
|
REVIEW: Carcinogenesis Induced by Foreign BodiesT. G. MoizhessInstitute of Carcinogenesis, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia; fax: (495) 324-1205; E-mail: moizhess@crc.umos.ru |
Received December 20, 2007
This review deals with the contemporary investigations of carcinogenesis induced by foreign bodies. The main attention is given to the interactions of macrophages with an implanted foreign body and their possible role in tumorigenesis.
KEY WORDS: carcinogenesis, foreign body, inflammation, macrophages, cytokines, free radicalsDOI: 10.1134/S0006297908070043
Abbreviations: AAMph) alternatively activated macrophages; C) carcinogenic; CAMph) classically activated macrophages; DMBA) 7,12-dimethylbenz(a)anthracene; FB) foreign body; LPS) lipopolysaccharides of Gram negative bacteria; MF) Millipore cellulose membrane filters; MIF) macrophage migration inhibitory factor; MMP) matrix metalloproteases; Mph) macrophages; NC) non-carcinogenic; PVC) polyvinyl chloride; SCT) subcutaneous connective tissue; SMC) smooth muscle cells; TLR) Toll-like receptors.
Carcinogenesis induced by foreign bodies (FB carcinogenesis), that is
the development of rat and mouse sarcomas in the immediate vicinity of
implanted FB in the form of plates of very distinct polymers or other
material, is considered to be one of the most enigmatic phenomena in
experimental oncology. This phenomenon was discovered in the 1940s [1]. Later numerous investigations [2] accumulated a number of experimental facts that
characterize this kind of carcinogenesis. The most intriguing was the
observation showing that the physical form of the implant is most
significant for tumor induction. In particular, it was found that
highly tumorigenic polymeric plates exhibited lower carcinogenicity
after perforation, while disintegration into small fragments resulted
in almost complete loss of carcinogenicity. Rather many hypotheses were
proposed to explain the mechanism of FB carcinogenesis but none of them
was generally acknowledged. Let us consider the long-known facts and
try to compare them with recent results.
The implantation of an FB into tissues results in an inflammatory reaction. Its role seems to be important for understanding the mechanism of FB carcinogenesis. After implantation of a sterile FB, its surface is covered with plasma proteins. This is followed by adhesion of blood neutrophils, lymphocytes, and monocytes releasing a number of biologically active products (chemoattractants, cytokines, growth factors, etc.). (Early events resulting in the ejection of cells from blood vessels and chemotaxis towards the FB and the role of blood coagulation systems and complement in the inflammation reaction are described in detail in an article by G. I. Abelev [3].)
Following adhesion to the implant surface, monocytes differentiate into macrophages, which often fuse and form multinucleated giant cells. Macrophages and giant cells very quickly become prevalent on the FB surface; this situation remains stable in appearance for many months. During a month a capsule of connective tissue is formed around the implant, which in the course of time may undergo some structural changes concerning thickness, blood vessel supply, and the ratio of the number of cells and the quantity of collagen fibers (the extent of fibrosis).
Due to the impossibility of elimination of the foreign body, the FB reaction is transformed into a chronic form, the main participants of which are macrophages and giant cells attached to the surface of the FB as well as free ones inhabiting the space between the implant surface and the internal surface of the capsule. The long-term presence of an FB in a tissue in some cases results in emergence of sarcomas in its immediate vicinity.
The goal of this review is to analyze possible mechanisms of FB carcinogenesis from the point of view of current understanding of cell transformation processes and factors leading to tumor development. In particular, the main attention will be given to the role of macrophages in these processes.
LOCALIZATION OF PRE-TUMOR CELLS IN THE PLATE-CAPSULE
COMPLEX
As a rule, tumors induced by FB implantation emerge within the plate-capsule complex. No tumors appear if the plate and capsule are removed together within 11-12 months after implantation [4]. It was also shown [5] that in the case of polystyrene plate removal and capsule preservation, no tumor appeared for six months, whereas later they developed in a significant number of cases. When cellophane was used as the FB, tumors developed even if plates were removed within four months. Thus, different materials induce tumors at different rates.
A question important for understanding the mechanism of FB-induced carcinogenesis remained open, namely, when and where, within the capsule or on the plate, do the first cells giving rise to tumors appear?
It has been shown [6, 7] that the tumor precursor cells (belonging to the same clone) [8], initially appearing in the connective tissue capsule and remaining in it, were found later in the monolayer on the plate. Despite the presence of such cells, the capsule without plate does not produce tumor development, except in cases when the plate is removed in the last stage of the latent period. Transplantation of a plate with the cell monolayer on its surface to a syngeneic recipient gives rise to a tumor consisting of donor cells.
Thus, the plate-capsule complex is responsible for tumor development. One should know which part of this complex is the main factor responsible for emergence of neoplastic cells. Some researchers assumed fibrosis of the capsule to be responsible for cell transformation, while by others the presence of the plate was considered to play the leading role in this event due to the interactions of cells with the hard surface of the plate or to vital activity of macrophages attached to the plate and surrounding the latter.
Brand et al. [9] considered the direct contact of cells with the implant surface to be necessary for completion of pre-neoplastic maturation. A number of experimental data were interpreted in favor of this hypothesis [10-13]. Cells of a nine-month-old capsule and the monolayer on the plate were cultivated separately for a number of passages and then introduced subcutaneously to syngeneic mice in the form of suspension and on the plate [10]. Introduction of cells on the plate in all cases resulted in emergence of tumors, whereas tumors developed much more rarely after introduction of cell suspensions. Similar experiments with cell culture lines (3T3, 10T1/2) gave similar results [11-13]: tumors developed only from cells introduced on the plate. Boone et al. [12] drew a direct analogy between spontaneous malignization in vitro and FB-induced carcinogenesis by assuming a similar mechanism of cell malignization due to the contact with the hard smooth surface.
However, it should be noted that the above-described experimental results might have an alternative explanation: the plate functions as a skeleton supporting the existence of the capsule and the whole complex responsible for FB carcinogenesis and presents the surface for its occupation by macrophages that, according to another hypothesis, play the key role in this process.
MACROPHAGES. PATHS OF ACTIVATION
According to current concepts, activation of macrophages can follow several paths that briefly reduce to the following. The so-called Toll-like receptors (TLR) for recognition of molecular structures, constantly associated with a certain group of microorganisms and recognized by their “own” TLR, are initially and conservatively present on cells of myelomonocytic series. (For example, lipopolysaccharides of Gram negative bacteria (LPS) are the ligand for TLR-4.) The binding of TLR to the corresponding ligand is the first step in the inborn immunity reaction. After ligand binding all TLR form dimers and undergo conformational changes that are necessary for liberation of sites for interaction with cellular adapter molecules and launching the signal transduction cascade. At a certain level of signal transduction, transcription factor NF-kappaB is translocated into the cell nucleus and directly binds promoter sites of a number of genes of molecules including cytokines that activate and regulate the development of the inflammatory reaction. At some steps of this cascade, other transcription factors, in particular AP-1, also involved in initiation of genes encoding inflammatory molecules, are activated as well [14]. Following activation of the pro-inflammation reaction, anti-inflammatory signals are activated, which results in development of repair processes in damaged tissue due to production of anti-inflammatory cytokines IL-4, IL-13, and IL-10.
It was also shown that a number of TLR are also able to recognize endogenous ligands emerging after tissue damage. An example is fibronectin domain A, synthesized only in response to tissue damage, or heat shock proteins that in the norm are present in the cell cytoplasm and become accessible for TLR only after cell damage or death [14].
Two macrophage (Mph) populations participate in the development of acquired immunity (upon interaction of pathogenic microorganisms with antigen-presenting cells): classically activated macrophages (CAMph) producing ROS, NO, and a set of anti-inflammatory cytokines, expression of which is induced by Th1-lymphocytes (inflammatory cells) using INF-gamma with involvement of LPS or using TNF receptor binding (so-called Th1-response); alternatively activated macrophages (AAMph) expressing anti-inflammatory cytokines in response to IL-4, IL-13, and IL-10, secreted by Th2 lymphocytes (helpers) (Th2 response), and to glucocorticoids. Some authors [15, 16] distinguish the action of the last two inducers as independent kinds of activation characterized by more pronounced effect, suppressive towards pro-inflammatory reaction (inhibition of production of inflammation stimulating cytokines, ROS, and NO, and induction of repair and synthesis processes, in particular due to secretion of TGF-beta).
The mechanism of the different behavior of CAMph and AAMph is largely explained by the different ways of L-arginine utilization: INF-gamma enhances the NO-synthase activity (NOS2) in macrophages, interaction of which with arginine results in NO generation and inhibition of arginase. In contrast, IL-4 and IL-13 inhibit NOS2 and, correspondingly, NO generation, and promote arginase-dependent formation of L-ornithine that is then transformed in parallel ways to proline (collagen precursor) by ornithine aminotransferase and by ornithine decarboxylase to polyamines stimulating fibroblast proliferation. Together these reactions mediate the fibrotic process [15].
Most likely specific immunity mechanisms are not involved in implantation of a sterile foreign body. This is confirmed in work [17] where it was shown that lymphocytes around an implanted material do not synthesize Th1 and Th2 cytokines. Long ago data appeared showing the expression of IL-1 by macrophages upon adhesion to plastic; it was shown that Mph from plate surfaces with different physicochemical properties expressed both pro-inflammatory (IL-1, TNF-alpha) and anti-inflammatory (IL-10, TGF-beta) cytokines [18].
Thus, independently of the nature of the induction of inflammation, with live pathogens (via TLR binding/antigen presentation) or sterile foreign bodies (via the inflammatory cell adhesion to the hard surface and/or via degradation products of cells damaged during implantation), the inflammatory reaction proceeds in two stages--cytotoxic and repair. The first stage is characterized by generation of oxy- and nitrogenous free radicals and a number of pro-inflammatory cytokines. The second stage suppresses cytotoxic effect of the first stage products and promotes repair of damaged tissues and production of extracellular matrix.
It remains not fully clear whether the above-described Mph phenotypes (CAMph and AAMph) remain irreversibly different or are able to change in response to various stimuli [19]. The latter possibility is supported by the following observations: the same Mph culture is capable of simultaneous production in vitro of both pro- and anti-inflammatory products. Expression of anti-inflammatory products is enhanced with time and inhibits generation of pro-inflammatory ones (the Mph activity is switched from destructive to productive) [20, 21]. It was also shown in the latter work that quantitative modulation of this process is possible depending on the properties of the substrate.
Perhaps different types of inflammation can proceed in various ways: via switching intracellular programs or by changing different cell populations.
Below we shall consider pro- and anti-inflammatory Mph as two cell populations and shall not decide beforehand the question concerning the irreversibility of distinctions between them.
Multinucleated cells usually covering large areas of the surface of an implanted material and formed upon fusion of Mph are divided into several types by morphological features, in particular, by arrangement of nuclei: chaotic, or parietal (horse shoes) [22]. It was shown that in vitro cells of the first type (giant FB cells) are formed in response to IL-4, while cells of the second type (Langhans cells) are formed in response to INF-gamma [23]. A similar effect of IL-4 was also confirmed in vivo [24]. Giant cells of both types can often be seen on the surface of a single implant. It was shown that the FB giant cells are able to express cytokines: TNF-alpha at the early stage after implantation and TGF-beta later [25]. Different materials are able to induce different extent of Mph fusion (from three nuclei to 50 and more). It is not clear to what extent this phenomenon is significant for carcinogenesis.
TUMORIGENICITY AND CHEMISTRY OF FB-MATERIAL
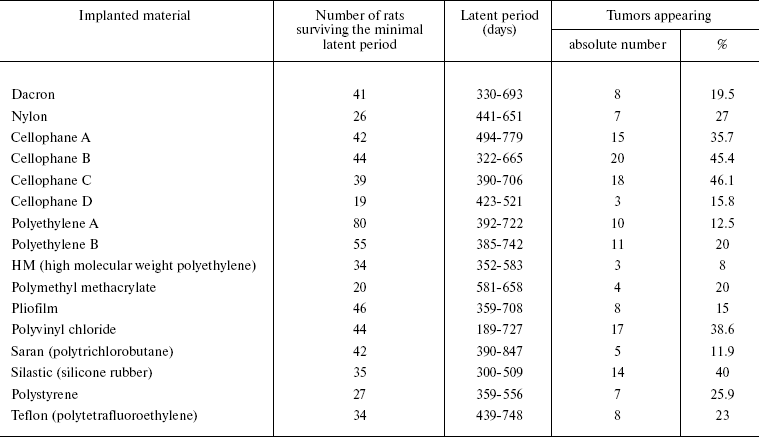
Now we shall consider available data on the relationship of implant tumorigenicity with its different properties and on the interactions of macrophages with the implant. In 1960, Nothdurft supposed on the basis of results of numerous investigations that any material is able to cause tumor development after sufficiently long presence in tissues [26]. However, the nature of the material influences the frequency of tumors induced by its implantation. Oppenheimer et al. [27] described results of subcutaneous implantation into rats of 16 types of plastic materials in the form of smooth continuous plates of equal shape and size (Table 1). The frequency of tumor emergence in response to implantation of these materials varies within a rather broad interval. Elimination of contaminants does not decrease the tumorigenicity of the material. Pustogarova [28] also compared tumorigenicity of polycaprolactam, polyethylene, and glass plates of the same shape and size upon subcutaneous implantation into rats; polycaprolactam gave rise to almost double the number of tumors compared to glass and polyethylene. Tumorigenicity of polycaprolactam plates covered in advance by a paraffin layer was three times lower. Plates of different metals also exhibited different tumorigenicity upon subcutaneous implantation into rats [29].
Table 1. Results of subcutaneous
implantation of different polymers into rats [27]
Carter et al. [30] tried to find correlation between some physicochemical properties of implanted material and its tumorigenicity. Three types of plates were prepared: with excess of anions or cations and with equal numbers of both. On implantation into rats of each of these types of plates, the highest yield of tumors was observed for cationic plates (9/14, i.e. in nine of 14 animals); it was lower for anionic (3/16) and the lowest (1/15) in animals with neutral plates. The authors noted that despite rather broad structural variety of capsules, most often inert fibrotic capsules were found around neutral plates. The most obvious distinction from the latter was formation of ectopic bone foci in four out of five cationic and in two out of 13 anionic capsules in which no tumors developed. The bone was usually well formed, sometimes containing portions of normal bone marrow. The results of this work allow one to draw at least two conclusions. First, the thick fibrous inert capsule, the presence of which was often noted by many authors as positively correlated with implant carcinogenicity, is not the crucial feature of carcinogenicity: neutral plates, most often surrounded by such capsule, caused almost no tumors, whereas cationic plates surrounded by the most “reactive” capsules (they included large portions of granulation tissue) appeared to be quite carcinogenic. Second, the properties of the foreign body influence both the capsule structure and the ectopic bone formation in it, which suggests the existence of a specific inducer whose appearance is stimulated by implantation of cationic plates.
In connection with all of the above-said, it is interesting to note, that as shown in vitro [31], macrophages of three lines (mouse J774A.1 and RAW264.1, as well as human THP-1) are able to secrete the BMP-2 protein stimulating differentiation of mesenchymal stem cells to osteoblasts. Takebe et al. [32] showed that the properties of substrate (chemical and topographical characteristics of the surface) influence ability of Mph to secrete BMP-2. BMP-2 expression was inhibited by pro-inflammatory Mph stimulation using LPS, which resulted in secretion of TNF-alpha. This fact is indicative of the possibility of the inverse trans-differentiation of Mph from pro-repair phenotype to pro-inflammatory.
Andrews [33] implanted into mice usual MF (round with 13-mm diameter) with pore diameters of 0.45 and 0.22 µm and the same filters prepared without detergent addition, and due to this they acquired hydrophobicity. Results of implantation of the usual filters confirmed their low tumorigenicity described earlier. Implantation of hydrophobic filters gave statistically significant increase of the tumor incidence over that obtained after implantation of hydrophilic filters.
Later works appeared providing an idea concerning the ways that the above-mentioned properties of implants might influence the activity of surrounding macrophages. It was shown in vivo [18] that implants with hydrophobic, anionic, and cationic surfaces stimulated a strong inflammatory response in the early phase, whereas in the case of hydrophilic surface leukocytes (the population included neutrophils, lymphocytes, and Mph, because it was studied in the early stage after implantation) produced significantly lower amounts of both pro- and anti-inflammatory cytokines. Additionally, adhesivity and the extent of macrophage fusion with formation of multinucleated cells were studied on these materials. Hydrophilic and anionic plates exhibited the lowest adhesivity. They were also characterized by the lowest extent of macrophage fusion and the highest number of apoptotic cells [34].
However, different chemical composition of implants in experiments of the above-mentioned authors should be noted. Therefore, there is no assurance in the cause-and-effect association of the plate properties, parameters of inflammation caused by them, and tumorigenicity. Nevertheless, the described experiments once more illustrate the effect of the FB surface properties on the modulation of gene expression in macrophages. It would be interesting to test for carcinogenicity the implants elaborated by Anderson et al. [18] and to compare the results obtained with those of Carter et al. [30].
Usual plastics also differently influence the functional state of Mph. This can be confirmed by data concerning different spectra of proteins released into the medium by peritoneal macrophages during their cultivation in vitro on the different material surfaces (culture and microbiological Petri dishes as well as MF with different pore diameters were tested). Differences were found between materials, but they did not include the filter pore dimensions as we had hoped (A. A. Neyfakh, T. G. Moizhess, unpublished). Reaction of macrophages to five polymers in vitro and in vivo was studied [35, 36]. The studied polymers revealed different ability of Mph to secrete bioactive products (IL-1 and the factor stimulating fibroblast proliferation).
A series of works by Tang and Hu reviewed in [37] answers the question concerning distinctions in expression of bioactive products emerging even in the case of inert substrate. Their experimental data show that differences in reaction of Mph to various materials might be the result of depending on the properties of the material different extent of the exposure of plasma fibrinogen molecule epitope upon protein adsorption to the implant surface. This is due to the denaturation of fibrinogen upon contact with biomaterial that results in conformational changes in its molecule. It was shown that the interaction of these epitopes with integrin Mac-1 of macrophages caused accumulation of the latter on the implant surface and their activation resulting in expression of pro-inflammatory cytokines and induction of fibrogenesis. Owing to this, it is preferable to study the biocompatibility of material for prosthesis in vivo.
In in vitro studies preliminary adsorption of different proteins by the implant surface also resulted in modulation of the behavior of Mph [38].
TUMORIGENICITY OF IMPLANT AND ITS SHAPE
As noted above, the most puzzling fact seemed to be the dependence of carcinogenicity of FB on its shape. The following was found by implantation of a polymeric plate:
- the larger the area of the plate, the higher its carcinogenicity [39, 40];
- for equal area, the plate minced into small fragments almost completely cancels its carcinogenic capacity [27, 41, 42];
- perforation of the plate significantly decreases its carcinogenicity [27, 42];
- plates with smooth surface induced tumors more rapidly than similar plates with rough surface [43, 44];
- MF (Millipore cellulose membrane filters) with pore diameter <=0.1 µm are highly tumorigenic, while no tumors developed when pore diameter was >=0.22 µm [42, 45].
Considering these facts, authors were inclined to interpret them as evidence supporting the idea that the emergence of a tumor is the result of tissue dissociation caused by a continuous plate or MF with small pore diameter. This hypothesis was disproved by Ferguson's experiment [45]: NC (non-carcinogenic)-filters stuck to both sides of a plastic plate and thus becoming impervious retained their properties, i.e. they did not acquire carcinogenicity, whereas filters with the small pore diameters were still tumorigenic as before. Thus, the properties of the implant surface appeared to be crucial for carcinogenicity.
However, it is interesting to note that for implantation of double and especially triple filters (with pore diameter of 0.45 µm) tumors formed in a large number of mice (7/30 and 16/30, respectively) [46]. The authors interpreted this result as being due to the enlarged area of implant surface.
Karp et al. [47] used the light and electron microscopy to study cells attached to C- (carcinogenic) and NC-implant surfaces. The macrophage-like cells were the only cell type discovered by them. To study these cells, C-plates of polyvinyl chloride (PVC) as well as C- and NC-MF with different pore diameters were introduced subcutaneously into mice. They compared cells attached to C- and NC-implants and noted that for NC-MF with pore diameters 0.22 and 0.45 µm, Mph covering their surface invaded the filter pores by their cytoplasmic processes. The lower cell surface contacting the foreign body on C-filters and plates was smoothed out. These cells, unlike those inhabiting MF with a large pore diameter, were free of phagolysosomes. However, collagen capsules around C-implants were thicker than those around NC, which is indicative of active collagen production in the case of C-plates. The presence of a thicker capsule could be also explained by low production of the collagen-degrading enzymes like collagenase and matrix metalloproteases (MMP), but active production of the latter by macrophages on the implant surface was found by Jones et al. [48]. The expression of MMP is specific for AAMph; this was shown during investigation of the gene expression profiles of CAMph and AAMph [49].
All these data suggest that the C- and NC-foreign body surfaces are inhabited by Mph in different functional states emerging in response to cell interaction with the substrate and, in particular, depending on the implant surface topography.
It was noted in a number of cases that the rougher the plate, the higher the synthetic activity of Mph on its surface [50-52]. This correlates with the above-noted lengthening of the latent period of tumors induced by rough plates [43, 44].
When the phenomenon of the loss of carcinogenicity of a minced plate is considered, the increased length of the border is obvious (the same is true of a perforated plate). Thus, for example, perimeter (7 cm) of a 2 × 1.5 cm plate, usually used in experiments on mice, is 7 times longer upon disintegration of the plate into 0.2 × 0.3 cm fragments. If cells at the implant border (the 1-1.5 mm wide strip along the border is suggested) differ from those in the middle, this gives us cause to look for the reason of different carcinogenicity of foreign bodies under consideration in the difference between cells inhabiting their surface. It was noted that the cell density at the border of PVC plates significantly exceeds their density over the remaining area; cells at the border often have polarized shape; an increased portion of neutrophils and lymphocytes was found among these cells (the situation is characteristic of acute inflammation). If the plate was covered with paraffin, in 1.5 months its borders (about 1 mm width) became free of paraffin, which, most likely, was the result of phagocytosis (unpublished data of the author).
Ziche and Gullino [53] compared the ability of cells inhabiting small and large area plates to secrete angiogenic factor. To make equal areas of the large (C) and small (NC) plates, they cut off the large plate margins and so studied cells located in its central part. Then, after mincing of both types of plates, their fragments were introduced into rabbit's eyes. The C-plate fragments caused intensive vascularization of the surrounding corneal tissue, whereas fragments of the small plate fragments were characterized by low vascularization activity. (Unfortunately, these authors did not test cells at the border of the C-plate. It is quite possible that the properties of these cells could be comparable with properties of cells inhabiting the small plate.) This confirms the hypothesis of different properties of cells located in the plate center and at its periphery. The angiogenic factor whose action was detected in this work was not identified. The ability of macrophages to express several such factors (VEGF, FGF) is now known; recently, a pro-angiogenic effect of IL-10 secreted by AAMph has been found [54].
THICKNESS OF CAPSULE SURROUNDING AN IMPLANT AND IMPLANT
CARCINOGENICITY
Comparative morphological investigation of the structure of the capsule surrounding an implant led some researchers [40, 41, 47] to conclude that the extent and continuance of chronic capsule fibrosis most constantly correlate positively with the frequency of tumor emergence near the implanted plates. High extent of fibrosis means a significant thickness of the capsule and prevalence in its composition of non-cellular component (collagen fibers) over cellular (fibroblasts, fibrocytes). Carcinogenic plates are surrounded by a thick capsule, while the capsule around a non-carcinogenic plate is thin. It is long known that under chronic inflammation macrophages are able to affect fibroblasts by stimulation of enhanced collagen production, which in some cases is responsible for pathological fibrosis of various organs [55].
Brand et al. ascribed an important role in FB-induced carcinogenesis to capsular fibrosis and noted the similarity between the response to foreign body and reaction to invasion of the helminth Schistosoma mannosi [56]. Eggs of these parasites stimulate formation of granulomas with deposition of a large amount of collagen. In some cases, tumors emerge at the site of these granulomas. Now it is known that these granulomas consist of anti-inflammatory AAMph producing proline and TGF-beta factor that stimulates collagen production by fibroblasts. Numerous giant FB cells are formed upon Mph fusion caused by IL-4 and IL-13, which are inducers of the alternative activation of Mph. It was shown that systemic introduction of antibodies against INF-gamma, an inducer of the classic Mph activation, does not change parameters of the organism's reaction to the foreign body invasion, and a hypothesis concerning induction of this reaction by IL-4 was proposed [57]. All these data support the hypothesis that foreign bodies accumulate AAMph on their surface (N. A. Glushankova, personal communication). As it was shown [18] that pro- and anti-inflammatory cytokines were present simultaneously in the seat of early inflammation caused by plate implantation, this also indicates the simultaneous presence of different Mph populations. Stages of inflammation are accompanied by a change in the ratio of different cytokines, which suggests the change of Mph populations. Evidently one can conclude that the capsule thickness is indirectly defined by macrophage activities and is indicative of the ratio of Mph populations surrounding the FB. If this is true, then anti-inflammatory Mph are prevalent on carcinogenic plates. In any case, the capsule thickness varies over a rather wide range depending on the implanted material [58]. This may be indicative of the effect of the surface properties of the FB on induction of different types of macrophage activation.
The role of anti-inflammatory Mph in FB-induced carcinogenesis may be associated with the well established fact that AAMph are frequently present in tumors and their harmful effect on prognosis: they prevent antitumor immunity due to IL-10 production [59]. A similar mechanism may be involved in generation of FB-induced sarcomas.
EFFECT OF GONADECTOMY ON FREQUENCY OF SARCOMAS INDUCED BY FOREIGN
BODIES
There are enough data in the literature concerning the relationship of FB-induced carcinogenesis with sex hormones. As shown by Olshevskaya on mongrel male rats [60], gonadectomy carried out a month ahead of cellophane plate (2 × 3 cm) implantations resulted in formation of sarcomas in 5/21 rats (23.8%) compared to 26/49 (53.2%) tumors in the control. Gonadectomy a month after plate implantation had practically no effect on tumor frequency (16/28, 57.1%). Brand et al. [61] showed that in Balb/c female mice, in which FB sarcomas usually develop earlier than in males, gonadectomy significantly lengthened the latent period of tumors. In males, castration caused no changes in the FB sarcoma latent period length and frequency. Lanari et al. [62] studied the effect of medproxyprogesterone acetate on FB carcinogenesis in Balb/c mice and found a significant decrease in the tumor frequency in males (7/34 compared to 14/30 in the control) after subcutaneous introduction of the hormone into the side opposite to that with the implanted plate. No significant effect was registered in female mice. Authors of works [60] and [62] noted positive correlation between lowering the tumor frequency and lower extent of fibrosis of the plate-surrounding capsules.
It is interesting to compare the described data with results obtained during studies of hormone effects on macrophage functions. Earlier it was assumed that estrogens were powerful Mph stimulators (estrogens cause increase in the number of blood monocytes and macrophages in peritoneal fluid along with enhancement of their proliferative activity), whereas androgens exhibit the opposite effect. Yurina and Radostina [63] studied the Mph ultrastructure in the seat of aseptic inflammation and observed features of increased synthetic and secretory activities of the macrophages of rat males in response to synestrol (a synthetic non-steroid estrogen): the increase in the number and dimensions of lysosomes as well as of sectional area of endoplasmic reticulum and Golgi apparatus.
Current studies of the effects of sex hormones on the functional state of Mph have shown that estrogen [64, 65] and testosterone [66] inhibit production of such pro-inflammatory cytokines as TNF-alpha. INF-gamma, IL-12, IL-6, and IL-1beta, but stimulate production of anti-inflammatory cytokines such as IL-10, IL-4, and TGF-beta. However, progesterone [67] inhibited production by dendritic cells of pro-inflammatory cytokines but did not influence expression of IL-10. In work [68], progesterone, unlike estradiol and androgen, did not inhibit IL-1 production in granuloma tissue of castrated animals, i.e. it did not exhibit anti-inflammatory effect. Data of Olshevskaya [60] show that castration preceding implantation and thus enhancing the intensity of initial inflammation (shown experimentally in [69]) simultaneously results in weakening of the carcinogenic effect of the implant. The lower extent of capsule fibrosis around such implants might be caused by weakening of the collagen-generating function of macrophages due to stimulation of pro-inflammatory component of the FB reaction. The indications of the Mph activity increase in response to synestrol action, noted by Yurina and Radostina [63], are probably due to synthesis of anti-inflammatory products.
Results of Brand et al. [61] concerning ovariectomized female mice also correlate with stimulation of the pro-inflammatory function of Mph shown in [68]. The relationship of decreased tumor frequency caused by progesterone in the experiment by Lanari et al. [62] as well as the absence of the effect of androgen level decrease on FB-induced carcinogenesis in castrated males in the experiment by Brand et al. [61] with differences in inflammation intensity is still unclear and requires further investigation.
WHY DO FOREIGN BODIES INDUCE TUMORIGENESIS?
The hypothesis that presently seems to be the most probable was proposed in the work [70]. According to this hypothesis, sarcomas emerge at the site of FB implantation due to the effect of products of respiratory burst that happens in neutrophils and macrophages of an inflammation focus. In the acute phase of inflammation at the high level of TNF-alpha expression, there is active generation of oxygen and nitrogen oxide free radicals [71] exhibiting cytotoxic and mutagenic effects [72-75]. As proved by results of many investigations, the latter are able to initiate tumor emergence and progression [76-83]. Cytokines and growth factors secreted by macrophages are also involved in these processes. The promoter effect of TNF-alpha and IL-1beta was shown on a model of two-stage chemical carcinogenesis of skin: TNF-alpha- and IL-1beta-deficient mice are resistant to skin carcinogenesis [84-87]. Antibodies to TNF-alpha inhibit the development of skin tumors [88].
Co-cultivation of two lines of human mammary gland cancer cells with macrophages results in their increased invasiveness caused by TNF-alpha-dependent induction of MMP9 in macrophages [89]. The latter is also notable for its ability to activate VEGF (vascular endothelium growth factor) in foci of a nascent tumor, thus promoting its development [90].
IL-1beta and TNF-alpha promoted in vitro transformation by erionite of human immortalized mesothelial cells [91]. TNF-alpha and IL-1beta are the main cytokines secreted by macrophages upon inhalation of asbestos. In mesothelium, TNF-alpha signaling via NF-kappaB_activation prevents cell death, allowing mesothelial cells to survive genetic damage induced by asbestos and free radicals - macrophage products [92]. The authors of this work concluded that pro-inflammatory cytokine TNF-alpha is a critical mediator of tumor promotion. However, it is not clear whether TNF-alpha is a promoter in FB carcinogenesis, because it is not known what is the continuance of its expression after FB implantation compared to the length of the latent period of FB-induced sarcomas. However, the presence of collagen capsule that can be a substrate for monocytes makes probable increased TNF-alpha expression by them [93].
In our experiments [94], a factor exhibiting promoter properties was found in medium conditioned by cells attached to a carcinogenic plate. It significantly stimulated formation of pre-sarcoma cell colonies in a semi-liquid medium containing methylcellulose (a test for tumor transformation). The nature of this factor is not clear.
Ryan et al. [95] described a nonstandard example of the promoter effect of small (4-7 mm) pieces of commercial contraceptive intrauterine helices (Lippes Loops and Cu-7) implanted intraperitoneally into female mice of A line on skin carcinogenesis induced by applications of 7,12-dimethylbenz(a)anthracene (DMBA). In mice with FB intraperitoneal implantation, 17 and 25 skin tumors emerged (correspondingly to the above-mentioned materials) compared to four tumors caused by DMBA alone. Tumors that emerged in response to the combination with FB exhibited more pronounced malignancy and shorter latent period. The authors supposed this was caused by a humoral promotion mechanism involving autoantibodies.
The role of p53 (and other tumor suppressors) in FB carcinogenesis is important but poorly studied. We shall return to this question below during consideration of NC-FB.
WHY DO SMALL PLATES (AND OTHER NON-CARCINOGENIC FB) NOT CAUSE
TUMORS?
Two explanations are possible a priori: they are incapable of initiation or cells initiated by them are eliminated. The second possibility is supported by results of work [96] in which implantation of small (0.5 × 1.0 cm) (and thus non-carcinogenic) plates into mice with knockout of gene p53 caused emergence of tumors in many cases (30/38, 79%). As expected, no tumors appeared in the wild-type mice. In this case, the enhanced formation of oxidative and nitrative stress markers was noted in tissue surrounding the plate. So, small plates probably initiate the emergence of transformed cells via exposure to free radicals; such initiated cells are not eliminated near the small plate in the absence of the protective mechanism of p53. Owing to its activity in intact mice, they do not survive.
The inverse dependence of FB tumorigenicity on the degree of initial acute inflammation caused by FB was noted long ago. In Nothdurft's experiment on rats [26] glasses, causing the most pronounced inflammatory reaction compared to other implants, were the least carcinogenic. This was also confirmed in our unpublished experiments. Slide pieces of 2 × 1.5 cm caused 20% (8/40) rate of tumor formation upon implantation into mice compared to 45% (5/11) in the case of PVC plates. However, the frequency of cells with chromosomal aberrations in the early (seven days after implantation) capsule around glass exceeded that around PVC plate. In many cases other NC-FB also cause a more pronounced inflammatory reaction compared to highly carcinogenic ones. In particular, this is true of reaction to implantation of minced plates compared to unbroken ones. A similar relationship between inflammation intensity and tumorigenicity was found during investigation of the organism's reaction to implantation of exogenous collagen. Intensive reaction to implanted collagen was described by Taira et al. [97]. Eierman et al. [93] showed that monocytes grown on a collagen support were characterized by a high level of expression of the pro-inflammatory cytokine TNF-alpha. On the other hand, comparative investigation of tumorigenicity of porous polyethylene plates and of similar plates with the surface modified by covalent-bond-immobilized collagen [98] showed that the high tumorigenicity of polyethylene (11/24) was almost completely lost after its combination with collagen (1/24). Increased ROS and NO production is sometimes able to suppress tumor formation (as shown for NO [99]) and it can be a factor responsible for lowered carcinogenicity in the case of more intensive inflammation.
In the experiment by Ferguson [100], subcutaneous implantation into mice of diffusion chambers made of MF with pores of 0.45 µm was accompanied by accumulation inside the chambers of a liquid able to lyse tumor cells in vitro upon addition to their culture medium. This result points to an additional possible factor influencing the level of FB carcinogenicity and probably explains the extremely low carcinogenicity of MF (1/40, according to our unpublished results). The nature of this factor was not fully clarified, but most likely it is due to the ability of Mph to kill tumor cells both on contact interaction and via secreted products (IL-1, TNF-alpha) [101].
PROMOTER PROPERTIES OF NON-CARCINOGENIC FB
It was shown [102] that non-carcinogenic small plates can serve as promoters upon implantation into animals after preliminary exposure to an initiating agent such as a chemical carcinogen, ionizing radiation, or FB in carcinogenic form (preliminary implantation of an unbroken plate). In the first two cases subcutaneous sarcomas emerged in mice only after additional implantation of a minced plate in immediate nearness to the implant. In the third case the implanted initially unbroken plate was cut out several months later together with the capsule, disintegrated, and implanted into a syngeneic mouse-recipient with a chromosomal marker. Sarcomas emerging in the place of repeated implantation originated from the donor cells, i.e. cells initiated by an unbroken plate continued to live and developed into a tumor. Thus, under our experimental conditions transplanted initiated cells do not undergo elimination in an intact organism (it should be noted that in the case of implantation of small plates, the initiated cells again undergo the genotoxic effect of the acute inflammation products). A logically possible explanation for the absence of elimination can be that all three initiating agents inactivate p53 function or that the initiated cells acquire the ability to escape apoptosis. Let us note in this connection that according to the data available in the literature, the pro-inflammatory macrophage migration inhibitory factor (MIF), production of which by Mph increases in some inflammatory diseases, is able to inhibit apoptosis mediated by the p53 gene, due to which cells damaged by free radicals can escape death [103]. The question concerning MIF production in FB reaction is interesting.
As already mentioned, slide pieces of 2 × 1.5 cm caused 20% (8/40) appearance of tumors upon implantation into mice. However, if 3.5 months later the highly carcinogenic PVC plate in its capsule was replaced by a glass one, then 65% of animals developed tumors (17/26) compared to 45% (5/11) in the control. A similar result was obtained if the PVC plate in its capsule was replaced after 3.5 months by a plate covered by a paraffin layer (14/27 or 51% against 11/75 or 14.7% in the case of implantation of the paraffin-covered PVC plate and 9/20 or 45% in the case of PVC plate implantation without replacement) (unpublished data of the author). These results suggest that in the case of repeated implantation, acute inflammation exerts a stimulating effect on previously developed initiated cells. NC-MF also exhibit promoter properties in the case of the C-plate replacements [102]. Comparison with the above-mentioned experiment by Ferguson [100] suggests the resistance of cells initiated by preceding C-FB implantation to tumoricidal agents as well.
Results of the above-mentioned experiments with implantation of minced plates as a promoter after the initiating effect of a chemical carcinogen or radiation raised the question whether initiation of subcutaneous connective tissue cells is direct (upon contact with the initiating agent) or mediated (via an effect of the initiator on the organism's response to FB implantation). To answer this question, experiments were carried out in which the chemical carcinogen N-nitroso-N-ethylurea (NEM) or general gamma-irradiation (as in [102]) were used as initiators. Transplantations of subcutaneous connective tissue (SCT) (scrape from the internal side of the skin patch of 3 × 4 cm) from donor CBA mice, either intact or “initiated” two months before transplantation, were performed. Recipients were mice F1 (CBA × C57Bl), also either intact or initiated two months before implantation, into which minced plates were implanted simultaneously with donor SCT. Transplantation of SCT from an irradiated donor together with minced plates into normal recipient resulted in formation of 5 out of 25 (20%) sarcomas from donor cells [104], which is indicative of the direct initiating effect of irradiation on SCT cells revealed using NC-FB as promoter.
Similar experiments with NEM as an initiator gave different results (unpublished data of the author). Variants of transplantations are given in Table 2.
Table 2. Variants of transplantations in
experiment with NEM

When both donor and recipient got the carcinogen (variant 1), the number of tumors in the place of implantation exceeded that in other combinations by more than one order of magnitude (14/31, i.e. 45%). In this case, 3/14 tumors were of donor origin, the rest emerging from the recipient cells. However, no tumors appeared after transplantation into an intact animal of SCT treated by NEM (variant 2), which shows that NEM does not exhibit any direct initiating effect. In variant 3 implantation of minced plates together with SCT from a normal donor to the NEM-treated recipient produced practically no tumors (1/21), and this result should be compared with results of experiments described in [102, 105] where similar implantation of minced plates without SCT gave a large number of tumors in the place of implantation (11/28). This is indicative of an unexpected inhibitory effect of normal SCT on tumor development. Results of these experiments suggest that NEM influences malignization of SCT cells only under conditions of systemic change by this carcinogen of the organism's reaction, and most likely of inflammatory reaction to implantation of minced plates.
The immunosuppressive effect of chemical carcinogens has been known for a long time [106]. In particular, it was shown in [107] that 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone (NNK) (a chemical carcinogen from the same class of nitrosamines as NEM) inhibits the secretion by macrophages of pro-inflammatory cytokines and enhances production of anti-inflammatory IL-10. We failed to find data directly concerning NEM, but it is reasonable to assume that it exerts a similar effect on the character of inflammatory reaction. Therefore, it seems reasonable to suppose that in the experiments under discussion the mechanism of the NEM initiating effect is not direct genotoxic damage of SCT cells, but rather lowering the intensiveness of the acute phase of the inflammatory reaction to the level of inflammation caused by the whole plate, and under these conditions minced plates acquire carcinogenic properties.
In this context, the inhibitory effect of normal SCT may be explained in connection with the above-mentioned stimulating effect of exogenous collagen on the acute phase of inflammation. Collagen from SCT treated with NEM is evidently devoid of such stimulating activity.
MORPHOLOGY AND ORIGIN OF TUMORS INDUCED BY SUBCUTANEOUSLY
IMPLANTED FB
According to determination of histological type of emerging tumors via morphological features, a significant part of FB-induced tumors are classified as poorly differentiated (spindle-cell and polymorphocellular sarcomas) or fibrosarcomas. Among other tumors are liposarcomas, osteogenic sarcomas, mesenhymomas, reticulosarcomas, histiocytoma, plasmocytoma, myxosarcoma, and rhabdomyosarcomas [27]. In addition to other tumors, Johnson et al. [108] described hemangiosarcoma and sarcomas containing segments with differentiation towards cartilage and bone, as well as leiomyomatosis and myxoid regions. This led the authors to hypothesize the unique origin of the FB-induced tumors: all tumors originate from a single type of pluripotent cells able to differentiate, depending on stimuli, to a broad spectrum of tissue elements of internal medium (i.e. according to current concepts, from stem cells).
Structural features in common were found during electron-microscopic investigation of tumors of different morphological types [108]. Tumors with morphological features of fibro-, osteo-, leiomyo-, mixo-, hemangiosarcomas, as well as poorly differentiated sarcomas were studied. Common for all of them is the presence of specific amorphous or slightly fibrillar, argyrophilic intercellular structure of the basal plate type as well as clusters of microfilaments 60 Å in diameter in most cells. Based on these morphological features, the authors suggest cells of small blood vessels, that feed capsule, for the role of tumor precursors.
It was shown [109] that cells inhabiting the cavity of connective-tissue capsule and the surface of triacetate C-plates (14 months after subcutaneous implantation into CBA mice) and grown in vitro were not stained by antibodies to Willebrand factor. Staining by antibodies to alpha-SM-actin revealed the presence of clearly defined filament bundles. In parallel, cultures of the first three passages of FB sarcomas were stained for alpha-SM-actin, which gave negative result. The absence of this marker in sarcoma cells may be indicative of the loss of its expression during transformation.
Thus, our data show that cells, most likely pre-tumor, have a marker specific of microvessel cells--pericytes and smooth-muscle cells. Identical cells but devoid of tumorigenic potential are present in capsules surrounding non-carcinogenic FB. This shows that such cells are normal participants of the reaction to FB implantation and undergo neoplastic transformation only in microenvironmental conditions specific for C-FB. It should be also taken into account that there are two additional cell types that are able to inhabit the capsule and express alpha-SM-actin. These are myofibroblasts, routinely detected in granulation tissue [110], and endothelial cells capable of trans-differentiation caused by TGF-beta [111, 112] which in our case is most likely secreted by macrophages covering the surface of the foreign body.
It is shown in this review that all known factors influencing the frequency of tumors emerging upon FB implantation, such as chemical and physical properties of the implant (surface charge, hydrophilicity, hydrophobicity), structure (smoothness, roughness, porosity) of its surface, implant size and shape (area, integrity), and hormonal status of the organism at the moment of implantation also influence the functional state of Mph, namely, qualitative and quantitative composition of secreted bioactive products. This, in turn, largely defines parameters of an organism's reaction to FB invasion and suggests that products released by activated neutrophils and Mph (neutrophils at the earliest steps of reaction to FB and Mph at rest) play the key role in the emergence of tumors in the place of FB implantation. At the stage of acute inflammation, it is generation of the highly reactive oxygen and nitrogen oxide free radicals exhibiting cytotoxic and mutagenic effects playing the crucial role in the initiation of the malignization process. According to available observations there is an inverse relationship between intensiveness of the acute inflammation and the level of its initiating effect. The mechanism of this phenomenon remains unclear. Hypothetically, one of the possible factors is the enhanced cytotoxic effect of free radicals resulting in enhanced cell death caused by vitally incompatible damage to their genetic apparatus. At the stage of chronic inflammation, promotion can be provided by cytokines and growth factors secreted by macrophages.
Experiments with transplantation of initiated cells and generation of tumors in a syngeneic donor led to a hypothesis concerning the enhanced resistance of these cells to toxic effect of oxygen and nitrogen stress products, but this hypothesis requires experimental confirmation. The nature of normal cells incorporated in the connective-tissue capsule and undergoing malignization caused by the microenvironment developed in the plate-capsule complex under the influence of macrophages is of considerable interest.
The currently rapidly developing branch of biology dealing with the elaboration of the optimally durable materials for prosthesis gives information concerning initial stages of an organism's reaction to implantation of various materials. However, there is the problem of safety in the use of artificial materials for invasion into the organism for prosthesis. Descriptions of cases of emergence of human tumor around prostheses, such as vascular ones, are known from the literature [113-116]. For better understanding of the mechanisms of FB carcinogenesis, there is not enough information on the behavior of macrophages and giant cells surrounding the implant at later stages.
All above-said serves as a good reason to intensify studies in the field of FB-induced carcinogenesis, which is of considerable scientific interest and significant practical value.
The author is grateful to Ju. M. Vasiliev, G. A. Belitskii, and L. M. Khromykh for help and attention to this work.
This work was supported by the Russian Foundation for Basic Research (grant 05-04-4822a), by the Leading Scientific Schools program (grant NSh-6003.2006.4), and by a grant of the Innovation Center “PROTEK”.
REFERENCES
1.Terner, F. C. (1941) J. Natl. Cancer
Inst., 2, 81-83.
2.Brand, K. G., Johnson, K. H., and Buoen, L. C.
(1976) CRC Crit. Rev. Toxicol., 4, 353-394.
3.Abelev, G. I. (1996) Soros Obrazovat.
Zh., 10, 28-32.
4.Oppenheimer, B. S., Oppenheimer, E. T., Stout, A.
P., Willhite, M., and Danishevsky, I. (1958) Cancer, 11,
204-207.
5.Oppenheimer, E., Willhite, M., Stout, A.,
Danishevsky, I., and Fishman, M. (1964) Cancer Res., 24,
379-390.
6.Brand, K. G., Buoen, L. C., and Brand, I. (1971)
J. Int. Cancer Inst., 47, 829-834.
7.Moizhess, T. G., and Prigozhina, E. L. (1973)
Byul. Eksp. Biol. Med., 9, 92-94.
8.Thomassen, M. J., Buoen, L. C., and Brand, K. G.
(1975) J. Natl. Cancer Inst., 54, 203-207.
9.Brand, K. G., Buoen, L. C., Johnson, K. H., and
Brand, I. (1975) Cancer Res., 35, 279-286.
10.Buoen, L. C., Brand, I., and Brand, K. G. (1975)
J. Natl. Cancer Inst., 55, 721-723.
11.Boone, Ch., and Jacobs, J. B. (1976) J.
Supramol. Struct., 5, 131-137.
12.Boone, Ch., Takeshi, N., Eaton, S. D., and
Paranipe, M. (1979) Science, 204, 177-179.
13.Barret, J. C. (1980) Cancer Res.,
40, 91-94.
14.Simbirtsev, A. S. (2005) Immunologiya,
26, 368-377.
15.Gordon, S. (2003) Nat. Rev. Immunol.,
3, 23-35.
16.Yona, S., and Gordon, S. (2007) Immunol. Cell
Biol., 85, 81-82.
17.Baldwin, L., Flanagan, B. F., McLaughlin, P. J.,
Parkinson, R. W., Hunt, J. A., and Williams, D. F. (2003)
Biomaterials, 23, 3007-3014.
18.Brodbeck, W. G., Voskerician, G., Ziats, N. P.,
Nakayama, Y., Matsuda, T., and Anderson, J. M. (2003) J. Biomed.
Mater. Res., 64, 320-329.
19.Mosser, D. M. (2003) J. Leuk. Biol.,
73, 209-212.
20.Zubova, S. G., and Okulov, V. B. (2001)
Immunologiya, 5, 18-22.
21.Anderson, J. M., and Jones, J. A. (2007)
Biomaterials, 28, 5114-5120.
22.Chambers, T. J., and Spector, W. G. (1982)
Immunobiology, 161, 283-289.
23.McNally, A. K., and Anderson, J. M. (1995) Am.
J. Pathol., 147, 1487-1499.
24.Kao, W. J., McNally, A. K., Hiltner, A., and
Anderson, J. M. (1995) Biomed. Mater. Res., 29,
1267-1275.
25.Hernandez-Pando, R., Bornstein, Q. L., Leon, A.
D., Orozco, E. H., Madrigal, V. K., and Cordero, E. M. (2000)
Immunology, 100, 352-358.
26.Nothdurft, H. (1960) Abhl. Deutsch Akad. Wiss.
Berl. Klasse Med., 3, 80-98.
27.Oppenheimer, B. S., Oppenheimer, E. T.,
Danishevsky, I., Stout, A. P., and Eirich, F. R. (1955) Cancer
Res., 15, 333-340.
28.Pustogarova, G. A. (1969) Voprosy Onkol.,
15, 75-77.
29.Oppenheimer, B. S., Oppenheimer, E. T.,
Danishevsky, I., and Stout, A. P. (1956) Cancer Res., 16,
439-441.
30.Carter, R. L., Roe, F. J., and Peto, R. (1971)
J. Int. Cancer Inst., 46, 1277-1289.
31.Champagne, C. M., Takebe, J., Offenbacher, S.,
and Cooper, L. F. (2002) Bone, 30, 26-31.
32.Takebe, J., Champagne, C. M., Offenbacher, S.,
Ishibashi, K., and Cooper, L. F. (2003) J. Biomed. Mater. Res.,
64, 207-216.
33.Andrews, E. J. (1972) J. Natl. Cancer
Inst., 48, 1251-1254.
34.Brodbeck, W. G., Patel, J., Voskerician, G.,
Christenson, E., Shive, M. S., Nakayama, Y., Matsuda, T., Ziats, N. P.,
and Anderson, J. M. (2002) Proc. Natl. Acad. Sci. USA,
99, 10287-10292.
35.Miller, K. M., Rose-Caprara, V., and Anderson, J.
M. (1989) J. Biomed. Mater. Res., 22, 713-731.
36.Miller, K. M., Huskey, R. A., Bigby, L. F., and
Anderson, J. M. (1989) Biomaterials, 10, 187-196.
37.Tang, L., and Hu, W. (2005) Exp. Rev. Med.
Dev, 2, 493-500.
38.Bonfield, T. L., Colton, E., Marchant, R. E., and
Anderson, J. M. (1992) J. Biomed. Mater. Res., 26,
837-850.
39.Alexander, P., and Khorning, E. (1961) in
Mechanisms of Carcinogenesis [Russian translation], Izd-vo
Inostrannoi Literatury, Moscow, pp. 25-41.
40.Brand, K. G. (1976) Natl. Cancer Inst.,
57, 973-976.
41.Olshevskaya, L. V. (1961) Byul. Eksp. Biol.
Med., 42, 79-84.
42.Moizhess, T. G., and Vasiliev, Ju. M. (1989)
Int. J. Cancer, 4, 449-453.
43.Bates, R. B., and Klein, M. (1966) J. Natl.
Cancer Inst., 37, 145-151.
44.Brand, K. G., Buoen, L. C., and Brand, I. (1975)
J. Natl. Cancer Inst., 55, 319-322.
45.Ferguson, D. J. (1977) Cancer Res.,
37, 4367-4371.
46.Iomhair, M. M., and Lavelle, S. M. (1997)
Technol. Health Care, 5, 331-334.
47.Karp, R., Johnson, K. H., Buoen, L. C., Brand,
I., and Brand, K. G. (1973) J. Natl. Cancer Ins.,
51, 1275-1285.
48.Jones, J. A., McNally, A. K., Chang, D. T., Qin,
L. A., Meyerson, H., Colton, E., Kwon, I. K., Matsuda, T., and
Anderson, J. M. (2007) J. Biomed. Mater. Res., 84,
158-166.
49.Sandler, N. G., Mentink-Kane, M. M., Cheever, A.
W., and Wynn, T. A. (2003) J. Immunol., 171,
3655-3667.
50.Takebe, J., Ito, S., Champagne, C. M., Cooper, L.
F., and Ishibashi, K. (2007) J. Biomed. Mater.
Res. A, 80, 711-718.
51.Refai, A. K., Textor, M., Brunette, D., and
Waterfield, J. D. (2004) J. Biomed. Mater. Res., 70,
194-205.
52.Tan, K. S., Qian, L., Rosado, R., Flood, P. M.,
and Cooper, L. F. (2006) Biomaterials, 27, 5170-5177.
53.Ziche, M., and Gullino, M. (1981) Cancer
Res., 41, 5060-5063.
54.Kelly, J., Khan, A., Yin, J., Thomas, A.,
Ferguson, T. A., and Apte, R. S. (2007) J. Clin. Invest.,
117, 3421-3426.
55.Mayanskii, D. N. (1991) Chronic
Inflammation [in Russian], Meditsina, Moscow.
56.Brand, K. G. (1979) Acta. Trop.,
36, 203-214.
57.Khouw, I. M., van Wachem, P. B., van der Worp, R.
J., van der Berg, T. K., de Leij, L. F., and van Luyn, M. J. (2000)
J. Biomed. Mater. Res., 49, 297-304.
58.Junge, K., Klinge, U., Klosterhalfen, B.,
Mertens, P. R., Rosch, R., Schachtrupp, A., Ulmer, F., and Schumpelick,
V. (2002) J. Invest. Surg., 15, 319-328.
59.Garcia-Hernandez, M. L., Hernandez-Pando, R.,
Gariglio, P., and Berumen, J. (2002) Immunology, 105,
231-243.
60.Olshevskaya, L. V. (1987) Voprosy
Onkol., 33, 69-74.
61.Michelich, V. J., and Brand, K. G. (1980) J.
Natl. Cancer Inst., 64, 807-808.
62.Lanari, C., Molinolo, A. A., and Pasqualini, C.
D. (1986) J. Natl. Cancer Inst., 77, 157-162.
63.Yurina, N. A., and Radostina, A. I. (1990)
Morphofunctional Heterogeneity and Interaction of Connective Tissue
Cells [in Russian], RUDN Publishers, Moscow.
64.Salem, M. L. (2004) Curr. Drug. Targets
Inflamm., Allergy, 3, 97-104.
65.Matalka, K. Z. (2003) Neuro Endocrinol.
Lett., 24, 185-191.
66.Corrales, J. J., Ameida, M., Burgo, R., Mories,
M. T., Miralles, J. M., and Orfao, A. (2006) J. Endocrinol.,
189, 595-604.
67.Butts, C. L., Shukair, S. A., Duncan, K. M.,
Bowers, E., Horn, C., Belyavskaya, E., Tonelli, L., and Sternberg, E.
M. (2007) Int. Immunol., 19, 287-296.
68.Da Silva, J. A., Colville, A., Nash, P., Spector,
T. D., Scott, D. L., and Willougnby, D. A. (1993) Arthritis
Rheum., 36, 1007-1013.
69.Da Silva, J. A., Colville, A., Nash, P., Spector,
T. D., Scott, D. L., and Willougnby, D. A. (1993) Ann. Rheum.
Dis., 52, 285-291.
70.Shaikhutdinov, E. M., and Lyu, B. N. (1989)
Uspekhi Sovrem. Biol., 107, 289-300.
71.Shvartsburd, P. M. (2006) Voprosy
Onkol., 52, 137-144.
72.Fulton, A., Loveless, S., and Heppner, G. (1984)
Cancer Res., 44, 4308-4311.
73.Kim, H. W., Murakami, A., Williams, M. V., and
Ohigashi, H. (2003) Carcinogenesis, 24, 235-241.
74.Kim, H. W., Murakami, A., Williams, M. V.,
and Ohigashi, H. (2004) Biosci. Biotechnol. Biochem., 68,
238-242.
75.Weitzman, S., and Stossel, T. (1981)
Science, 212, 546-547.
76.Troll, W., Witz, G., Goldstein, B., Stone, D.,
and Sigimura, T. (1982) Carcinogenesis, 7, 593-597.
77.Lewis, J., and Adams, D. (1987) Environ.
Health Perspect., 76, 19-27.
78.Weitzman, S., Weitberg, A., Clarc, E., and
Stossel, T. (1985) Science, 227, 1231-1233.
79.Yamashina, K., Miller, R., and Heppner, G. (1986)
Cancer Res., 16, 2396-2401.
80.Fox, H. B., Togni, P., Bernard, H., and Babior,
D. (1985) Immunol. Today, 6, 327-328.
81.Kawanishi, S., Hiraku, Y., Pinlaor, S., and Ma,
N. (2006) Biol. Chem., 387, 365-372.
82.Hussain, S. P., Hofseth, L. J., and Harris, C. C.
(2003) Nat. Rev. Cancer, 3, 276-285.
83.Kensler, T., Bush, D., and Kozumbo, W. (1983)
Science, 221, 75-77.
84.Suganuma, M., Okabe, S., Marino, M., Sakai, A.,
and Fujiki, H. (1999) Cancer Res., 59, 4516-4518.
85.Rollings, B., Pasparakis, M., Kollias, G., and
Balkwill, F. (1999) Nat. Med., 5, 828-831.
86.Arnott, C. H., Scott, K. A., Moore, R. J., Hewer,
A., Phillips, D. H., Parker, P., Balkwill, F. R., and Owens, D. M.
(2002) Oncogene, 21, 4728-4738.
87.Krelin, Y., Voronov, E., Dotan, S., Elkabets, M.,
Reich, E., Fogel, M., Huszar, M., Iwakura, Y., Segal, S., Dinarello, C.
A., and Apte, R. N. (2007) Cancer Res., 67,
1062-1071.
88.Scott, K. A., Moore, R. J., Arnott, C. H., East,
N., Thompson, R. G., Scallon, B. J., Shealy, D. J., and Balkwill, F. R.
(2003) Mol. Cancer Ther., 2, 445-451.
89.Hagemann, T., Robinson, S. C., Schulz, M.,
Trumper, L., Balkwill, F. R., and Binder, C. (2004)
Carcinogenesis, 25, 1543-1549.
90.Bergers, G., Brekken, R., McMahon, G., Vu, T. H.,
Itoh, T., Tamaki, K., Tanzava, K., Thorpe, P., Itohara, S., Werb, Z.,
and Hanahan, D. (2000) Nat. Cell Biol., 2, 737-744.
91.Wang, Y., Faux, S. P., Hallden, G., Kirn, D. H.,
Houghton, C. E., Lemoine, N. R., and Patrick, G. (2004) Int. J.
Oncol., 25, 173-178.
92.Carbone, M., and Bedrossian, C. W. (2006)
Semin. Diagn. Pathol., 23, 56-60.
93.Eierman, D. F., Johnson, C. E., and Haskill, J.
S. (1989) J. Immunol., 142, 1970-1976.
94.Moizhess, T. G., Stromskaya, T. P., and
Stavrovskaya, A. A. (1989) Tsitologiya, 31, 1114.
95.Ryan, W. L., Stenback, F., and Curtis, G. L.
(1981) Cancer Lett., 13, 299-302.
96.Tazawa, H., Tatemichi, M., Sawa, T., Gilibert,
I., Ma, N., Hiraku, Y., Donehower, L. A., Ohgaki, H., Kawanishi, S.,
and Ohshima, H. (2007) Carcinogenesis, 28, 191-198.
97.Taira, M., Araki, Y., Nakao, H., Takahashi, J.,
Hyon, S. H., and Tsutsumi, S. (2003) J. Oral Rehabil.,
30, 106-109.
98.Kinoshita, Y., Kuzuhara, T., Kobayashi, M., and
Ikada, Y. (1995) J. Long Term Eff. Med. Implants, 5,
275-284.
99.Hussain, S. P., Trivers, G. E., Hofseth, L. J.,
He, P., Shaikh, I., Mechanic, L. E., Doja, S., Jiang, W., Subleski, J.,
Shorts, L., Haines, D., Laubach, V. E., Wiltrout, R. H., Djurickovic,
D., and Harris, C. C. (2004) Cancer Res., 64,
6849-6853.
100.Ferguson, D. J., and Urban, J. L. (1980)
Cancer Res., 40, 1255-1262.
101.Suslov, A. P. (1990) Macrophages and
Anti-tumor Immunity, in Advances in Science and Technology. Ser.
Oncology [in Russian], VINITI, Moscow.
102.Moizhess, T. G., and Vasiliev, Ju. M. (1989)
Int. J. Cancer, 44, 449-453.
103.Hofseth, L. J., Saito, S., Hussain, S. P.,
Espey, M. G., Miranda, K. M., Araki, Y., Jhappan, C., Hiqashimoto, Y.,
He, P., Linke, S. P., Quezado, M. M., Zurer, I., Rotter, V., Wink, D.
A., Appella, E., and Harris, C. C. (2003) Proc. Natl. Acad. Sci.
USA, 100, 143-148.
104.Moizhess, T. G. (1997) Eksp. Onkol.,
19, 250-252.
105.Moizhess, T. G., and Vasiliev, Ju. M. (1992)
Vestnik RONTs, 3, 51-54.
106.Deitchman, G. I. (2004) in Carcinogenesis
(Zaridze, D. G., ed.) [in Russian], Meditsina, Moscow, p. 467.
107.Therriault, M. J., Proulx, L. I., Castonguay,
A., and Bissonnette, E. Y. (2003) Clin. Exp. Immunol.,
137, 232-238.
108.Johnson, K. H., Ghobrial, H. C., Buoen, L. C.,
Brand, I., and Brand, K. G. (1973) Cancer Res., 33,
3139-3150.
109.Moizhess, T. G., and Aleksandrova, A. Yu.
(2000) Second Russ. Congr. on Pathophysiology [in Russian],
Moscow, p. 263.
110.Hinz, B. (2007) J. Invest. Dermatol.,
127, 526-537.
111.Frid, M. G., Kale, V. A., and Stenmark, K. R.
(2002) Circ. Res., 90, 1189-1199.
112.Deissler, H., Lang, G. K., and Lang, G. E.
(2006) Int. J. Mol. Med., 18, 577-582.
113.Weiss, W. M., Riles, T. S., Gouge, T. H., and
Mizrachi, H. H. (1991) J. Vasc. Surg., 14, 87-91.
114.Fufe, B. S., Quintana, C. S., Kaneko, M., and
Griepp, R. B. (1994) Ann. Thorac. Surg., 58,
1752-1754.
115.Ben-Izhak, O., Vlodavsky, E., Ofer, A., Engel,
A., Nitecky, S., and Hoffman, A. (1999) Am. J. Surg. Pathol.,
23, 1418-1422.
116.Alexander, J. J., Moawad, J., and Cai, D.
(2006) Vasc. Endovasc. Surg., 40, 509-515.