect on tumor cells, the latter resume their proliferation [44].
Initially, trastuzumab was used for the treatment of patients with metastatic breast cancer. In patients with HER-2 hyperexpression (immunohistochemistry index 2+/3+), trastuzumab efficiency was estimated as 15% (early pretreated patients) [45] and 35% in chemotherapy-naïve patients [46]. The addition of trastuzumab to cytotoxic chemotherapy improved both objective tumor response rate and patients' survival (Table 1) [47, 48].
Table 1. Efficiency of trastuzumab as a
first-line therapy in patients with metastatic breast cancer and
overexpression of HER-2 [46-48]
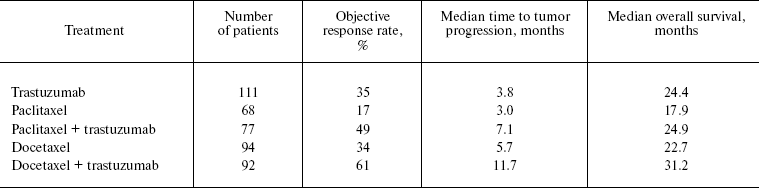
Unexpectedly, in patients with operable HER-2-hyperexpressing breast cancer the gain from trastuzumab plus adjuvant therapy was achieved unusually quickly. Randomized studies (with total number of patients >13,000) even with very short observation (follow-up periods 2-3 years) showed that trastuzumab added to adjuvant chemotherapy (simultaneously or immediately after its completion) diminished relative risk of progression by ~50%. With a short follow-up period, this resulted in modest, but statistically significant improvement of overall survival in comparison with a group of patients who obtained only adjuvant chemotherapy [49, 50].
Based on these clinical findings, combinations of trastuzumab with chemotherapy were indicated for patients with localized and disseminated HER-2-overexpressing breast tumors. At the same time, the use of trastuzumab (monotherapy) as a first-line treatment of disseminated breast cancer enables control over tumor growth in only one-third of patients. Though combined treatment (trastuzumab plus chemotherapy) results in significant “gain” in survival, the objective effect is observed in much less than 100% of patients and it is always “terminated” (soon or later, tumor begins to progress). The use of trastuzumab in combination with adjuvant therapy diminishes, but does not eliminate relapses, suggesting that natural or acquired drug resistance is present (unless rebuttal evidence is available).
The presence of HER-2 can be proved and determined using registered diagnostic tests and has prognostic significance. Since the registration of trastuzumab, it was axiomatic that its administration had to be preceded by validation of HER-2-positivity of respective tumors using immunohistochemical (IHC) methods and/or fluorescent or chromogenic in situ hybridization (FISH or CISH). The positivity criteria were developed on the basis of laboratory and early clinical trials. Overexpression, i.e. drastic increase in the population of specific receptors, was “corroborated” by IHC, while gene amplification was validated using FISH or CISH. The specific criteria for trastuzumab indication included overexpression of HER-2 estimated as 3+ and corroborated by semiquantitative IHC tests (intensive staining of >10% of tumor cell membranes). If overexpression was estimated as 2+ in IHC, gene amplification had to be confirmed by FISH or CISH data. In FISH, HER-2-positivity was determined as a ratio of the copy number of the HER-2-coding gene to the copy number of the 17th chromosome and was >=2. In the absence of the 17th chromosome signal, the presence of four HER-2 gene copies was estimated as HER-2 positivity. If, according to CISH, >50% of tumor cells contained more than five copies of the encoding gene, the tumors were considered to be HER-2 positive. The cases estimated as 1+ and 0 in the IHC test were considered to be HER-2-negative and not subject to trastuzumab therapy. Nevertheless, the prognostic value of HER-2-positivity was low: the objective antitumor trastuzumab effect was noted only in 35% of patients selected by these criteria, whereas in the remaining 65% of patients signs of clinical improvement were absent [46]. Also, the mechanism of remission and life prolongation on trastuzumab treatment of patients with HER-2 overexpression (by IHC data) remained unclear, because even in such patients the majority of tumor cells may lack targets for trastuzumab on membrane surface. What, then, is the mechanism whereby trastuzumab acts on tumor cells and what are the criteria for selection of patients for trastuzumab therapy?
These questions are being raised with increasing frequency in many recent publications. Adjuvant trials preceding trastuzumab registration utilized two approaches to estimation of the HER-2 status. In two trials (NSABP B-31 and NCCTG N983), trastuzumab indication demanded HER-2 overexpression estimated as 3+ (IHC) and HER-2-positive in FISH [50]. Validation or disproval of HER-2 overexpression (and/or gene amplification) was made on the basis of a local laboratory test results. However, further analyses demanded their validation and expert evaluation by a central laboratory. In the active phase of allocation of patients into cohorts, the incidence of false-positive results from local laboratories was very high: ~20% of cases estimated as HER-2-positive by a local laboratory were estimated negative by a central laboratory. The results of the B-31 trial reported by the NSABP group [51] brought into challenge the “false positivity” of many HER-2 assays by local laboratories. Thus, 207 out of 1795 FISH samples (11.5%) analyzed at a local laboratory were found to be false-positive by a central laboratory, while 299 out of 1787 IHC samples (16.7%) were “rejected”. Moreover, 174 out of 1795 samples (9.7%) were found to be HER-2-negative in both tests. Quite unexpectedly, the survival gain from trastuzumab therapy in patients whose tumors were estimated as HER-2-negative by the central laboratory was the same as in patients whose HER-2 status was estimated positive by both laboratories (Table 2)!
Table 2. Decrease in risk of progression of
disease or death after indication of trastuzumab depending on HER-2
status (data from central laboratory) [51]
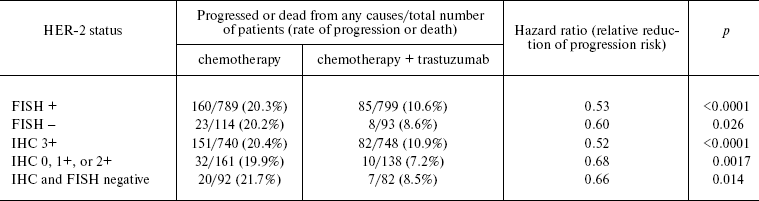
What are the implications of these findings for clinical practice? First, we have to remember that all patients included into studies so far possessed tumors estimated as HER-2-positive by local laboratory tests. Thus, no one case can be regarded as never being recognized as HER-2-positive. Second, it is necessary to remember that the boundary of positiveness used for clinical purpose is to some extent arbitrary. We also cannot rule out the possibility of nonuniform distribution of HER-2-positive cell clusters in tumors. So, it is possible that in a local laboratory a sample with overexpression/amplification was analyzed in contrast to a sample analyzed in a central laboratory. Notwithstanding, this situation calls into question the utility of routine clinical protocols for estimating therapeutic efficiency of trastuzumab therapy (at least for adjuvant purposes) and its classification as a targeted-action remedy despite the fact that early studies of patients with metastatic breast tumors demonstrated low (if any) activity of trastuzumab with respect to HER-2-negative tumors.
Immeasurably more important are “fundamental” problems raised in these studies. How does the effect of trastuzumab on very small amount of target cells (that cannot even be detected by ICH or FISH methods) result in changes in prognosis and overall course of the disease? What are the targets for trastuzumab action and how is this action translated into survival increase (i.e. control over the whole tumor)? Maybe a very small number of HER-2-expressing tumor cells (not detected by IHC and FISH methods) worsen prognosis through micrometastasis formation and trastuzumab “corrects” the situation at early stages of the disease? However, it is not clear then, how does all of these match the data about correlation between HER-2 status of primary tumor and of metastases (in case of HER-2-negative tumors, HER-2 expression in metastases is usually not observed)?
Lack of toxicity related to the basic mechanism of trastuzumab action and low or zero level of nonspecific toxicity. Similar to bevacizumab, trastuzumab is devoid of toxicity associated with classical cytostatic drugs, but it has a number of adverse side effects. Its toxicity is manifested in inhibition of myocardial contractility, which narrows the range of applications and demands systematic monitoring (e.g. echocardiography, etc.) aimed at early diagnostics of complications and minimization of fatal toxicity risk [47, 49, 50]. The cardiotoxic effect of trastuzumab varies widely from clinically unimportant symptoms revealed upon echocardiographic examination to severe, sometimes fatal manifestations of cardiac insufficiency. The frequency of this complication depends on many factors, such as a chest left-half irradiation in anamnesis, anthracycline intake, preexisting cardiac pathologies, etc. Up to 16% of patients receiving trastuzumab as adjuvant therapy had to withdraw from treatment because of disturbances in myocardial contractility; 2% of patients (cf. 0.4% in the placebo group) developed severe cardiac insufficiency despite systemic monitoring. In patients with metastatic breast cancer receiving combined therapy (trastuzumab plus anthracyclines), the risk of symptomatic cardiac failure was as high as 28%.
INHIBITION OF EGFR TYROSINE KINASE (ERLOTINIB AND
GEFITINIB)
Effect on EGFR critical for tumor survival and not compromising for normal tissues and whole body organs. Beyond doubt, erlotinib and gefitinib provide the most illustrative example of a discrepancy between the basic principles of targeted therapy and their clinical applications. The choice of EGFR as a target for antitumor therapy was rationalized by the fact that EGFR overexpression or overproduction of its coding mRNA is characteristic of many epithelial tumors (lung, gastrointestinal tract, breast, pancreas, bladder, etc.) [30, 31]. In the foregoing sections, the mechanism of action of EGFR was considered with the example of anti-HER-2 therapy. EGFR is involved in many processes relevant to tumor cell survival, e.g. proliferation, stimulation of angiogenesis, inhibition of apoptosis, invasion, metastasis, etc., and, last but not least, unfavorable prognosis [52-54]. In vitro and in vivo studies showed that inhibition of EGFR is accompanied by suppression of proliferative activity of tumor cells and a decrease in their viability [54]. Based on these findings and taking into account the secondary role of this receptor in vital activity of normal cells, EGFR is considered to be a promising candidate target for anticancer therapy [55].
In addition to blockade of the extracellular domain of EGFR by monoclonal antibodies, it seemed also expedient to affect the intracellular domain of EGFR with a group of drugs represented by gefitinib (Iressa) and erlotinib (Tarceva), inhibitors of EGFR tyrosine kinase C (TK-C). By competing with ATP for the intracellular ATP-binding domain of the receptor, gefitinib and erlotinib prevent the phosphorylation of tyrosine residues of intracellular proteins and block signal transmission to cell nuclei. Blockade of signaling pathways responsible for signal transduction to transcription factors at the TK-C level takes place independently of ligand binding and EGFR dimerization. Both drugs are designed for oral administration.
Monotherapy with gefitinib and erlotinib was studied in patients with disseminated NSCLC in whom tumor progression continued after completion of standard treatment. Both drugs demonstrated high efficiency in nonrandomized studies of this prognostically unfavorable population: antitumor responses were recorded in 12-19% of patients [56, 57]. Direct comparison of gefitinib and placebo as second- and third-line treatments carried out within the framework of randomized ISEL studies established a notably higher response rate in the gefitinib arm (8 and 1%, respectively); however, overall survival was not different in both groups (5.6 and 5.1 months, respectively) [58]. In a similar protocol (BR.21), effects of erlotinib and placebo were compared in a cohort of NSCLC patients after one or two lines of treatment with platinum derivatives [59]. In this study, median overall survival in the erlotinib-treated group significantly exceeded that in the placebo group (6.7 and 4.7 months, respectively; p < 0.0001). Based on these findings, erlotinib was indicated as a drug of choice for patients with metastatic NSCLC refractory to first- and second-line treatment.
Contrary to the preclinical data on the synergy of EGFR TK-C low molecular weight inhibitors and chemotherapy, the use of gefitinib or erlotinib within chemotherapeutic combinations with other drugs was not recommended for treatment of patients with metastatic non-small cell lung cancer. Their addition to standard drug combinations (cisplatin + gemcitabine or carboplatin + paclitaxel) failed to improve therapeutic efficiency in four randomization placebo-controlled trials (INTACT1 and 2, TRIBUTE, TALENT). The results of these studies were disappointing, especially after successful outcome of monotherapy (gefitinib or erlotinib) in a prognostically less favorable group of cancer patients [60-63].
The analysis of clinical findings prompts a conclusion that gefitinib and erlotinib are efficient in very small cohorts of NSCLC patients. The gain from their application in very limited group of patients with erlotinib or gefitinib sensitive tumors is lost among an immense diversity of randomized tests, while lack of improvement in other categories of cancer patients is apparent. Therefore, selection of cancer patients in whom this strategy might give the most beneficial therapeutic results, especially in earlier steps of systemic treatment, is a currently central task.
The presence of EGFR can be proved and determined using registered diagnostic tests and has prognostic significance for therapy. Indication of EGFR inhibitors to all patients with NSCLC was rationalized by the speculation that EGFR overexpression is the only prerequisite to realization of their therapeutic effects. By analogy with histochemical estimation of the HER-2 status, attempts to establish a correlation between EGFR expression and therapeutic efficiency of low molecular weight EGFR inhibitors failed. In patients with EGFR overexpression, the efficiency of gefitinib was higher than in its absence (8.2 and 1.5%, respectively) [64]. However, signs of clinical improvement were found in only 1 out of 12 gefitinib-treated patients (8%) with EGFR overexpression, suggesting low predictive value of EGFR overexpression test. In ISEL, amplification of the EGFR gene was concomitant with a higher degree of tumor regression (16 and 3%, respectively) and better survival [64]. However, despite theoretical implications, the presence of EGFR in all epithelial cells and its overexpression in the overwhelming majority of NSCLC patients were insufficient for predicting its therapeutic efficiency. In these studies, EGFR overexpression was rather prognostic (i.e. prognosing the outcome irrespective of the treatment schedule) than predicting (i.e. forecasting the response to implemented therapy).
These findings stimulated a search for clinical factors able to predict the therapeutic efficiency of EGFR low molecular weight inhibitors [64-66]. Randomized trials demonstrated that antitumor effects of gefitinib and erlotinib used as monotherapy were more pronounced in females than in males (25 vs. 8%), in nonsmokers than in smokers (31 vs. 8%), in representatives of the Asian race than in Europeans or Americans (27 vs. 11%), and in patients with adenocarcinomas than in patients with squamous cell carcinomas (19 vs. 7%) [9]. Patients with bronchioloalveolar cancer and adenocarcinomas with the predominance of the bronchioloalveolar component were especially responsive to anticancer therapy.
What is the reason for higher sensitivity of such patients to low molecular weight EGFR inhibitors? In 2004, several independent investigators reported on beneficial antitumor effects of gefitinib and erlotinib in patients with mutations in the EGFR gene and structural disturbances in tyrosine kinase-binding domains of EGFR [67-69]. Meta analysis of clinical findings of a large (>900) cohort of NSCLC patients demonstrated that signs of clinical improvement were more apparent in patients with EGFR mutations (77%), while in patients without mutations this parameter was as low as 10% [70]. In the overall NSCLC population, EGFR mutations did not exceed 10%, and 90% of those were localized in exons 19 or 21. Somatic mutations in EGFR can be regarded as a key mechanism of tumor progression responsible for excessive activation of the mutant receptor after its binding to the ligand, on one hand, and as a factor promoting effective and stable binding of anticancer drugs to EGFR tyrosine kinase, on the other hand. Under these conditions the activity of the mutant receptor is blocked by gefitinib and erlotinib more efficiently than that of nonmutant EGFR. High detection frequency of mutant EGFR receptors in the NSCLC cohorts (females, Japanese, nonsmoking patients, those with adenocarcinoma) explains, at least partly, higher sensitivity of these patients to gefitinib and erlotinib.
It may seem at first glance that the factor prognosing the therapeutic efficiency of EGFR low molecular weight inhibitors has been found at last and these drugs can be prescribed for a very small contingent of patients with mutations. However, BR.21 studies showed that survival in the cohort of patients with EGFR mutations was the same as in the placebo and erlotinib-treated groups, which points to the lack of erlotinib effect on survival, despite high frequency of tumor regression [65]. This paradox can be attributed to the beneficial prognostic effect of EGFR mutations. Patients with mutations manifest better survival irrespective of gefitinib and erlotinib treatment or their antitumor activity. Therefore, EGFR TK-C low molecular weight inhibitors can be prescribed to all NSCLC patients irrespective of the presence or absence of mutations.
Lack of toxicity related to the basic mechanism of action of EGFR inhibitors and low or zero level of nonspecific toxicity. To the main clinical manifestations of toxic effects of EGFR low molecular weight inhibitors, one can relate skin lesions (eruption), diarrhea, nausea, and weakness. Numerous studies showed that patients with skin eruptions usually manifest better survival than those in whom skin complications are absent. Pronounced toxic effects (grades III and IV) were found in only 2-6% of cases. Other complications, viz., interstitial pneumonitis (1-5%), are rare and more characteristic of populations in which representatives of the Asian race are predominant. In randomization tests, pneumonitis in the drug-treated groups occurred as frequently as in the placebo group [56-63].
DISCUSSION
What conclusions can be made from the analysis of the most popular anticancer drugs and what is the state-of-the-art in targeted therapy of solid tumors? Till now their mechanisms of action and areas of applications are still obscure, and we gradually come to realize that their safety is far from being absolute. The first steps on the way to practical realization of this strategy did not give very encouraging results and were contradictory to theoretical implications.
Undoubtedly, bevacizumab increases survival in patients with colorectal cancer and NSCLC (relative increase in overall survival by ~30 and ~20%, respectively). However, contrary to theoretical implications, modern antiangiogenic drugs fail to promote long-term transfer of tumors into the “dormant” state in 100% of patients, since in absolute terms the gain in median overall survival does not exceed 2-5 months and varies widely among different patients. The most possible effect of therapy is not “distributed equally”: in some patients the gain is more significant, while others do not gain at all or their survival decreases due to toxicity. The universality of anti-VEGF therapy is also doubtful: in many tumors (kidney, melanoma, pancreas, breast, etc), bevacizumab effectiveness is either absent or very low. Taking into account the need for adequate blood supply of any tumor whose size exceeds the “critical” size (1-2 mm3), the existence of alternative (with the exception of VEGF) angiogenesis-stimulating pathways is the most plausible rationale of bevacizumab failures, although extreme diversity of nontiangiogenic effects of VEGF should not be ruled out either. If VEGF really inhibits the proliferation of VEGFR-1-overexpressing cancer cells, its inactivation may give opposite results. Furthermore, judging from an ever increasing body of evidence on VEGF expression in tumor cells, direct effect of the drug on tumor vasculature cannot be regarded as the only mechanism of bevacizumab antitumor activity [71]. Improvement of the therapeutic effect of chemotherapy in combination with bevacizumab due to additional direct antitumor effect of anti-VEGF therapy provide a more logical explanation than structural “normalization” of the tumor vasculature, especially if we take into consideration low antitumor effect of bevacizumab monotherapy [72]. According to present-day criteria, bevacizumab cannot be attributed to targeted-action drugs, since its mechanism of action follows the principles of classical chemotherapy (the drug is prescribed empirically to all patients with a definite type of tumor and with unpredictable outcome). Moreover, bevacizumab has a number of specific and nonspecific toxic effects.
Trastuzumab proved to be an efficient remedy for breast cancer owing to its ability to increase survival. Over decades, this effect was unattainable for the overwhelming majority of other cytostatic drugs. But do its mechanisms of action, design, and commercialization principles differ from those of conventional anticancer drugs? Does this drug meet the goals of targeted therapy? Have we come to the comprehension of processes associated with malignant growth and ways to their targeted control, e.g. through blockade of HER-2? Our present experience does not provide an explicit answer to these questions.
Clinical application of low molecular weight EGFR inhibitors as targeted therapy is an “anti-example” rather than an example. Preclinical studies of their ability to inhibit normal EGFR TK-C followed by four-year wide clinical application revealed that antitumor activity of low molecular weight EGFR inhibitors is specifically directed against the mutant receptor with a minimum (or zero) effect on normal receptors expressed in the majority of NSCLC and other malignant epithelial tumors. However, if such effect really takes place, it can hardly be established in clinical trials. And again we deal with a situation when the mechanism of action and range of clinical applications of a drug introduced into routine clinical practice remain obscure.
It is possible that we are searching in a “wrong” place. EGFR inhibitors can adequately attack target receptors in both drug-responding and -nonresponding patients, while the efficiency of one or another drug is independent of quantitative and/or qualitative changes in the tumor receptor activity. It is quite probable that the clue should be sought in the a priori existence (or nonexistence) of alternative stimulating pathways. If such mechanisms really exist, even effective blockade of EGFR receptors will not have any effect on tumor growth. If not (in patients with congenital deficiency of alternative EGFR-stimulating mechanisms in epithelial cells of different origin), the therapeutic effect can be achieved. In such patients, signs of clinical improvement are often concomitant with skin lesions. Indeed, skin eruptions in patients gaining from treatment with EGFR inhibitors can hardly be ascribed to mutations in tumor cell receptors or lack of smoking experience. Needless to say, corroboration of this hypothesis will require more extensive and costly expertise than a search for EGFR mutations or pharmacokinetic analysis of the drugs, but it can shed additional light on different aspects of the problem and culminate in the development of more precise prediction of efficiency. Moreover, validation of this hypothesis can provide new tools for predicting the efficiency of anticancer drugs whose effects are specific against a concrete (or even unique!) mechanism initiating tumor growth.
The attempt to prescribe combinations of “targeted-action” drugs to all patients within a population in the hope that their empiric indication will help overcome drug resistance of tumor cells in the overwhelming majority of patients may have an opposite effect. Blockade of some stimulating pathways in tumor cells may not produce a summation effect, but may overcome the tolerance threshold in normal cells. In such situations, the toxicity of targeted therapy can overcome the toxicity of chemotherapeutic drugs, but without significant gains in efficiency. Perhaps this was the case when panitumumab (anti-EGFR monoclonal antibodies), bevacizumab, and chemotherapy were used as first-line therapy in patients with colorectal cancer where the decrease in survival was concomitant with significant increases in general toxicity instead of synergism (PACCE data).
Of course, the above-said should not be regarded as comprehensive and unambiguous characterization of targeted therapy at large. Some anticancer drugs (e.g. tamoxifen, imatinib, etc.) are more conforming to this definition than others. In the general sense, contemporary targeted therapy has lost its significance as a radically new branch of clinical oncology whose fundamental principles allow unambiguous task-oriented approach to selection of low-toxicity drugs and successful and predictable outcome for individual patients. Thus, the majority of agents initially declared as targeted-action drugs preserve, in one degree of another, all the disadvantages of conventional remedies, but it is not the main thing. The major problem is in traditional approaches to the design and approval of anticancer drugs and estimation of their clinical efficiency. Only in rare cases was it possible to depart, at least partly, from empirical approaches to the prescription of new drugs, but it is still too premature to speak about their indication as treatment of choice for a particular patient. We think it meaningless to raise the questions about mechanisms of action and applications of drugs after completion of clinical trials and commercialization, especially if we take into consideration the lack of cooperation between clinicians gathering clinical information and experimentalists giving it an adequate interpretation, and lack of interest in narrowing the range of drug indications in manufacturers sponsoring the research. The clinical tools for validation of experimental hypotheses (randomized trials) are exact, but demand considerable investment, effort, and involvement of hundreds and thousands of patients. The clinical goals (time and sequence of drug prescription, their combinations, etc.), stipulated as primary goals in the majority of contemporary studies, are the same as 60 years ago, i.e. are based on empirical principles. Considering that commercial production of pharmaceutical agents for clinical trials is rapidly increasing, many of contemporary trials with “targeted-action” drugs have a real chance to be not completed at the time when more recently designed drugs appear.
REFERENCES
1.US Food and Drug Administration, Office of
Combination Products (2003) Annual Report to Congress Federal Food,
Drug, and Cosmetic Act as Amended by the Medical Device User Fee Act of
2002, National Press Office, Rockville, MD.
2.Brien, S. E., Zagzag, D., and Brem, S. (1989)
Neurosurgery, 25, 715-719.
3.Folkman, J. (1990) J. Natl. Cancer Inst.,
82, 4-6.
4.Fukumura, D., Xavier, R., Sugiura, T., Chen, Y.,
Park, E. C., Lu, N., Selig, M., Nielsen, G., Taksir, T., Jain, R. K.,
and Seed, B. (1998) Cell, 94, 715-725.
5.Peichev, M., Naiyer, A. J., Pereira, D., Zhu, Z.,
Lane, W. J., Williams, M., Oz, M. C., Hicklin, D. J., Witte, L., Moore,
M. A. S., and Rafii, S. (2000) Blood, 95, 952-958.
6.Rafii, S. (2000) J. Clin. Invest.,
105, 17-19.
7.Folberg, R., Hendrix, M. J., and Maniotis, A. J.
(2000) Am. J. Pathol., 156, 361-381.
8.Chang, Y. S., di Tomaso, E., McDonald, D. M.,
Jones, R., Jain, R. K., and Munn, L. L. (2000) Proc. Natl. Acad.
Sci. USA, 97, 14608-14613.
9.Dickson, P. V., Hamner, J. B., Sims, T. L., Fraga,
C. H., Ng, C. Y. C., Rajasekeran, S., Hagedorn, N. L., McCarville, M.
B., Stewart, C. F., and Davidoff, A. M. (2007) Clin. Cancer
Res., 13, 3942-3950.
10.Shaked, Y., Emmenegger, U., Man, S., Cervi, D.,
Bertolini, F., Ben-David, Y., and Kerbel, R. S. (2005) Blood,
106, 3058-3061.
11.Colleoni, M., Rocca, A., Sandri, M. T., Zorzino,
L., Masci, G., Nole, F., Peruzzotti, G., Robertson, C., Cinieri, S., de
Braud, F., Viale, G., and Goldhirsch, A. (2002) Ann.
Onc., 13, 73-80.
12.Burstein, H. J., Spigel, D., Kindsvogel, K.,
Parker, L. M., Bunnell, C. A., Partridge, A. H., Come, S. E., Ryan, P.
D., Gelman, R., and Winer, E. P. (2005) Breast Cancer Res.
Treat., 94 (Suppl. 1), S6 (abstract 4).
13.Senger, D. R., Galli, S. J., Dvorak, A. M.,
Perruzzi, C. A., Harvey, V. S., and Dvorak, H. F. (1983)
Science, 219, 983-985.
14.Iliopoulos, O., Levy, A. P., Jiang, C., Kaelin,
W. G., Jr., and Goldberg, M. A. (1996) Proc. Natl. Acad. Sci.
USA, 93, 10595-10599.
15.Ferrara, N., Gerber, H. P., and LeCouter, J.
(2003) Nat. Med., 9, 669-676.
16.Hurwitz, H., Fehrenbacher, L., Novotny, W.,
Cartwright, T., Hainsworth, J., Heim, W., Berlin, J., Baron, A.,
Griffing, S., Holmgren, E., Ferrara, N., Fyfe, G., Rogers, B., Ross,
R., and Kabbinavar, F. (2004) N. Engl. J. Med., 350,
2335-2342.
17.Giantonio, B. J., Catalano, P. J., Meropol, N.
J., O'Dwyer, P. J., Mitchell, E. P., Alberts, S. R., Schwartz, M. A.,
and Benson, A. B., 3rd (2007) J. Clin. Oncol., 25,
1539-1544.
18.Miller, K. D. (2003) Clin. Breast Cancer,
3, 421-422.
19.Miller, K. D., Chap, L. I., Holmes, F. A.,
Cobleigh, M. A., Marcom, P. K., Fehrenbacher, L., Dickler, M.,
Overmoyer, B. A., Reimann, J. D., Sing, A. P., Langmuir, V., and Rugo,
H. S. (2005) J. Clin. Oncol., 23, 792-799.
20.Sandler, A. B., Gray, R., Perry, M. C., Brahmer,
J., Schiller, J. H., Dowlati, A., Lilenbaum, R., and Johnson, D. H.
(2006) N. Engl. J. Med., 355, 2542-2550.
21.Burger, R. A., Sill, M. W., Monk, B. J., Greer,
B. E., and Sorosky, J. I. (2007) J. Clin. Oncol., 25,
5165-5171.
22.Cannistra, S. A., Matulonis, U., Penson, R.,
Wenham, R., Armstrong, D., Burger, R. A., Mackey, H., Douglas, J.,
Hambleton, J., and McGuire, W. (2006) J. Clin. Oncol.,
24, 257S.
23.Shalaby, F., Rossant, J., Yamaguchi, T. P.,
Gertsenstein, M., Wu, X. F., Breitman, M. L., and Schuh, A. C. (1995)
Nature, 376, 62-66.
24.Fong, G. H., Zhang, L., Bryce, D. M., and Peng,
J. (1999) Development, 126, 3015-3025.
25.Zhukova, L., Zhukov, N., and Lichinitser, M.
(2003) Byul. Eksp. Biol. Med., 135, 478-481.
26.Chung, G. G., Yoon, H. H., Zerkowski, M. P.,
Ghosh, S., Thomas, L., Harigopal, M., Charette, L. A., Salem, R. R.,
Camp, R. L., Rimm, D. L., and Burtness, B. A. (2006) Cancer,
106, 1677-1684.
27.Seto, T., Higashiyama, M., Funai, H., Imamura,
F., Uematsu, K., Seki, N., Eguchi, K., Yamanaka, T., and Ichinose, Y.
(2006) Lung Cancer, 53, 91-96.
28.Carrivick, L., Rogers, S., Clark, J., Campbell,
C., Girolami, M., and Cooper, C. (2006) J. R. Soc. Interface,
3, 367-381.
29.Fan, F., Wey, J. S., McCarty, M. F., Belcheva,
A., Liu, W., Bauer, T. W., Somcio, R. J., Wu, Y., Hooper, A., Hicklin,
D. J., and Ellis, L. M. (2005) Oncogene, 24,
2647-2653.
30.Mendelsohn, J., and Baselga, J. (2003) J.
Clin. Oncol., 21, 2787-2799.
31.Olayioye, M. A., Neve, R. M., Lane, H. A., and
Hynes, N. E. (2000) EMBO J., 19, 3159-3167.
32.Graus-Porta, D., Beerli, R. R., Daly, J. M., and
Hynes, N. E. (1997) EMBO J., 16, 1647-1655.
33.Citri, A., Skaria, K. B., and Yarden, Y. (2003)
Exp. Cell Res., 284, 54-65.
34.Slamon, D. J., Clark, G. M., Wong, S. G., Levin,
W. J., Ullrich, A., and McGuire, W. L. (1987) Science,
235, 177-182.
35.Toikkanen, S., Helin, H., Isola, J., and Joensuu,
H. (1992) J. Clin. Oncol., 10, 1044-1048.
36.Papaldo, P., Fabi, A., Ferretti, G., Mottolese,
M., Cianciulli, A. M., di Cocco, B., Pino, M. S., Carlini, P., di
Cosimo, S., Sacchi, I., Sperduti, I., Nardoni, C., and Cognetti, F.
(2006) Ann. Onc., 17, 630-636.
37.Chia, S., Speers, C., and Kang, A. (2003)
Proc. Am. Soc. Clin. Oncol., 22, 6.
38.Sliwkowski, M. X., Lofgren, J. A., Lewis, G. D.,
Hotaling, T. E., Fendly, B. M., and Fox, J. A. (1999) Semin.
Oncol., 26 (Suppl. 12), S60-S70.
39.Arnould, L., Gelly, M., Penault-Llorca, F.,
Benoit, L., Bonnetain, F., Migeon, C., Cabaret, V., Fermeaux, V.,
Bertheau, P., Garnie, J., Jeannin, J. F., and Coudert, B. (2006) Br.
J. Cancer, 94, 259-267.
40.Gennari, R., Menard, S., Fagnoni, F., Ponchio,
L., Scelsi, M., Tagliabue, E., Castiglioni, F., Villani, L., Magalotti,
C., Gibelli, N., Oliviero, B., Ballardini, B., da Prada, G., Zambelli,
A., and Costa, A. (2004) Clin. Cancer Res., 10,
5650-5655.
41.Cooley, S., Burns, L. J., Repka, T., and Miller,
J. S. (1999) Exp. Hematol., 27, 1533-1541.
42.Stockmeyer, B., Beyer, T., Neuhuber, W., Repp,
R., Kalden, J. R., Valerius, T., and Herrmann, M. (2003) J.
Immunol., 171, 5124-5129.
43.Saez, R., Molina, M. A., Ramsey, E. E., Rojo, F.,
Keenan, E. J., Albanell, J., Lluch, A., Garcia-Conde, J., Baselga, J.,
and Clinton, G. M. (2006) Clin. Cancer Res., 12,
424-431.
44.Pietras, R. J., Pegram, M. D., Finn, R. S.,
Maneval, D. A., and Slamon, D. J. (1998) Oncogene, 17,
2235-2249.
45.Cobleigh, M. A., Vogel, C. L., Tripathy, D.,
Robert, N. J., Scholl, S., Fehrenbacher, L., Wolter, J. M., Paton, V.,
Shak, S., Lieberman, G., and Slamon, D. J. (1999) J. Clin.
Oncol., 17, 2639-2648.
46.Vogel, C. L., Cobleigh, M. A., Tripathy, D.,
Gutheil, J. C., Harris, L. N., Fehrenbacher, L., Slamon, D. J., Murphy,
M., Novotny, W. F., Burchmore, M., Shak, S., Stewart, S. J., and Press,
M. (2002) J. Clin. Oncol., 20, 719-726.
47.Slamon, D. J., Leyland-Jones, B., Shak, S.,
Fuchs, H., Paton, V., Bajamonde, A., Fleming, T., Eiermann, W., Wolter,
J., Pegram, M., Baselga, J., and Norton, L. (2001) N. Engl. J.
Med., 344, 783-792.
48.Marty, M., Cognetti, F., Maraninchi, D., Snyder,
R., Mauriac, L., and Tubiana-Hulin, M. (2005) J. Clin.
Oncol., 23, 4265-4274.
49.Piccart-Gebhart, M. J., Procter, M.,
Leyland-Jones, B., Goldhirsch, A., Untch, M., Smith, I., Gianni, L.,
Baselga, J., Bell, R., Jackisch, C., Cameron, D., Dowsett, M., Barrios,
C. H., Steger, G., Huang, C., Andersson, M., Inbar, M., Lichinitser,
M., Lang, I., Nitz, U., Iwata, H., Thomssen, C., Lohrisch, C., Suter,
T. M., Rãuschoff, J., Suto, T., Greatorex, V., Ward, C., Straehle,
C., McFadden, E., Dolci, M. S., and Gelber, R. D. (2005) N. Engl. J.
Med., 353, 1659-1672.
50.Romond, E. H., Perez, E. A., Bryant, J., Suman,
V. J., Geyer, C. E., Davidson, N. E., Tan-Chiu, E., Martino, S., Paik,
S., Kaufman, P. A., Swain, S. M., Pisansky, T. M., Fehrenbacher, L.,
Kutteh, L. A., Vogel, V. G., Visscher, D. W., Yothers, G., Jenkins, R.
B., Brown, A. M., Dakhil, S. R., Mamounas, E. P., Lingle, W. L., Klein,
P. M., Ingle, J. N., and Wolmark, N. (2005) N. Engl. J. Med.,
353, 1673-1684.
51.Paik, S., Kim, C., Jeong, J., Geyer, C. E.,
Romond, E. H., Mejia-Mejia, O., Mamounas, E. P., Wickerham, D.,
Costantino, J. P., and Wolmark, N. (2007) Proc. Am. Soc. Clin.
Oncol., 25, 5s (abstract 511).
52.Brabender, J., Danenberg, K. D., Metzger, R.,
Schneider, P. M., Park, J., Salonga, D., Holscher, A. H., and
Danenberg, P. (2001) Clin. Cancer Res., 7, 1850-1855.
53.Baselga, J. (2000) Signal, 1,
12-21.
54.Woodburn, J. R. (1999) Pharmacol. Ther.,
82, 241-250.
55.Baselga, J. (2002) The Oncologis, 7
(Suppl. 4), 2-8.
56.Fukuoka, M., Yano, S., Giaccone, G., Tamura, T.,
Nakagawa, K., Douillard, J., Nishiwaki, Y., Vansteenkiste, J., Kudoh,
S., Rischin, D., Eek, R., Horai, T., Noda, K., Takata, I., Smit, E.,
Averbuch, S., Macleod, A., Feyereislova, A., Dong, R., and Baselga, J.
(2003) J. Clin. Oncol., 21, 2227-2229.
57.Perez-Soler, R., Chachoua, A., Hammond, L. A.,
Rowinsky, E. K., Huberman, M., Karp, D., Rigas, J., Clark, G. M.,
Santabárbara, P., and Bonomi, P. (2004) J. Clin. Oncol.,
22, 3238-3247.
58.Thatcher, N., Chang, A., Parikh, P., Pemberton,
K., and Archer, V. (2005) Lancet, 366, 1527-1537.
59.Shepherd, F. A., Rodrigues Pereira, J., Ciuleanu,
T., Tan, E. H., Hirsh, V., Thongprasert, S., Campos, D.,
Maoleekoonpiroj, S., Smylie, M., Martins, R., van Kooten, M., Dediu,
M., Findlay, B., Tu, D., Johnston, D., Bezjak, A., Clark, G.,
Santabarbara, P., and Seymour, L. (2005) N. Engl. J. Med.,
353, 123-132.
60.Giaccone, G., Herbs, R. S., Manegold, C.,
Scagliotti, G., Rosell, R., Miller, V., Natale, R. B., Schiller,
J. H., von Pawel, J., Pluzanska, A., Gatzemeier, U., Grous, J., Ochs,
J. S., Averbuch, S. D., Wolf, M. K., Rennie, P., Fandi, A., and
Johnson, D. H. (2004) J. Clin. Oncol., 22, 777-784.
61.Herbst, R. S., Giaccone, G., Schiller, J. H.,
Natale, R. B., Miller, V., Manegold, C., Scagliotti, G., Rosell, R.,
Oliff, I., Reeves, J. A., Wolf, M. K., Krebs, A. D., Averbuch, S. D.,
Ochs, J. S., Grous, J., Fandi, A., and Johnson, D. H. (2004) J.
Clin. Oncol., 22, 785-794.
62.Herbst, R. S., Prager, D., Hermann, R.,
Fehrenbacher, L., Johnson, B. E., Sandler, A., Kris, M. G., Tran, H.
T., Klein, P., Li, X., Ramies, D., Johnson, D. H., and Miller, V. A.
(2005) J. Clin. Oncol., 23, 5892-5899.
63.Gatzemeier, U., Pluzanska, A., Szczesna, A.,
Kaukel, E., Roubec, J., de Rosa, F., Milanowski, J.,
Karnicka-Mlodkowski, H., Pesek, M., Serwatowski, P., Ramlau, R.,
Janaskova, T., Vansteenkiste, J., Strausz, J., Manikhas, G. M., and von
Pawel, J. (2007) J. Clin. Oncol., 25, 1545-1552.
64.Bell, D. W., Lynch, T. J., Haserlat, S. M.,
Harris, P. L., Okimoto, R. A., Brannigan, B. W., Sgroi, D. C., Muir,
B., Riemenschneider, M. J., Iacona, R., Krebs, A. D., Johnson, D. H.,
Giaccone, G., Herbst, R. S., Manegold, C., Fukuoka, M., Kris, M. G.,
Baselga, J., Ochs, J. S., and Haber, D. A. (2005) J. Clin.
Oncol., 23, 8081-8092.
65.Tsao, M. S., Sakurada, A., Cutz, J. C., Zhu, C.,
Kamel-Reid, S., Squire, J., Lorimer, I., Zhang, T., Liu, N.,
Daneshmand, M., Marrano, P., da Cunha Santos, G., Lagarde, A.,
Richardson, F., Seymour, L., Whitehead, M., Ding, K., Pater, J., and
Shepherd, F. A. (2005) N. Engl. J. Med., 353,
133-144.
66.Hirsch, F. R., Varella-Garcia, M., Bunn, F. A.,
Franklin, W. A., Dziadziuszko, R., Thatcher, N., Chang, A., Parikh, P.,
Rodrigues Pereira, J., Ciuleanu, T., von Pawel, J., Watkins, C.,
Flannery, A., Ellison, G., Donald, E., Knight, L., Parums, D., Botwood,
N., and Holloway, B. (2006) J. Clin. Oncol., 24,
5034-5042.
67.Lynch, T. J., Bell, D. W., Sordella, R.,
Gurubhagavatula, S., Okimoto, R. A., Brannigan, B. W., Harris, P. L.,
Haserlat, S. M., Supko, J. G., Haluska, F. G., Louis, D. N.,
Christiani, D. C., Settleman, J., and Haber, D. A. (2004) N. Engl.
J. Med., 350, 2129-2139.
68.Paez, J. G., Janne, P. A., Lee, J. C., Tracy, S.,
Greulich, H., Gabriel, S., Herman, P., Kaye, F. J., Lindeman, N.,
Boggon, T. J., Naoki, K., Sasaki, H., Fujii, Y., Eck, M. J., Sellers,
W. R., Johnson, B. E., and Meyerson, M. (2004) Science,
304, 1497-1500.
69.Pao, W., Miller, V., Zakowski, M., Doherty, J.,
Politi, K., Sarkaria, I., Singh, B., Heelan, R., Rusch, V., Fulton, L.,
Mardis, E., Kupfer, D., Wilson, R., Kris, M., and Varmus, H. (2004)
Proc. Natl. Acad. Sci. USA, 101, 13306-13311.
70.Sequist, L. V., Bell, D. W., Lynch, T. J., and
Haber, D. A. (2007) J. Clin. Oncol., 25, 587-595.
71.Epstein, R. J. (2007) Cancer Met.
Rev., 26, 443-452.
72.Cobleigh, M. A., Langmuir, V. K., Sledge, G. W.,
Miller, K. D., Haney, L., Novotny, W. F., Reimann, J. D., and Vassel,
A. (2003) Semin. Oncol., 30 (Suppl. 16),
117-124.