Mitochondria-Targeted Plastoquinone Derivatives as Tools to Interrupt
Execution of the Aging Program.
2. Treatment of Some ROS- and
Age-Related Diseases (Heart Arrhythmia, Heart Infarctions, Kidney
Ischemia, and Stroke)
L. E. Bakeeva1, I. V. Barskov2, M. V. Egorov3, N. K. Isaev1,2, V. I. Kapelko4, A. V. Kazachenko5, V. I. Kirpatovsky5, S. V. Kozlovsky3, V. L. Lakomkin4, S. B. Levina2, O. I. Pisarenko4, E. Y. Plotnikov1, V. B. Saprunova1, L. I. Serebryakova4, M. V. Skulachev3, E. V. Stelmashook2, I. M. Studneva4, O. V. Tskitishvili4, A. K. Vasilyeva1, I. V. Victorov6, D. B. Zorov1, and V. P. Skulachev1,3,7*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-0338; E-mail: skulach@belozersky.msu.ru2Department of Brain Research, Institute of Neurology, Russian Academy of Medical Sciences, Pereulok Obukha 5, 105064 Moscow, Russia
3Center for Mitoengineering, Lomonosov Moscow State University, 119991 Moscow, Russia
4Institute of Experimental Cardiology, Cardiology Research Center, 3-ya Cherepkovskaya ul. 15A, 121552 Moscow, Russia
5Institute of Urology, 3-ya Parkovaya ul. 51, 105425 Moscow, Russia
6Serbsky State Research Center of Social and Forensic Psychiatry, Kropotkinsky Pereulok 23, 119992 Moscow, Russia
7Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received December 29, 2007; Revision received August 14, 2008
Effects of 10-(6′-plastoquinonyl) decyltriphenylphosphonium (SkQ1) and 10-(6′-plastoquinonyl) decylrhodamine 19 (SkQR1) on rat models of H2O2- and ischemia-induced heart arrhythmia, heart infarction, kidney ischemia, and stroke have been studied ex vivo and in vivo. In all the models listed, SkQ1 and/or SkQR1 showed pronounced protective effect. Supplementation of food with extremely low SkQ1 amount (down to 0.02 nmol SkQ1/kg per day for 3 weeks) was found to abolish the steady heart arrhythmia caused by perfusion of isolated rat heart with H2O2 or by ischemia/reperfusion. Higher SkQ1 (125-250 nmol/kg per day for 2-3 weeks) was found to decrease the heart infarction region induced by an in vivo ischemia/reperfusion and lowered the blood levels of lactate dehydrogenase and creatine kinase increasing as a result of ischemia/reperfusion. In single-kidney rats, ischemia/reperfusion of the kidney was shown to kill the majority of the animals in 2-4 days, whereas one injection of SkQ1 or SkQR1 (1 µmol/kg a day before ischemia) saved lives of almost all treated rats. Effect of SkQR1 was accompanied by decrease in ROS (reactive oxygen species) level in kidney cells as well as by partial or complete normalization of blood creatinine and of some other kidney-controlled parameters. On the other hand, this amount of SkQ1 (a SkQ derivative of lower membrane-penetrating ability than SkQR1) saved the life but failed to normalize ROS and creatinine levels. Such an effect indicates that death under conditions of partial kidney dysfunction is mediated by an organ of vital importance other than kidney, the organ in question being an SkQ1 target. In a model of compression brain ischemia/reperfusion, a single intraperitoneal injection of SkQR1 to a rat (1 µmol/kg a day before operation) effectively decreased the damaged brain area. SkQ1 was ineffective, most probably due to lower permeability of the blood-brain barrier to this compound.
KEY WORDS: mitochondria-targeted antioxidant, plastoquinone, arrhythmia, heart, kidney, infarction, strokeDOI: 10.1134/S000629790812002X
Abbreviations: AI, area of infarction; CK, creatine kinase; DCF, 5-(-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate; ICF, intensity of the (heart) contractile function; LDH, lactate dehydrogenase; LV, the heart left ventricle; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium; RA, risk area; ROS, reactive oxygen species; SkQs, cationic derivatives of plastoquinone or methyl plastoquinone; SkQ1, 10-(6′-plastoquinonyl) decyltriphenylphosphonium; SkQR1, 10-(6′-plastoquinonyl) decylrhodamine 19; TMRE, tetramethylrhodamine ethyl ester; TTC, 2,3,5-triphenyltetrazolium chloride.
In the first paper of this series [1], we described
the synthesis, physicochemical properties, and anti- and prooxidant
activities of cationic derivatives of plastoquinone (SkQs). It was
shown that very low (nM) SkQ1 (10-(6′-plastoquinonyl)
decyltriphenylphosphonium) and SkQR1 (10-(6′-plastoquinonyl)
decylrhodamine 19) operate as highly active antioxidants in aqueous
solutions, lipid micelles, planar phospholipid membrane, liposomes,
isolated mitochondria, and cell cultures. Here we tried the same
penetrating cations as possible therapeutic tools to treat the most
common age-related disease, which seem to be mediated by reactive
oxygen species (ROS). Among them heart attack, arrhythmia, stroke, and
kidney ischemia were studied. The data suggest that (i) age-related
ischemic diseases of heart, brain, and kidney as well as
H2O2-induced heart arrhythmia are mediated by
intramitochondrial ROS and (ii) a pathogenic increase in level of these
ROS and its consequences can be prevented by SkQs.
MATERIALS AND METHODS
Ex vivo studies on rat heart arrhythmia. Male Wistar rats (300-350 g) received the standard laboratory diet supplemented with 20 g cottage cheese with SkQ1 bromide from 0.02 to 250 nmol/kg per day for 3 weeks. Rats of control groups received, instead of SkQ1, sodium bromide in the same doses.
All studies described in this paper were performed in accordance with institutional guidelines for animal research. Rats were anesthetized with urethane (1.7 g/kg body weight); the isolated hearts were perfused at 37°C through the aorta with modified Krebs-Henseleit solution containing 11 mM glucose and saturated with carbogen (95% O2 and 5% CO2). The perfusion was carried out with a pump at controlled rate of approximately 10 ml/min per gram. The pressure in a latex balloon introduced into the left ventricular cavity was continuously monitored with a Gould Statham P23Db electromanometer and Gould 2400S recorder (Gould Statham Inc., USA). The product of developed pressure and heart rate was assumed to be a measure for intensity of contractile function (ICF).
The protocol of experiments with application of hydrogen peroxide included an estimation of inotropic cardiac response to a two-fold increase in the perfusion rate. For 40 min, 0.1 mM H2O2 was introduced into coronary vessels. The protocol of experiments with ischemia/reperfusion consisted of preliminary 20-min perfusion (10 ml/min per gram) followed by 30-min total normothermic ischemia and 50-min reperfusion. Functional parameters were registered each 5 min during preliminary perfusion, and each 1 min during ischemia/reperfusion. To measure arrhythmia, the following four-point scale was applied: the heart contraction frequency in the range of 90-180/min, 1 point; that of 1-90/min or 450/min and higher, 2 points; fibrillations, 4 points.
Data were statistically processed using the Student t-test. The results are presented as average value ± standard error (mean ± SEM).
An in vivo study on rat heart infarction. Male Wistar 300-450 g rats were used. The animals were kept at temperature about 23.5°C and 12/12-h light/dark cycles. Commercial pellet diet and water were provided ad libitum. Two series of experiments were performed with different duration of nutritional supplementation of SkQ1. In the first series, SkQ1 was added for two weeks in three different doses: 25, 125, and 500 nmol/kg per day. In the second series, the animals were fed SkQ1 for three weeks in doses of 2.5 and 250 nmol/kg per day. Bromide salt of SkQ1 dissolved in 0.1 M KH2PO4 buffer, pH 6.0, was used. Control groups were fed a regular diet supplemented with NaBr dissolved in 0.1 M KH2PO4 buffer, pH 6.0, in a dose of 250 nmol/kg per day for two or three weeks, respectively. Each group consisted of 9-12 animals.
Rats were anaesthetized with 20% urethane (120 mg/kg body weight), tracheotomized, and ventilated with room air using a KTR-5 animal ventilator (Hugo Sacks Electronik, Germany). Blood pressure was monitored via a cannula introduced into the left carotid artery using a Mingograph-804 (Siemens Elema, Germany). The left ventricle was perforated and the left anterior descending coronary artery was ligated (using atraumatic needle 5-0) to model a regional ischemia. Appearance of ischemia was verified by epicardial cyanosis. Removing the ligature allowed reperfusion of the ischemia area. Arterial blood pH, pO2, and pCO2 were maintained within physiological range and monitored by an ABL-30 acid-base analyzer (Radiometer, Denmark).
After operation, hemodynamic parameters were stabilized during 30 min. Durations of ischemia and reperfusion were 40 and 60 min, respectively. Blood samples were withdrawn from a venous catheter before ischemia and after reperfusion. At the end of the experiment, the coronary artery was re-occluded and the heart was stained through the jugular vein with 2 ml of 1% Evans blue dye (Sigma-Aldrich Co., USA) to estimate the region of myocardial perfusion and risk area (RA, unstained area). Then the heart was quickly excised and the left ventricle (LV) was frozen at -20°C to determine the size of infarction.
The frozen LV was sectioned into 1.5-2 mm slices. The slices were incubated in 1% TTC (2,3,5-triphenyltetrazolium chloride) solution in 0.1 M phosphate buffer, pH 7.4, for 10 min at 37°C. The stained slices were scanned and the area of infarction (AI, pale in color), RA (dark red), and an intact (non-ischemic) LV region (blue) were estimated using image analysis with Imagecal software. The weight of LV was measured, and in each group the ratio of RA/LV (weight) and AI/RA were determined [2].
The damage to myocardial tissue was measured by increase in activity of LDH (lactate dehydrogenase) and heart isoform of CK (creatine kinase) in blood plasma. Blood samples (about 0.5 ml) were collected from the vein before occlusion and 1 h after reperfusion. The plasma was separated from the cells by centrifugation (10 min at 2000 rpm, 10°C) and stored at -20°C. Activities of LDH and CK were measured using BioSystems kits (BioSystems S.A., Spain). The absorbance was read at 340 nm using a Yanako UO-2000 spectrophotometer. Concentration of metabolites in myocardial tissue was measured enzymatically according to standard protocols.
All values are expressed as mean ± SEM. Differences between groups were compared using ANOVA (SigmaStat 2.0; Jandel Corporation, USA). A p value of <0.05 was assumed to be statistically significant.
For the electron microscopy studies of rat heart, the tissue was fixed with 3% glutaraldehyde in phosphate buffer, pH 7.4, at 4°C for 2 h and then with buffered 1% osmium tetroxide for 1.5 h. The fixed material was dehydrated in alcohol solutions (50-80 and 96% alcohol) with increasing alcohol concentrations, 70% alcohol being saturated with uranyl acetate. Then the material was embedded into Epon-812 epoxide resin, and slices were prepared using a Leica microtome. Slices were stained with lead by the method of Reynolds. The resulting preparations were investigated and photographed using a HU-12B electron microscope (Hitachi, Japan).
Primary renal tubular epithelium cell cultures. Kidneys were aseptically excised from 3-7-day-old rats, then minced and placed into Hanks solution, pH 7.4. After several washes, the minced tissue was placed into 0.1% collagenase solution and incubated for 20-30 min at 37°C. Large pieces were removed, and cells were sedimented at 1000g for 3 min. The pellet was resuspended in DMEM/F-12 (1 : 1) containing 10% fetal calf serum and seeded in culture plates and glass-bottom dishes. Cells were cultivated in a CO2 (5%) incubator for 1-2 days before the experiments. Cell viability was evaluated by a standard MTT-test. For cultivation, 96-well plates were used.
Kidney ischemic model. Experiments were performed on outbred white male rats (200-250 g) fed ad libitum. Rats were anesthetized with diethyl ether of pentobarbital (50 mg/kg, intraperitoneal). To induce post-ischemic acute renal failure, renal vascular fascicle was clamped for 90 min by a microvascular clip. Then circulation was restored by removing the clip. The absence of blood flow was evaluated visually by the change of the kidney color. For the evaluation of ROS production, 200-µm thick renal cortex tissue slices were cut with a blade and placed into Hanks solution supplemented with 10 mM Hepes-NaOH, pH 7.4, and 10 µM DCF (5-(-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate). After incubation for 10 min at room temperature, the slices were washed for 5 min with Hanks solution. In certain protocols, cells were co-loaded with 1 µM TMRE (tetramethylrhodamine ethyl ester) for 10 min at room temperature to simultaneously study the mitochondrial membrane potential and ROS production.
In some cases, right side laparoscopic nephrectomy was performed and the other kidney was exposed to ischemia/reperfusion. After the surgery, the abdominal wall was closed with suturing.
The animals were observed for 2 weeks after the procedure with periodic evaluation of renal functioning. For single-kidney animals, the critical time for the survival is the first week (especially 3-4th day after application of ischemia/reperfusion when uremia develops).
On the 3rd and 7th days after ischemia, blood samples were taken from the animal. On 2-3rd and 6-7th days, the animals were placed in individual cages for the collection of urine. Several biological markers were measured in the blood specimens using an ADVIA 1200 automatic analyzer (Bayer, Germany) and hematological parameters were estimated using a CellTac blood analyzer (Nihon Kohben, Italy) and by counting various types of cells in blood samples.
Primary cell culture from brain cortex. Primary brain cortical cultures were prepared from brains of fetal rats (17-18th gestational day). The tissue was digested with trypsin/EDTA (0.05/0.02% w/v in PBS, 15 min, 37°C) followed by mechanical dissociation. Cortical cells were seeded on poly-L-lysine-coated 96-well plates (120,000 cells/cm2) and grown in neurobasal medium with B-27 serum-free supplement (Gibco, Germany) and 2 mM L-glutamine. Glutamate (25 µM) was added during the initial 3 days of cultivation. The cultures were maintained at 36.5°C in a CO2-incubator (5% CO2, 95% air). The medium was changed starting from day 4 by replacing half of the medium twice a week. Serum-free primary cortical cultures were used for the experiments after day 9.
Cortical cultures were exposed to 25-150 nM SkQR1 for 5 days in neurobasal medium with B-27 serum-free supplement. Then SkQR1-treated or untreated cortical cultures were exposed to 0.2 mM H2O2 for 20 min in balanced salt solution of the following content: 154 mM NaCl, 5 mM KCl, 35 µM Na2HPO4, 2.3 mM CaCl2, 1 mM MgCl2, 3.6 mM NaHCO3, 5.6 mM glucose, 10 mM Hepes, pH 7.4, 20°C. The MTT assay was used to assess the viability of cortical neurons. The percentage of viable cells was quantified by MTT reduction after incubation with 0.5 mg MTT/ml for 30 min. The viability of untreated control cultures was set to 100%, while the viability of treated cultures was expressed as percentage of the MTT light absorption of treated cultures compared to that of the control cultures.
The one-way ANOVA with Bonferroni post test was used for statistical analysis. Levels of p < 0.05 were considered as statistically significant. The results are given as mean ± SEM. All data were obtained from at least two independent experiments with 12-17 different cultures in each.
Brain ischemia model. A compression ischemic brain model [3-5] was accomplished with male Wistar rats of 130-180 g. Before surgery, the rats were injected intraperitoneally with chloral hydrate (330 mg/kg). The animal's head was fixed in a Medicor steriotaxis device. The head skin was shaved and a median longitudinal cut over the skull skin was made. On the location of a planned skull trephining, the periosteum was removed. With the use of a cylinder milling cutter 5 mm in diameter, the trephining opening was made aside from the median line by 1 mm. When drilling, to avoid a thermocoagulation effect, the drilling location was constantly wetted with physiological saline solution. The cranial dura matter was cleaved without touching the brain surface. Bleeding was stopped by a hemostatic sponge.
For making a compression ischemia, the Teflon rod was used which was moving inside a glass wall tube fitted into the trephining opening [6]. When pulled out of the glass tube, pressure of the Teflon rod was achieved up to 40 mm Hg, and it was applied to the brain for 15 min. After this, the rod was removed from the wound, the skin was mended, and the suture was treated with 2% iodine solution in 20% alcohol. Rectal temperature was monitored before the surgery and for 24-48 h after it.
For morphometric study, the animals under anesthesia were transcardially perfused with modified fixing solution of Telesnitsky [7] (formalin/ethanol/acetic acid ratio, 2 : 7 : 1). After 20 min, the brain was removed and placed sequentially into 70 and 50% ethanol, following by a distilled water wash. The 100-µm brain slices were prepared in a vibratome, mounted on a glass slides covered with gelatin, stained with 1% solution of Methylene Blue, dehydrated with ethanol, and sealed with a synthetic sealer (Entellan). Microphotographs of the slices were made with a high-resolution slide scanner. The images were analyzed with Image Analysis System software (ImageJ, USA).
RESULTS
Very low amounts of SkQ1 prevent appearance in isolated heart of a steady arrhythmia induced by H2O2 treatment or by ischemia/reperfusion. Cardiac contractile indices of SkQ1-supplemented rats were found to be almost the same as in the control group both at usual and high perfusion rate. In fact, a three-week SkQ1 application in broad range of dosages (0.02-250 nmol/kg per day) did not affect maximal level of cardiac contractile function (Table 1).
Table 1. Effect of SkQ1 feeding for three
weeks on key parameters of contractile function of isolated rat heart
at high perfusion rate

*ICF is the product of developed pressure and the heart
contraction frequency (heart rate).
Addition of hydrogen peroxide to the perfusate resulted in a raised heart contraction frequency (heart rate) by 12% during the first 15 min. However, then the ICF index decreased due to profound drop in developed pressure. Perfusion pressure in the initial stage of the hydrogen peroxide treatment decreased, but later it increased to the initial level or even exceeded it. Such dynamics were found to be inherent in both control and SkQ1-supplemented groups (not shown in figures and tables).
The 40-min H2O2 treatment was shown to result in arrhythmias in the majority (64%) of hearts from the control animals (Table 2). Cardiac automatism of rats that received SkQ1 in doses 0.02, 0.2, and 2.0 nmol/kg per day was found to be more resistant to H2O2 action, the number of experiments with regular rhythm being 1.5-2.0 times higher than in the control group. SkQ1 was found to decrease the cases of fibrillation by factor 2.5-4.0 (Table 2). The total arrhythmia index during 25-40 min was 3-4 times lower in SkQ1 groups with dosage 0.02-0.20 nmol/kg per day. Favorable SkQ1 effect disappeared when the SkQ1 dosage was increased to 250 nmol/kg per day (Table 2).
Table 2. Effect of prolonged in vivo
SkQ1 treatment on stability of cardiac rhythm after 40-min
H2O2 treatment

Note: Asterisks designate statistically significant (* p < 0.05;
** p < 0.01) difference between the SkQ1 and control groups.
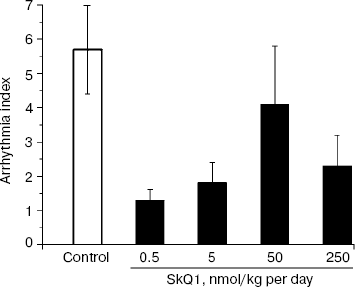
Ischemia/reperfusion of isolated hearts was studied in the next series of experiments. Contractile function indices during preliminary perfusion were similar in the control and SkQ1-supplemented groups. The ischemia resulted in a sharp drop in developed pressure and slower decrease in the heart rate. The heart rate was restored at the onset of reperfusion. However, this period was characterized by development of arrhythmia. Hearts of the SkQ1 groups (0.5 and 5 nmol/kg per day) were characterized by fewer arrhythmias in the first 5 min and much faster rhythm normalization in the range of 10-30 min (Fig. 1). Total index of arrhythmia for a 30-min period in these series was approximately 3 times lower than in the group without SkQ1 (Fig. 2). In the group receiving 250 nmol SkQ1/kg per day, the rhythm normalization occurred only by the end of the 30-min period. The heart rate by the end of reperfusion was significantly higher in the groups receiving 0.5 and 250 nmol SkQ1/kg per day (267 ± 11 and 261 ± 12 min-1, respectively, vs. 223 ± 13 in the control group, p < 0.05).
Fig. 1. Feeding rats with SkQ1 during 3 weeks decreases arrhythmia in isolated rat heart induced by ischemia/perfusion.
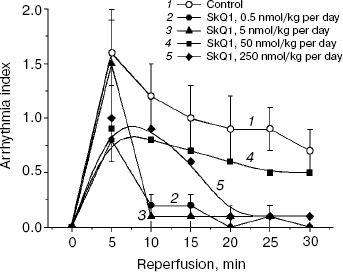
SkQ1 decreases myocardial infarction region. Two or three week supplementation of different SkQ1 doses with a regular diet did not affect the mean arterial pressure and the heart rate in the steady state in vivo. These indices were on average 102 ± 2 mm Hg and 225 ± 5 beats/min, respectively, and did not significantly differ from the corresponding values in the control groups.Fig. 2. Dose dependence of the SkQ1 effect on arrhythmia induced by 50-min reperfusion of ischemic hearts. Values of the arrhythmia index for 0.5 and 5 nmol SkQ1/kg per day are significantly lower than for the control (p < 0.01 and 0.05, respectively).
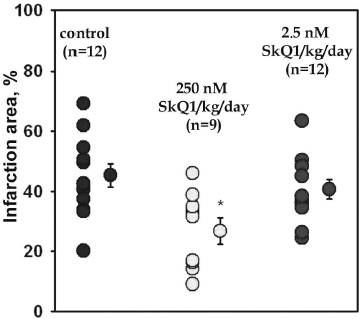
Experiments on rats after in vivo ischemia/reperfusion (an infarction model) showed no measurable differences in myocardial ischemic zone (RA/LV, %) between the control and SkQ1-fed groups. This index was 34.0 ± 2.7% (n = 24) for two control groups and 34.5 ± 1.9% (n = 48) for five SkQ1-fed groups. Myocardial infarct size (AI/RA, %) did not depend on duration of NaBr supplementation and was on average 42.2 ± 3.0% for both control groups. On the other hand, this parameter was clearly affected by SkQ1 (Figs. 3 and S1)1. A significant reduction in AI/RA was found after SkQ1 supplementation at a dose of 250 nmol/kg per day for 3 weeks and at a dose of 125 nmol/kg per day for 2 weeks (down to 26.8 ± 4.3 and 29.3 ± 5.5%, respectively) compared with the controls. Lower (≤25 nmol) and higher (500 nmol) SkQ1 doses were without measurable effect.
1 Here and further, for figures designated with “S”, see Supplementary Information presented in electronic version of the article.
The plasma LDH and CK activities did not differ significantly between the controls and SkQ1-fed groups in the steady state (Table 3), a fact indicating that SkQ1 supplementation at a dose of 250 nmol/kg per day for 2 weeks did not damage the outer cell membrane of cardiomyocytes. Myocardial infarction was associated with a profound increase in both plasma LDH and CK activities at the end of reperfusion. These activities were markedly reduced when rats were fed SkQ1 (on average, by 25 and 37% for LDH and CK, respectively, Table 3).Fig. 3. Dose-dependent reduction of myocardial infarction size in rats in vivo induced by SkQ1 feeding. Control, 250 nmol NaBr/kg per day for 2 or 3 weeks (n = 24); 1) 2.5 nmol SkQ1/kg per day for 3 weeks (n = 12); 2) 25 nmol SkQ1/kg per day for 2 weeks (n = 11); 3) 125 nmol SkQ1/kg per day for 2 or 3 weeks (n = 9); 4) 250 nmol SkQ1/kg per day for 3 weeks (n = 11); 5) 500 nmol SkQ1/kg per day for 2 weeks (n = 10). * p < 0.05; ** p < 0.02 versus control.
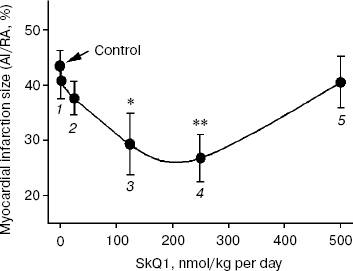
Table 3. Effects of SkQ1 on plasma lactate
dehydrogenase and creatine kinase activities measured before coronary
occlusion and after 40-min regional ischemia followed by 60-min
reperfusion
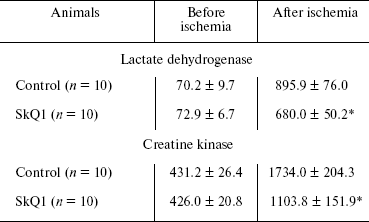
Note: Control, 250 nmol NaBr/kg per day for 2 weeks; SkQ1,
250 nmol SkQ1/kg per day for 2 weeks.
*p < 0.05 versus control.
In Table 4, data are shown indicating that in vivo pretreatment with SkQ1 decreases effects of ischemia/reperfusion on content of adenine nucleotides, creatine, creatine phosphate, and lactate in the risk area.
Table 4. SkQ1 partially reverses effect of
ischemia/reperfusion on levels of metabolites (µmol/g dry weight)
in the risk area of the left ventricle
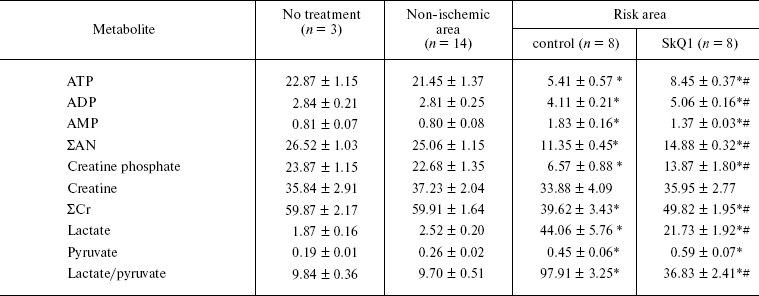
Note: Conditions of the experiments were the same as in Table 3. ΣAN, ATP + ADP + AMP. ΣCr, creatine
phosphate + creatine.
*p < 0.05 vs. no treatment and non-ischemic area.
#p < 0.05 vs. no SkQ1.
Figure 4 shows electron micrographs of hearts of control rats (no ischemia/reperfusion) (a) and of risk area after ischemia/reperfusion (b, c). In (c), rats received SkQ1 for 2 weeks. It is clearly seen that in a rat without SkQ, the ischemia/reperfusion procedure results in severe damage to all the structural parameters of cardiomyocytes. In the infarction area, one can see extensive disruption of actomyosin fibrils, Z-discs, and mitochondria. In numerous mitochondria, continuity of both the outer and inner membranes disappears, other mitochondria are swollen, and matrix becomes transparent. The majority of these pathological changes do not occur in hearts of the SkQ1-fed rats. In particular, no disrupted mitochondrial profiles are seen. In fact, the only obvious effect of ischemia/reperfusion in this case was a decrease in the electron density of the matrix space.
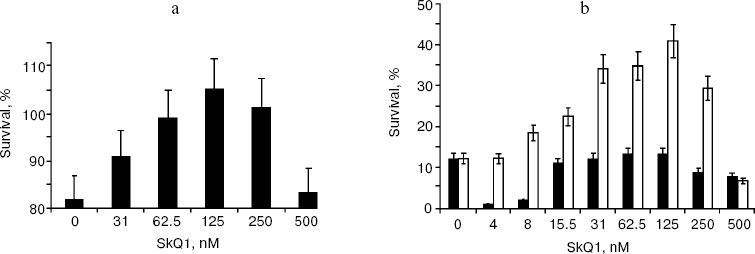
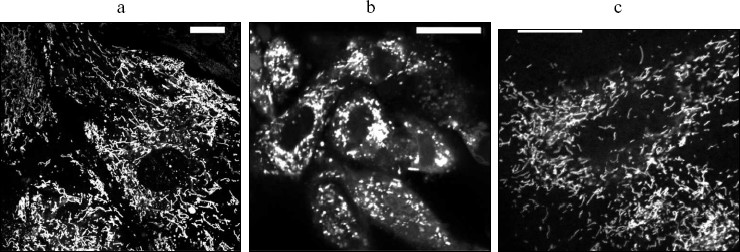
SkQs save life of rats after kidney ischemia. At first, we used culture of kidney epithelial cells as a model to test possible protective properties of SkQs in this tissue. As one can see in Fig. 5, preincubation with SkQ1 increases survival of these cells after 7- or 24-h anoxia followed by 24-h reoxygenation. Optimal concentrations of SkQ1 were between 30 and 250 nM. Figure 6 shows that SkQ1 prevents fission of elongated mitochondrial filaments to small roundish mitochondria (“the thread-grain transition” [8]), induced by the anoxia/reoxygenation procedure.Fig. 4. Electron microscopy of sections of 3-month-old rat hearts before (a) and after (b, c) ischemia/reperfusion. a, b) Control rats; c) rats fed with 250 nmol SkQ1/kg per day for 2 weeks.

Fig. 5. Protective effect of preincubation with SkQ1 on kidney epithelial cell survival after exposure to 7-h (a) or 24-h (b) anoxia followed by 24-h reoxygenation. Preincubation time, 1 day (a and b, black columns) or 5 days (b, white columns).
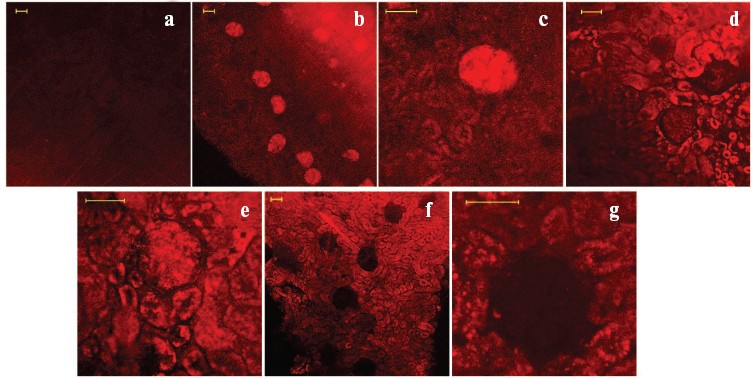
In the next series of experiments, we were following the distribution of SkQR1, a fluorescent SkQ1 derivative, in kidney slices after either intravenous or intraperitoneal injection into a rat. The fluorescence was clearly seen in glomerular zones of the kidney 60 min after injection into the vena cava inferior (cf. Figs. 7a and 7, b and c; see color insert). After 2 h, fluorescence was decreased in these regions with parallel appearance of the fluorescence in the kidney tubular epithelial cells (Fig. 7, d and e). In 3 days, further accumulation of SkQR1 in epithelium and almost complete loss of fluorescence in glomerular zones took place (Fig. 7, f and g). Incubation of the kidney slice taken from the rat 120 min after intraperitoneal injection of SkQR1 in the presence of mitochondrial fluorescent dye (Mitotracker Green) revealed colocalization of the two dyes in the kidney cells (not shown in figures).Fig. 6. Effect of SkQ1 on mitochondrial structure of kidney epithelial cells. Cells were stained with 200 nM TMRE. a) Control cells; b) cells exposed to 24-h anoxia followed by 24-h reoxygenation; c) cells were pretreated with 60 nM SkQ1 for 5 days before 24-h anoxia/24-h reoxygenation. Bar, 20 µm.
After a 40-min ex vivo kidney ischemia followed by reoxygenation, the ROS production (detected by DCF fluorescence in kidney slices) was found to be significantly elevated (Fig. 8, b, d-f). Earlier we demonstrated that this elevated DCF fluorescence originated from mitochondria [9]. A single intraperitoneal injection of SkQR1 (1 µmol/kg) to the animal a day before ischemia caused partial normalization of the ROS level (Fig. 8, c and d). Injection of the same amount of SkQ1 or MitoQ (Fig. 8, e and f) had no statistically significant effect.Fig. 7. Pharmacokinetics of SkQR1 distribution over kidney compartments after its intravenous injection: a) 10 min after injection of 300 µl of 1 mM SkQR1 to vena cava inferior; b, c) 60 min after injection of 200 µl of 1 mM SkQR1; d, e) 120 min after injection of 200 µl of 0.1 mM SkQR1; f, g) 3 days after injection of 200 µl of 0.1 mM SkQR1. Bar, 50 µm.
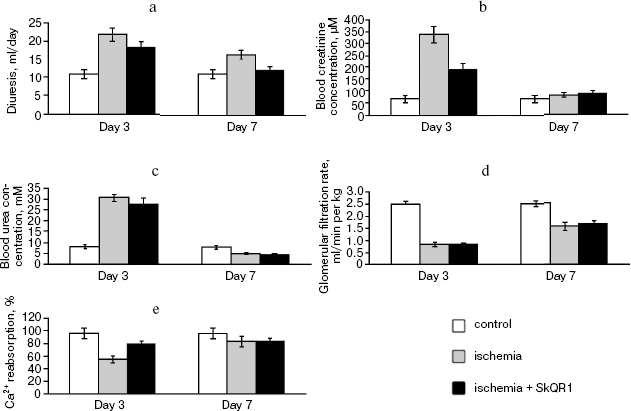
Some markers reporting on the distortion of kidney function after ischemia/reoxygenation are presented in Fig. 9. These experiments were done on animals having a single kidney (the other was removed before the ischemia). This model recently applied, e.g. by Serviddio et al. [10] was chosen to put higher load on kidney function since single-kidney ischemia in animals carrying two kidneys was shown to be without crucial effect on the organism (all animals survived after such a procedure). In the case of single kidney experiments, there was an obvious beneficial action of SkQR1 (partial normalization of diuresis, blood creatinine concentration, and Ca2+ reabsorption, facts showing that this compound protects to a certain degree the kidney tissue from a damage induced by ischemia/reperfusion) (Fig. 9). At the same time, the chosen amounts of SkQ1 did not have any influence on such a key marker as blood creatinine level (Fig. 10a). However, an effect was found when duration of the SkQ1 treatment was increased (Fig. 10c). A similar situation was observed when blood urea level was measured (Fig. 10d). As to Ca2+ reabsorption, favorable effect of SkQ1 was obtained when its dose was decreased from 1 µmol to 5 nmol/kg (Fig. 10b).Fig. 8. ROS generation in kidney slices detected by DCF fluorescence: a) control kidney; b) isolated kidney after 40-min ischemia followed by 10-min reperfusion (I/R); c) kidney from a rat receiving 1 µM SkQR1/kg per day before ischemia; d) average ROS production evaluated from six different experiments; e, f) as in (d) but instead of SkQR1 the same concentration of SkQ1 and MitoQ was injected, respectively. a-c) Bar, 100 µm.
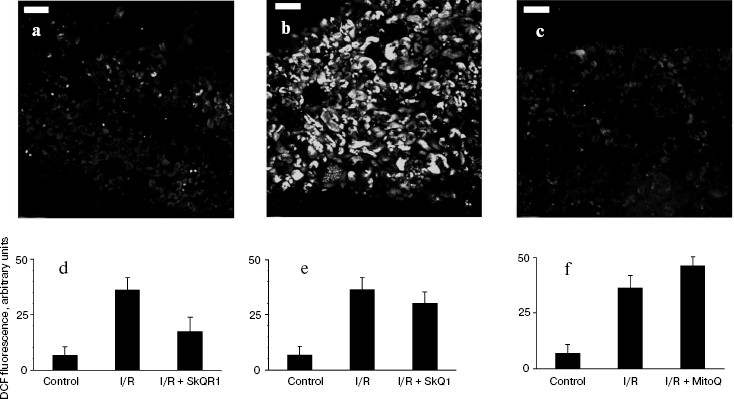
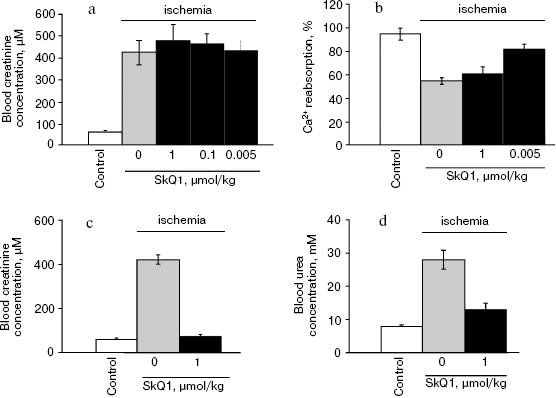
Fig. 9. Effects of SkQR1 (1 µmol/kg, intraperitoneally injected 1 day before 90-min ischemia) on kidney functioning: a) diuresis; b) blood creatinine concentration; c) blood urea concentration; d) glomerular filtration rate; e) Ca2+ reabsorption.
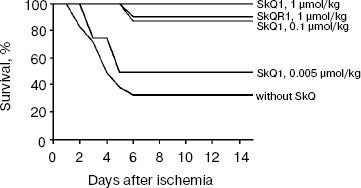
A single-kidney ischemia (90 min) followed by reoxygenation was extremely traumatic to the whole organism. The majority of the rats died on day 6 after this kind of treatment. However, preventive injection of SkQR1 or SkQ1 almost totally abolished the death of experimental animals (Fig. 11). The rats that survived on the critical day 6 completely restored their vital functions and were alive for at least half a year after ischemia.Fig. 10. Effects of different concentrations of SkQ1 (intraperitoneal injection 1 (a, b) or 3 days (c, d) before the ischemia) on kidney functioning. a, c) Blood creatinine concentration; b) Ca2+ reabsorption; d) blood urea concentration.
SkQR1 decreases damaged brain area after ischemia. To obtain brain ischemia, we used a compression model of hemorrhagic insult by application of a load to a particular brain area (see “Materials and Methods”). This model simulates the situation in the brain when the outflow blood is released into the brain cavity as a result of a hemorrhagic insult, causing the pressure on vital areas of the brain and, as a consequence, ischemia. Analysis of serial 100-µm brain sections revealed severe damage to the tissue, which could be decreased by intraperitoneal injection of SkQR1 (0.5-1 µmol/kg) one day before ischemia (Fig. 12). SkQ1 at a dose of 0.5 µmol/kg failed to substitute for SkQR1 (not shown).Fig. 11. Survival of single kidney rats exposed to 90-min ischemia followed by reoxygenation. Before the vascular clamp application, the second kidney had been excised. SkQ1 or SkQR1 were intraperitoneally injected 1 day before the ischemia.
Favorable effect of SkQR1 could be related to inhibition of ROS-induced apoptosis by this antioxidant. In Table 5, one can see how SkQR1 protects primary culture of rat cortical neurons against H2O2-induced apoptosis. The optimal SkQR1 concentration was 100 nM.Fig. 12. Compressive brain ischemia and protective effect of SkQR1 (1 µmol/kg, single intraperitoneal injection one day before the experiment): a, b) parietal and frontal parts of the rat brain, respectively. Methylene Blue staining of histological serial sections. Section thickness, 100 µm. Numbers of animals in (a) and (b) were 38 and 26, respectively.
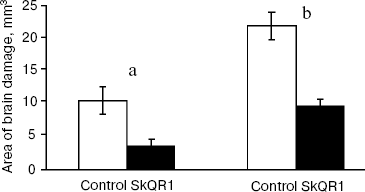
Table 5. Effect of SkQR1 on survival of
cortical neurons treated with 200 µM
H2O2 for 20 min

DISCUSSION
In this paper, we described favorable effects of SkQ1 and SkQR1 on models of several age-related pathologies, namely, heart and kidney infarction, heart arrhythmia, and stroke. In the literature, one can find numerous indications that all of these pathologies are somehow connected with an increase in the ROS level [11-14].
Heart arrhythmia in isolated heart induced by adding H2O2 to perfusate may well be a consequence of the inhibition by H2O2 of Ca2+ and Na+ pumps in cardiomyocytes [15] resulting in massive Ca2+ efflux from sarcoplasmic reticulum [15, 16]. As to ischemia/reperfusion, this procedure was shown to cause a strong increase in the free Ca2+ level in cardiomyocytes, shortening the contraction and delaying the relaxation [17]. Under these conditions, fibrillation was shown to occur [18]. Our data showed that such extremely low amounts of SkQ1 as 0.02 nmol (i.e. 12 ng/kg per day) strongly decrease the arrhythmia index and prevent fibrillation induced either by H2O2 perfusion or ischemia/reperfusion, pointing to key role of intramitochondrial ROS in these processes. The results are in line with data of Murphy and coauthors who showed that another mitochondria-targeted antioxidant, MitoQ, (i) arrests cytoplasmic [Ca2+] oscillations in pancreatic cells [19] and (ii) preserves cardiac contractile function and intactness of mitochondria during heart ischemia/reperfusion [20]. However, SkQ1 was effective in an amount 106 times smaller than that of MitoQ (cf. [19] and this paper). Similarly, myocardial protection by CoQ10 treatment in cases of cardiac operation [21] or infarction [22, 23] was observed at CoQ10 doses much higher than those of SkQs in our experiments even in the heart infarction model, although in this case effective concentrations of SkQ1 proved to be larger (125-250 nmol/kg per day) than for arrhythmia. The difference in SkQ1 amounts showing anti-arrhythmic and anti-infarction effects might indicate that different cell types are the SkQ1 targets in these two cases.
As we found when studying brain ischemia, a single intraperitoneal injection of SkQR1 showed strong protective effect, whereas that of SkQ1 was ineffective. Such a difference is most probably due to higher membrane permeability of the former [24], which might be of importance for crossing the blood-brain barrier. On the other hand, it does not mean that SkQ1 cannot cross this barrier under any conditions. As was shown in our group, SkQ1 added to the food for several weeks has pronounced therapeutic effect on cataract and certain retinopathies [25] and age-related changes in behavior of animals [25, 26]. The same explanation (higher SkQR1 permeability) can be used to account for efficiency of a single injection of SkQR1 but not of SkQ1 in prevention of the increase of ROS in kidney cells and creatinine concentration in blood after kidney ischemia/reperfusion if the injection was done a day before ischemia (cf. Figs. 8e, 9b, and 10a). In this case, an increase in the gap between the injection and ischemia to three days made SkQ1 efficient (Fig. 10c). However, the “anti-death” effect of 1 µmol SkQ1/kg was found to be observed even in the one-day gap experiments. This fact might indicate that SkQ1 saves the life of ischemic animals by a mechanism other than normalization of the kidney functioning. As to the SkQ1 target, it might be in such a case an unknown system of “biochemical suicide”, i.e. killing the organism which failed to maintain certain important parameters of homeostasis at the level guaranteeing intactness of DNA in reproductive organs (so-called “Bahys principle” [27]). This intriguing problem is now under investigation in our group. However, it is at present impossible to exclude another, trivial explanation of the above relationships. Perhaps, the SkQ1 effect is directed on specific cells amounting to only a small part of the kidney, which results in normalization of parameter(s) other than the creatinine level.
Thus, it is found that very low levels of mitochondria-targeted antioxidants of the SkQ series are of pronounced favorable effect in systems modeling ROS attack on heart, brain, and kidney, i.e. organs of the highest rate of aerobic metabolism. The described results, in combination with data on model lipid membranes, isolated mitochondria and cell cultures, as well as on whole organisms, published in four other papers of this issue [1, 25, 26, 28], suggest that this class of small synthetic compounds is promising as the most active mitochondria-addressed rechargeable antioxidants of possible therapeutic significance.
We are very grateful to Professor V. A. Sadovnichii, Rector of Moscow State University, for his interest in the project and encouragement. We would like to express our sincere gratitude for valuable advice to Professor G. Blobel. Generous support of Mr. O. V. Deripaska, which in fact made possible this study, is greatly appreciated.
Supported by Mitotechnology LLC, Lomonosov Moscow State University, the Vol'noe Delo Foundation (grant No. 99F-06), Russian Foundation for Basic Research (grant Nos. 08-04-01731 and 08-04-01667), and Russian Ministry of Education and Science (grant “Leading Scientific Schools” No. 5762.2008.4).
REFERENCES
1.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L. E.,
Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Yu.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Yu., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Yu., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1273-1287.
2.Pisarenko, O. I., Studneva, I. M., Serebriakova, L. I.,
Tskitishvili, O. V., and Timoshin, A. A. (2005) Kardiologiya,
45, 37-44.
3.Watanabe, S., Hoffman, J. R., Craik, R. L., Hand,
P. J., Croul, S. E., Reivich, M., and Greenberg, J. H. (2001)
Stroke, 32, 2615-2623.
4.Kundrotiene, J., Wagner, A., and Liljequist, S.
(2002) J. Neurotrauma, 19, 69-84.
5.Andersson, B., Bjelke, B., and Sykova, E. (2006)
Physiol. Res., 55, 339-348.
6.Andersson, D., Wu, X., Bjelke, D., and Sekova, E.
(2004) J. Neurosci. Res., 77, 901-912.
7.Andrews, R., and Muto, R. (1992) Neurol.
Res., 14, 12-18.
8.Skulachev, V. P., Bakeeva, L. E., Chernyak, B. V.,
Domnina, L. V., Minin, A. A., Pletjushkina, O. Y., Saprunova, V. B.,
Skulachev, I. V., Tsyplenkova, V. G., Vasiliev, J. M., Yaguzhinsky, L.
S., and Zorov, D. B. (2004) Mol. Cell. Biochem., 256/257,
341-358.
9.Plotnikov, E. Y., Kazachenko, A. V., Vyssokikh, M.
Y., Vasileva, A. K., Tcvirkun, D. V., Isaev, N. K., Kirpatovsky, V. I.,
and Zorov, D. B. (2007) Kidney Int., 72, 1493-1502.
10.Serviddio, G., Romano, A. D., Gesualdo, L.,
Tamborra, R., Di Palma, A. M., Rollo, T., Altomare, E., and Vendemiale,
G. (2008) Nephrol. Dial. Transplant., 23, 1504-1512.
11.McCord, J. M. (1985) N. Engl. J. Med.,
312, 159-163.
12.Baud, L., and Ardaillou, R. (1993) Br. Med.
Bull., 49, 621-629.
13.Moro, M. A., Almeida, A., Bolanos, J. P., and
Lizasoain, I. (2005) Free Rad. Biol. Med., 39,
1291-1304.
14.Webster, K. A. (2007) Trends Pharmacol.
Sci., 28, 492-499.
15.Goldhaber, J. I., and Liu, E. (1994) J.
Physiol., 477, 135-147.
16.Okabe, E., Tsujimoto, Y., and Kobayashi, Y.
(2000) Antioxid. Redox. Signal, 2, 47-54.
17.Meissner, A., and Morgan, J. P. (1995) Am. J.
Physiol., 268, 100-111.
18.Kojima, S., Wikman-Coffelt, J., Wu, S. T., and
Parmley, W. W. (1994) Am. J. Physiol., 266,
1473-1484.
19.Camello-Almaraz, M. C., Pozo, M. J., Murphy, M.
P., and Camello, P. J. (2006) J. Cell Physiol., 206,
487-494.
20.Adlam, V. J., Harrison, J. C., Porteous, C. M.,
James, A. M., Smith, R. A. J., Murphy, M. P., and Sammut, I. A. (2005)
FASEB J., 19, 1088-1095.
21.Taggart, D. P., Jenkins, M., Hooper, J.,
Hadjinikolas, L., Kemp, M., Hue, D., and Bennett, G. (1996) Ann.
Thorac. Surg., 61, 829-833.
22.Kalenikova, E. I., Gorodetskaya, E. A.,
Kolokolchikova, E. G., Shashurin, D. A., and Medvedev, O. S. (2007)
Biochemistry (Moscow), 72, 332-338.
23.Verma, D. D., Hartner, W., Levchenko, T., and
Torchilin, V. (2007) J. Mol. Cell. Cardiol., 42,
214-215.
24.Rokitskaya, T. I., Klishin, S. S., Severina, I.
I., Fedorov, I. I., Tashlitsky, V. N., Antonenko, Y. N., and Skulachev,
V. P. (2008) J. Membr. Biol., 222, 141-149.
25.Neroev, V. V., Archipova, M. M., Bakeeva, L. E.,
Fursova, A. Zh., Grigorian, E. N., Grishanova, A. Yu., Iomdina, E. N.,
Ivashchenko, Zh. N., Katargina, L. A., Khoroshilova-Maslova, I. P.,
Kilina, O. V., Kolosova, N. G., Kopenkin, E. P., Korshunov, S. S.,
Kovaleva, N. A., Novikova, Yu. P., Philippov, P. P., Pilipenko, D. I.,
Robustova, O. V., Saprunova, V. B., Senin, I. I., Skulachev, M. V.,
Sotnikova, L. F., Stefanova, N. A., Tikhomirova, N. K., Tsapenko, I.
B., Shchipanova, A. I., Zinovkin, R. A., and Skulachev, V. P. (2008)
Biochemistry (Moscow), 73, 1317-1328.
26.Anisimov, V. N., Bakeeva, L. E., Egormin, P. A.,
Filenko, O. F., Isakova, E. F., Manskikh, V. N., Mikhelson, V. M.,
Panteleeva, A. A., Pasyukova, E. G., Pilipenko, D. I., Piskunova, T.
S., Popovich, I. G., Roshchina, N. V., Rybina, O. Yu., Samoylova, T.
A., Saprunova, V. B., Semenchenko, A. V., Skulachev, M. V., Spivak, I.
M., Tsybul'ko, E. A., Tyndyk, M. L., Vyssokikh, M. Yu., Yurova, M. N.,
Zabezhinsky, M. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1329-1342.
27.Skulachev, V. P. (2007) Biochemistry
(Moscow), 72, 1385-1396.
28.Agapova, L. S., Chernyak, B. V., Domnina, L. V.,
Dugina, V. B., Efimenko, A. Yu., Fetisova, E. K., Ivanova, O. Yu.,
Kalinina, N. I., Khromova, N. V., Kopnin, B. P., Kopnin, P. B.,
Korotetskaya, M. V., Lichinitser, M. R., Lukashov, A. L., Pletjushkina,
O. Yu., Popova, E. N., Skulachev, M. V., Shagieva, G. S., Stepanova, E.
V., Titova, E. V., Tkachuk, V. A., Vasiliev, Y. M., and Skulachev, V.
P. (2008) Biochemistry (Moscow), 73, 1300-1316.
SUPPLEMENTARY INFORMATION
Download (PDF)
Fig. S1. SkQ1 decreases the infarction area after in vivo ischemia/reperfusion. SkQ1 was given to rats with food during three weeks. Number of animals: without SkQ1, 24; with SkQ1, 9-12; *, effect of SkQ1 is statistically significant (p < 0.05).