Mitochondria-Targeted Plastoquinone Derivatives as Tools to Interrupt
Execution of the Aging Program.
3. Inhibitory Effect of SkQ1 on Tumor
Development from p53-Deficient Cells
L. S. Agapova1, B. V. Chernyak2, L. V. Domnina2, V. B. Dugina2, A. Yu. Efimenko3, E. K. Fetisova2, O. Yu. Ivanova2, N. I. Kalinina3, N. V. Khromova1, B. P. Kopnin1, P. B. Kopnin4, M. V. Korotetskaya2, M. R. Lichinitser1, A. N. Lukashev5, O. Yu. Pletjushkina2, E. N. Popova2, M. V. Skulachev5, G. S. Shagieva2, E. V. Stepanova1, E. V. Titova2, V. A. Tkachuk3, J. M. Vasiliev1,2, and V. P. Skulachev2,5,6*
1Blokhin Russian Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, 115478 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-0338; E-mail: skulach@belozersky.msu.ru
3Cardiology Research Center, 3-ya Cherepkovskaya ul. 15a, 121552 Moscow, Russia
4Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia
5Mitoengineering Center, Lomonosov Moscow State University, 119991 Moscow, Russia
6Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received December 29, 2007; Revision received August 15, 2008
It was proposed that increased level of mitochondrial reactive oxygen species (ROS), mediating execution of the aging program of an organism, could also be critical for neoplastic transformation and tumorigenesis. This proposal was addressed using new mitochondria-targeted antioxidant SkQ1 (10-(6′-plastoquinonyl) decyltriphenylphosphonium) that scavenges ROS in mitochondria at nanomolar concentrations. We found that diet supplementation with SkQ1 (5 nmol/kg per day) suppressed spontaneous development of tumors (predominantly lymphomas) in p53-/- mice. The same dose of SkQ1 inhibited the growth of human colon carcinoma HCT116/p53-/- xenografts in athymic mice. Growth of tumor xenografts of human HPV-16-associated cervical carcinoma SiHa was affected by SkQ1 only slightly, but survival of tumor-bearing animals was increased. It was also shown that SkQ1 inhibited the tumor cell proliferation, which was demonstrated for HCT116 p53-/- and SiHa cells in culture. Moreover, SkQ1 induced differentiation of various tumor cells in vitro. Coordinated SkQ1-initiated changes in cell shape, cytoskeleton organization, and E-cadherin-positive intercellular contacts were observed in epithelial tumor cells. In Ras- and SV40-transformed fibroblasts, SkQ1 was found to initiate reversal of morphological transformation of a malignant type, restoring actin stress fibers and focal adhesion contacts. SkQ1 suppressed angiogenesis in Matrigel implants, indicating that mitochondrial ROS could be important for tumor angiogenesis. This effect, however, was less pronounced in HCT116/p53-/- tumor xenografts. We have also shown that SkQ1 and related positively charged antioxidants are substrates of the P-glycoprotein multidrug resistance pump. The lower anti-tumor effect and decreased intracellular accumulation of SkQ1, found in the case of HCT116 xenografts bearing mutant forms of p53, could be related to a higher level of P-glycoprotein. The effects of traditional antioxidant N-acetyl-L-cysteine (NAC) on tumor growth and tumor cell phenotype were similar to the effects of SkQ1 but more than 1,000,000 times higher doses of NAC than those of SkQ1 were required. Extremely high efficiency of SkQ1, related to its accumulation in the mitochondrial membrane, indicates that mitochondrial ROS production is critical for tumorigenesis at least in some animal models.
KEY WORDS: mitochondria-targeted antioxidant, tumorigenesis, tumor cellsDOI: 10.1134/S0006297908120031
Abbreviations: DCF, 2′,7′-dichlorodihydrofluorescein diacetate; FBS, fetal bovine serum; MDR, multiple drug resistance; NAC, N-acetyl-L-cysteine; PBS, phosphate-buffered saline; ROS, reactive oxygen species; SkQ1, 10-(6′-plastoquinonyl) decyltriphenylphosphonium; SkQR1, 10-(6′-plastoquinonyl) decylrhodamine 19.
As mentioned in a preceding paper [1], an increase
in oxidative stress is a key factor of the aging process. On the other
hand, elevation of reactive oxygen species (ROS) level plays an
important role in tumor development [2-5]. Such relationships might be at least partially
responsible for strong increase with age in probability of cancer. It
is shown that moderate increase in ROS content, which per se
does not kill a cell, leads to enhanced DNA oxidation and, as a result,
to genetic instability and accumulation of oncogenic mutations in
proto-oncogenes, tumor suppressor genes, and other genes whose
dysfunction causes neoplastic cell transformation and/or further tumor
progression [2, 3, 6]. In addition, changes in level of ROS, which
function as second messengers in many signal transduction pathways,
affect cell proliferation, migration, and viability that also
contribute to tumorigenesis [4, 5, 7-9]. It is
noteworthy that 1.5-3.0-fold increase in intracellular ROS content
represents an important component of oncogenic effects of such genomic
alterations, typical for human neoplasias, as inactivating mutations of
a tumor suppressor p53 or activating mutations of RAS and
MYC proto-oncogenes [6, 9-14]. Antioxidants
N-acetyl-cysteine (NAC) and some others:
- mitigate genetic instability caused by p53, Ras, and Myc dysfunctions [10-12];
- decrease growth rate of Ras-transformed cells [13];
- diminish intensity of Ras-induced morphological transformation and cell motility [9];
- inhibit ROS-dependent tumor angiogenesis induced by Ras and p53 dysfunctions [14, 15] or by some other oncogenic events [5, 16]. As a result, development of tumors (in particular in transgenic mice with p53 gene knockout (p53-/-)) [10, 17] or expressing enhanced levels of Myc protein [18] was shown to be significantly delayed.
It was proposed that changes of ROS level in various cell compartments can differentially affect probability of neoplastic transformation and tumorigenesis [7]. Since many signaling pathways are regulated by ROS production in mitochondria [3, 19-23], we decided to study whether a new mitochondria-targeted antioxidant SkQ1 (10-(6′-plastoquinonyl) decyltriphenylphosphonium) [1, 24-27] influences tumor development. For this purpose, we used two approaches--an analysis of the effect of diet supplementation with SkQ1 on spontaneous development of tumors (prophylactic action), and a study on the effect of SkQ1 on progression of already formed tumors (therapeutic activity). In the first case, we used mice with homozygous p53 gene knockout [28, 29] accompanied with a ROS up-regulation [10]. In the second part of work, we studied the growth of xenografts of a set of human carcinomas with different state of p53 tumor suppressor. Various p53 abnormalities were investigated based on the following considerations. A p53 dysfunction represents the most universal molecular alteration in various human tumors [17, 30-32] whose oncogenic effect is partially dependent on attenuation of antioxidant defense [10, 17, 32]. Several types of p53 abnormalities are characteristic for human tumors, including missense mutations leading to synthesis of proteins which fail to perform the wild-type p53 functions but acquire quite other activities [30, 31], full loss of p53 expression, or binding of p53 to its negative regulators such as E6 oncoprotein of human papilloma viruses, Mdm2, PARK, etc. [30-32].
In particular, we have studied xenografts of HCT116 colon carcinomas expressing wild-type p53 or expressing p53 proteins with amino acid substitutions at codons 248 and 273, which represent the hot-spots of mutations in colorectal cancers, or showing loss of p53 expression due to homozygous p53 gene knockout. In addition, we have analyzed xenografts of SiHa human HPV-16-associated cervical carcinoma in which p53 functioning is decreased due to its interaction with E6 viral oncoprotein. Finally, we have studied SkQ1 effects on cells cultured in vitro to understand possible mechanisms of inhibitory influence of SkQ1 on growth of some types of tumors.
MATERIALS AND METHODS
Cells. The following cell lines were used: HCT116 (ATCC No. CCL-247), human colon carcinoma bearing two wild-type p53 gene alleles; HCT116/p53-/-, the HCT116 subline with homozygous p53 gene knockout [33]; HCT116/R248W, HCT116/R273H, and HCT116/p53-/-/R248W [15]; derivatives of HCT116 cells expressing introduced by lentiviral gene transfer mutant p53 proteins with substitutions of Arg to Trp at codon 248 or Arg to His at codon 273; HaCaT, spontaneously immortalized human keratinocytes (L. Fontao, Department of Dermatology, University Hospital of Geneva); SiHa (ATCC No. HTB-35); human cervical carcinoma with integrated human papilloma virus type 16 genome (HPV-16, 1 to 2 copies per cell); K562 chronic myeloid leukemia cell line (ATCC No. CCL-243) and its multidrug resistant K562-MDR subline over-expressing active P-glycoprotein transporter [34]; murine embryonic 10(3) fibroblasts lacking expression of p53 and their derivative 10(3)-Ras expressing activated human N-RASAsp13 oncogene [35]; human embryonal lung fibroblasts MRC-5 and their clonal derivative MRC5-V2 transformed with SV40 virus [36]. The cells were cultured in DMEM (Dulbecco's Modified Eagle's Medium) (PanEko, Russia) supplemented with 10% FBS (fetal bovine serum) (5% FBS for SiHa and HaCaT), antibiotics penicillin and streptomycin (100 U/ml each), and glutamic acid. The cells were kept at 37°C in a humidified atmosphere of 5% CO2.
Intracellular ROS was measured as described earlier [37]. Briefly, the cells were incubated with 5 µM 2′,7′-dichlorodihydrofluorescein diacetate (DCF) (Molecular Probes, USA) for 30 min, washed with PBS, trypsinized, collected in 1 ml of PBS, transferred to polystyrene tubes with cell-strainer caps (Falcon, USA) containing 1 ml PBS, and analyzed with a FACScan cytofluorometer (Beckton Dickinson, USA) using Cell Quest 3.2 (Beckton Dickinson) software for acquisition and analysis. About 104 cells were analyzed in each sample. To detect the changes in ROS levels in splenocytes in vivo, the spleens from 1-2-month-old mice were used. The splenocytes were dispersed by pressing the spleens at the bottom of a DMEM-containing tissue culture dish, using the flat surface of a syringe plunger. Cells were left undisturbed for 1 min to let the splenic debris settle and then were collected, washed by centrifuging for 5 min at 400g, incubated with DCF, and analyzed as described above.
Western analysis. Whole cell extracts were lysed in ice-cold RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 1% NP-40, 0.1% SDS, 100 µM phenylmethylsulfonyl fluoride, 1 µM pepstatin A, and 1 µM antibiotic E64). Protein concentration in the extracts was determined with a protein assay system (BioRad, USA). A 20 µg protein sample was separated on a 10% SDS-polyacrylamide gel and transferred to poly(vinylidene fluoride) (PVDF) membrane (Amersham, USA) as described earlier [38]. The membranes were probed with antibodies specific to p53 (DO-7; Dako, USA), p63 (4A4; Dako), p21Cip/Waf1 (SX118; Dako), p27Kip1 (SX53G8; Dako), and E-cadherin (SHE78-7; Calbiochem, USA). Antibodies against alpha-tubulin (Sigma, USA) and gamma-tubulin (sc-53777; Santa Cruz Biotechnology, USA) were used for a loading control. Membranes were treated with secondary anti-mouse-HRP (Amersham). Filters were developed with ECL chemiluminescence reagents (Amersham) according to the manufacturer's protocol.
Expression of p53-responsive gene reporter was studied as described earlier [38]. Briefly, cells were transiently transfected using Lipofectamine reagent (Invitrogen, USA) with 1 µg pWWp-GL2 plasmid expressing luciferase under the control of wild-type p21Cip1/Waf1 gene promoter. After incubation with various doses of SkQ1 for 2-5 days, the expression was measured by luciferase activity using the Steady-Glo Luciferase Assay System (Promega, USA).
Intracellular accumulation of SkQ1. To study the accumulation of SkQ1 in the cells, we have employed its fluorescent analog SkQR1, where the triphenylphosphonium cationic group was substituted with rhodamine-19 [1, 24]. It was shown in an accompanying study that SkQR1 was selectively accumulated in mitochondria of various cells [1]. Intracellular accumulation was measured after incubation of the cells in complete medium at 37°C with 50 nM SkQ1 for 1.5-3 h and following incubation without SkQR1 for 0.5-2 h. The inhibitors of P-glycoprotein verapamil (50 µM) or pluronic L61 (60 µg/ml) were added simultaneously with SkQR1. Fluorescence was measured by FACS (Beckton Dickinson FACScan or Beckman-Coulter FC500). The protective effect of SkQR1 and SkQ1 was measured after incubation with 5 nM of the antioxidants for 6 days. Cells were treated with 100-300 µM H2O2 for 20 h and viability was analyzed by reduction of 3-(4,5-dmethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT).
Immunostaining. For immunofluorescence staining, cells were rinsed with DMEM containing 20 mM Hepes at 37°C, fixed with 2% paraformaldehyde in pre-warmed (37°C) DMEM for 30 min, followed by 5 min treatment with methanol at -20°C or 0.2% Triton X-100 for actin staining with phalloidin-TRITC (Sigma). The following mAbs against beta-catenin, E-cadherin, and paxillin (BD Transduction Laboratories, USA), actin, and vinculin (Sigma) were used. As secondary antibodies, goat anti-mouse antibodies conjugated with Alexa-488 (Molecular Probes) and rhodamine-conjugated antibodies against rabbit immunoglobulins (Jackson Labs, USA) were used. Images were acquired using an Axiovert microscope equipped with an oil immersion objective (Plan-Neofluar 100×/1.3; Carl Zeiss, Germany).
Cell morphometry. Cells were plated at single-cell density onto glass cover slips, and an inverted microscope with a phase-contrast objective and ImageJ software (National Institutes of Health (NIH), http://rsb.info.nih.gov/ij/) was used to trace cell outlines and measure single cell surface area. For focal adhesion morphometry, cells were fixed with 3% paraformaldehyde, then treated with 0.1% Triton X-100 and stained for vinculin or paxillin.
Animal studies. For spontaneous tumor frequency assay, we maintained families of p53+/- mice (C57BL/6 background, the colony is sustained at the Laboratory of Cytogenetics of Blokhin Russian Cancer Research Center since 2000 as described in [10]) on a diet supplemented with SkQ1 added to the drinking water to yield doses 0.5-50 nmol SkQ1/kg body weight per day (on average, a mouse drank about 5 ml of water per day). The p53-/- progeny were tested using PCR analysis of DNA as described in [10] and maintained with or without SkQ1 supplementation during all their life. The formation of tumors was monitored by daily inspection and confirmed by autopsy followed by histological examination of tumors. Nude mice assay was performed as described previously [15, 39]. Briefly, female D2&I nude mice (10 animals for each experimental group) were inoculated subcutaneously into the dorsal flank with 106 cells suspended in 100 ml of PBS. Some groups of mice were supplemented with SkQ1. Tumor sizes were measured every 3 days and their volumes were calculated as (width)2 × (length) × 0.5. After 3 to 5 weeks of observation, explanted tumors were isolated and analyzed immunohistochemically.
Angiogenesis in Matrigel implants. The effect of SkQ1 on angiogenesis was studied in Matrigel (Matrigel Basement Membrane Matrix; BD Biosciences, USA) implants. Matrigel (0.4 ml at 4°C) was subcutaneously injected into C57 mice (20-22 g). SkQ1 was daily injected intraperitoneally for 10 days. The wet weight of the implants was not affected by SkQ1 treatment. After extraction, implants were divided in two parts. In one part, hemoglobin content was measured with Drabkin reagent. The second part was frozen and 6 mm thick slices were prepared. Vessels were stained with monoclonal antibodies against endothelial antigen CD31 (Pharmingen, USA) and secondary anti-mouse IgG conjugated with Alexa-595 (Molecular Probes). Vessels were counted in four implants for every group.
Tumor vessel count. Tumor tissue was excised from mice killed by cervical dislocation, rinsed in ice-cold PBS, fixed with 10% neutral formalin for 24 h, and embedded into paraffin. Sections (5 µm) were immunostained with monoclonal antibodies to CD34 (Pharmingen), followed by detection with biotin-conjugated secondary antibodies. Bound secondary antibodies were detected with horseradish peroxidase (Dako). Sections were counterstained with hematoxylin and mounted. Vessels and capillaries (identified by positive staining for CD34 and appropriate morphology) were counted. Angiogenic properties were scored as average number of all microvessels, relatively large microvessels (lumen size =100 µm in length), and underdeveloped vessels without lumens. On average, immunohistochemistry quantification was performed by taking pictures from the maximum possible fields per tumor, and imaging at least three tumors per sample set, using AxioVision software (Carl Zeiss Imaging Systems).
RESULTS AND DISCUSSION
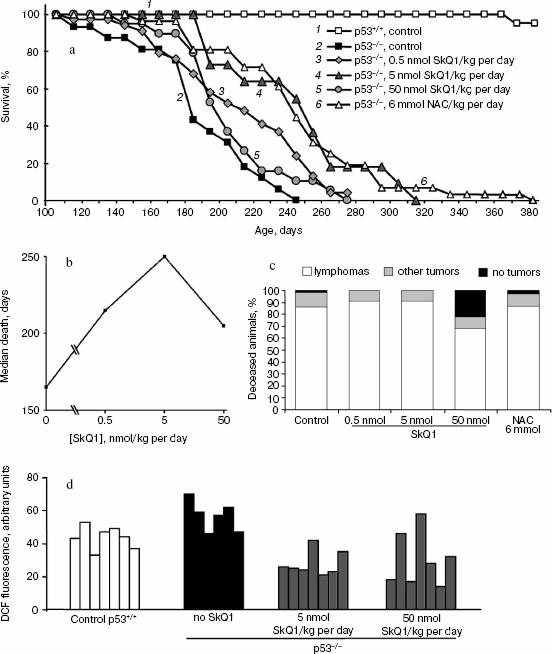
SkQ1 delays appearance of tumors in p53-/- mice. To study possible anti-oncogenic potential of SkQ1 we, first, studied possible effects of diet supplementation with various SkQ1 dosages on tumor development in the p53-/- mice. Such mice show increased levels of ROS [1, 10] and are highly predisposed to the spontaneous development of a variety of neoplasms, starting at about 3 months of age [17, 18]. The predominant types of malignancy are T- and B-cell lymphomas (70-90%). Other forms of tumors include osteosarcoma, rhabdomyosarcoma, hemangiosarcoma, germ cell tumors, and teratoma [17, 18]. We found that 5 nmol SkQ1/kg per day (obtained with drinking water) delayed tumor appearance, and the lifetime of animals was increased by 30-40% (Fig. 1, a and b). Simultaneously, about 2-fold decrease in intracellular level of ROS in mouse tissues, in particular in spleen cells, was observed (Fig. 1d). Fifty percent death for p53-/- mice obtaining no SkQ1 or 5 nmol SkQ1/kg per day occurred on days 180 or 250, respectively. Effect of SkQ1 on early death was especially demonstrative. On day 185, about 60% of the p53-/- mice without SkQ1 died, whereas there was not a single death among the animals who obtained SkQ1 (Fig. 1a). Such effect of extremely low dose of SkQ1 (5 nmol/kg per day) was comparable with the effect of very much higher dose (more than 1,000,000-fold!) of antioxidant NAC (6 mmol/kg per day) found in our previous studies [10, 17] and confirmed in the experiment shown in Fig. 1a. Lower (0.5 nmol/kg per day) and higher (50 nmol/kg per day) SkQ1 dosages were less effective in tumor prevention (Fig. 1, a and b).
Of note, the spectrum of tumors in mice that received 5 nmol SkQ1/kg per day was the same as in control animals, i.e. about 90% of animals in both groups died with signs of lymphomas as revealed by autopsy followed by histological examination of tumor tissues. As seen from Fig. 1c, the use of higher SkQ1 dosage (50 nmol SkQ1/kg per day) decreased the frequency of lymphomas to 68%, but increased the number of deceased animals that showed on autopsy no signs of any tumor development (21%, 4/19) (autopsy of all deceased p53-/- animals without SkQ1 revealed neoplasias).Fig. 1. Effect of various doses of SkQ1 added to the drinking water on lifetime of p53-/- mice (a, b), frequency of lymphomas and other tumors in animals (post mortem analysis) (c), and the ROS content in spleen cells (d). a) Survival curves of control p53+/+ mice (30 animals), p53-/- mice (17 animals), and p53-/- mice receiving 0.5 nmol SkQ1/kg per day (32 animals), 5 nmol SkQ1/kg per day (11 animals), 50 nmol SkQ1/kg per day (19 animals), or 6 mmol NAC/kg per day (26 animals). A log-rank test comparing the group of non-treated p53-/- mice and the groups treated with 5 nmol/kg per day of SkQ1 or NAC showed a two-sided distribution, p < 0.005; b) the median death as a function of SkQ1 dose; c) dead animals were analyzed on autopsy followed by histological examination of tumor tissues; d) total DCF fluorescence of 104 splenocytes obtained from individual spleens.
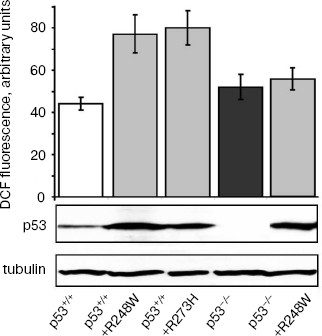
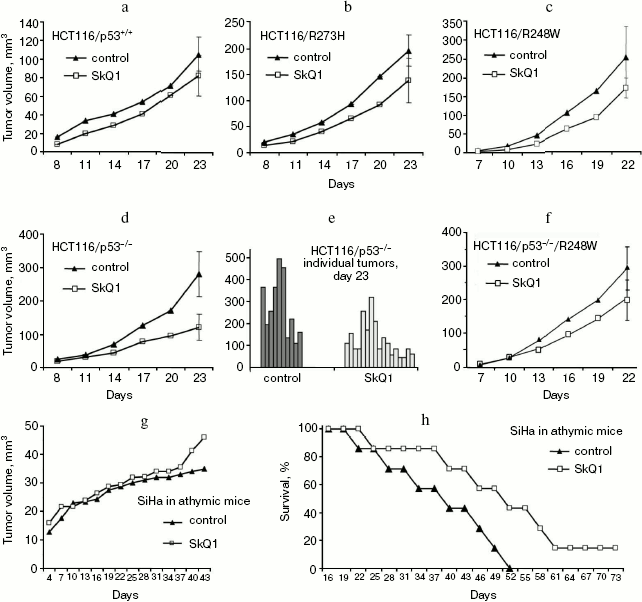
Effect of SkQ1 on growth of xenografts of human tumors in athymic mice. We have studied the effect of SkQ1 on growth of xenografts of human tumors in immunodeficient athymic mice. For this purpose, we used tumor cells differing in p53 status. In particular, we used a set of derivatives of HCT116 colon carcinoma mimicking all situations observed in human colon cancers, i.e. cells expressing wild-type p53 protein, cells with simultaneous expression of wild-type and dominant-negative mutant p53 proteins, and full loss of p53 expression. All the HCT116 cell derivatives with abnormal p53 expression/function showed some increase in ROS levels as compared with the original HCT116 cell line expressing wild-type p53 protein (Fig. 2). Diet supplementation with 5 nmol SkQ1/kg per day caused certain, but statistically non-significant, inhibition of the growth of HCT116 tumors expressing mutant and/or wild-type p53 proteins (Fig. 3, a-c). The inhibitory effect of the used SkQ1 dosage on the growth of HCT116/p53-/- xenografts (cells lacking expression of p53 protein) was more prominent and statistically significant, although individual tumors in both control and SkQ1-treated groups showed rather wide dispersal in their volumes (Fig. 3, d and e). Surprisingly, the xenografts of HCT116/p53-/- derivative that expressed additionally the R248W mutant p53 protein showed lower sensitivity to growth-suppressive effect of SkQ1 (Fig. 3f), although such cells practically did not differ from the original HCT116/p53-/- cells in intracellular ROS level (Fig. 2). (For possible explanation, see below.)
Fig. 2. ROS levels in HCT116 cells differing in p53 expression. Values of the total DCF fluorescence of 104 cells are shown. In each case, the average data of two experiments are presented. Western analysis indicates the p53 and gamma-tubulin contents in different HCT116 sublines.
In addition, we found that the lifetime of SkQ1-treated tumor-bearing immunodeficient mice increased compared with control SkQ1-untreated animals even in those cases when the treatment with SkQ1 showed no significant effect on the growth rate of the tumor xenografts. For example, the diet supplementation with 50 nmol SkQ1/kg per day did not inhibit at all the augmentation in volumes of SiHa human cervical carcinoma xenografts but significantly delayed the death of such animals (Fig. 3, g and h).Fig. 3. Effect of diet supplementation with SkQ1 on growth kinetics of HCT116 human colon carcinoma xenografts differing in p53 state and of SiHa human cervical carcinoma xenografts in athymic mice: a-f) SkQ1 dose, 5 nmol/kg per day; each experimental group contained 8-17 tumors; g, h) effect of SkQ1 (50 nmol/kg per day) on growth kinetics of SiHa xenografts (g) and survival time of animals bearing the tumors (h); each experimental group contained 14 tumors (7 animals, two xenografts per mouse).
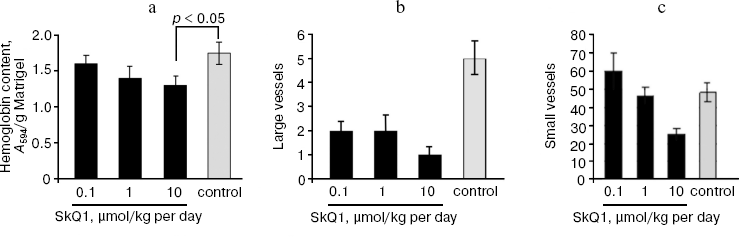
Effect of SkQ1 on tumor angiogenesis. As found earlier in our group, antioxidant NAC inhibited the growth rate of HCT116 xenografts mainly through the inhibition of tumor angiogenesis (tumors treated with NAC showed about 2-fold decrease in the number of typical microvessels and 2-3-fold increase in the number of underdeveloped vessels without lumens) [15]. This is why we decided to study whether SkQ1-induced inhibition of tumor xenograft growth is connected with suppression of tumor angiogenesis. We found that SkQ1 inhibited angiogenesis in vivo in a model of subcutaneous Matrigel implants (Fig. 4). When injected intraperitoneally, SkQ1 significantly decreased the load of implants with blood as well as the content of small blood vessels in the Matrigel, the effect being observed only at the highest dose of SkQ1 used (10 µmol/kg per day, 10 days) (Fig. 4, a and c). The content of relatively large (>40 µm) vessels was strongly decreased even at 0.1 µmol SkQ1/kg per day (Fig. 4b). Angiogenesis in Matrigel implants depends on invasion of the endothelial cells (or their precursors) and further development of the blood vessels. The latter process could be strongly controlled by transcription factor HIF-1alpha, which was activated (stabilized) under hypoxic conditions [40]. It was shown recently that stabilization of HIF-1alpha under hypoxia was mediated by ROS produced in mitochondria of endothelial cells, so this effect was inhibited by mitochondria-targeted antioxidant MitoQ [41]. Our data indicated that in vivo maturation of the vessels was suppressed by low doses of SkQ1, while their invasion was less sensitive.
Immunohistochemical analysis of HCT116/p53-/- tumor xenografts shown that, similar to treatment with NAC, diet supplementation with 5 nmol SkQ1/kg per day caused about 2-fold increase in the number of underdeveloped lumen-free blood vessels but, unlike NAC, caused only minor, statistically non-significant decrease in the number of typical vessels with lumens (Fig. 5).Fig. 4. Effect of SkQ1 on angiogenesis in Matrigel implants. a) Effect of intraperitoneal injection of SkQ1 for 10 days on hemoglobin content of implants. b) Effect of SkQ1 on the number of large (lumen diameter = 40 µm) vessels. c) Effect of SkQ1 on the number of small and capillary blood vessels. Vessels were counted in four implants for every group. In each section, 10-12 fields of vision were analyzed, and the mean values of number of vessels in a field of vision are presented.
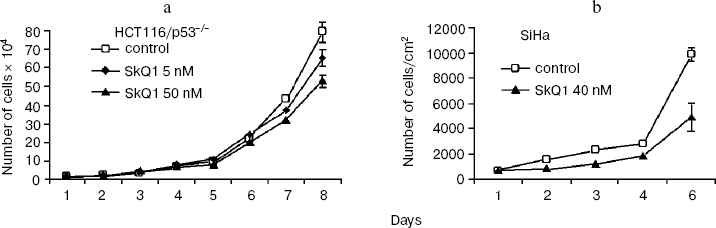
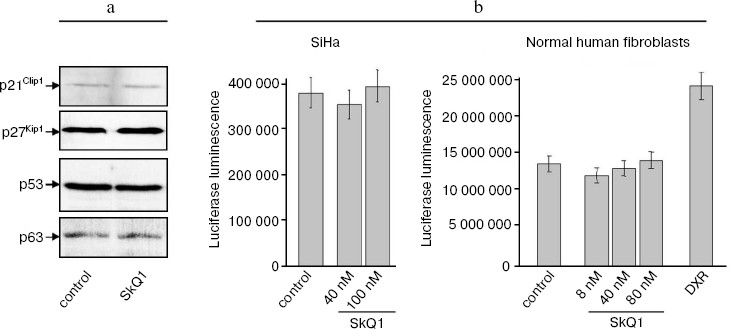
SkQ1 affects the growth and differentiation of tumor cells in vitro. To learn more about possible mechanisms responsible for SkQ1-induced inhibition of growth of HCT116/p53-/- tumor xenografts in athymic mice, we studied how SkQ1 affects the behavior of cell culture in vitro. First, it was found that SkQ1 is able to inhibit the rate of cell proliferation. In fact, growth of the HCT116/p53-/- cells in the culture was inhibited to some degree after incubation with 5-50 nM of SkQ1 for 5-7 days (Fig. 6a). A similar effect was observed in other tested cell cultures, in particular in SiHa cervical carcinoma cells (Fig. 6b). In the same experiments, it was found (Fig. 7a) that SiHa cells treated with 50 nM SkQ1 for 5 days have unchanged levels of the p21Cip1/Waf1 and p27Kip1 proteins, members of the Cip/Kip family of inhibitors of cyclin-dependent kinases. These proteins are known to mediate the growth inhibitory effect of a wide variety of exogenous and endogenous factors [42, 43]. To investigate changes in functional activity of the members of p53 protein family (p53, p63, p73) affecting cell cycle progression not only through activation of p21Cip1/Waf1, but also through some other signaling pathways [17, 32, 44, 45], we studied the effect of SkQ1 on an expression of p53-responsive reporter construct. This experiment showed no significant changes in transcriptional activity of p53 family members both in the cancer and normal cells treated with various doses of SkQ1 (Fig. 7b). In line with this observation, SkQ1-treated SiHa cells showed no changes in content of p53 and p63 proteins (Fig. 7a). Thus, SkQ1-induced growth inhibition seems to be independent of changes in activity of the p53-responsive genes.Fig. 5. Effect of diet supplementing with SkQ1 (5 nmol/kg per day) on angiogenesis in HCT116/p53-/- xenografts. The different types of blood vessels were counted in sections from 3-4 tumors in each group. In each section, 7-12 fields of vision were analyzed; averaged data are presented.
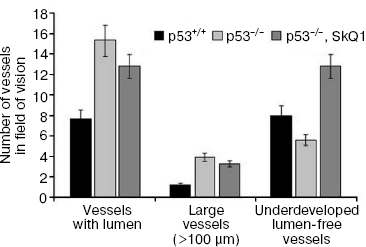
Fig. 6. Effect of SkQ1 on proliferation of the HCT116/p53-/- (a) and SiHa (b) cells in vitro. a) HCT116 cells were plated onto 24-well plates (2⋅104 cells/well) and 5 or 50 nM SkQ1 was added after 5 h. The cells were counted using a hemocytometer (two wells per each experimental point). b) SiHa cells were placed on cover slips in Petri dishes (diameter, 30 mm; 105 cell/dish) and 5 h later were treated with 40 nM SkQ1. Living cells were counted during 5 days. Average data of three experiments are presented.
Seemed possible that the SkQ1-induced inhibition of proliferation of carcinoma cells might be connected with the ability of SkQ1 to affect differentiation state of neoplastic epithelial cells and, as a result, to modulate pathways whose activity, on one hand, is dependent on expression of some differentiation markers and, on the other hand, can regulate cell cycle progression (the well-known example of such pathway is E-cadherin/beta-catenin/Cdk signaling [46, 47]). In fact, we found that treatment with SkQ1 induced in HCT116/p53-/- and SiHa carcinoma cells changed cell morphology and expression of E-cadherin, suggesting stimulation of epithelial cell differentiation.Fig. 7. Effect of SkQ1 on content of p21Cip1/Waf1, p27Kip1, p53, and p63 proteins (a) and expression of p53-responsive p53RE-Luc reporter construct transcription (b) in SiHa cervical carcinoma cells and normal human fibroblasts. a) 50 nM SkQ1 was added to SiHa cells for 5 days; Western analysis with corresponding antibodies was performed as described in “Materials and Methods”. b) SiHa carcinoma cells and normal fibroblasts were transfected with p53-responsive pWWp-GL2 reporter construct and treated with various doses of SkQ1 for 5 days, and then luciferase luminescence of cell lysates was estimated as described in “Materials and Methods”. As a positive control for p53-induced activation of reporter expression, a treatment with doxorubicin (DXR, 100 ng/ml for 5 days) was used.
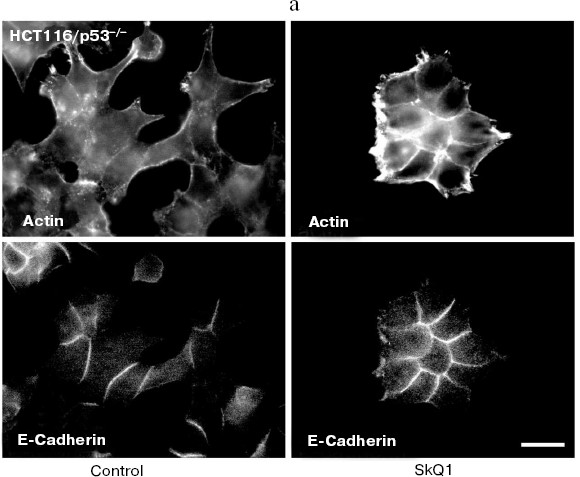
It is well established that in normal epithelial cells, actin bundles are present as circumferential rings in peripheral regions of the cell. A less abundant population of actin bundles also appears to terminate at sites of E-cadherin-positive cell-cell contacts [48]. E-Cadherin plays a causal role in the establishment and maintenance of the differentiated epithelial phenotype. Numerous cellular changes take place during epithelial malignancy, and disruption of E-cadherin-based cell-cell adhesion is a major event. The small GTPases of Rho family have been implicated in multiple steps of cellular transformation, including alterations of cell adhesions [49]. HCT116/p53-/- cells do not show normal epithelial type of cytoskeletal organization. Treatment of these cells with SkQ1 restored the normal epithelial features (Fig. 8a; see also colored inset, Fig. 8b). Actin cytoskeleton became more organized and filaments at the cell periphery and at cell-cell contacts were well pronounced. Immunostaining of E-cadherin revealed a formation of prolonged E-cadherin-positive contacts. As a result, the cells treated with SkQ1 formed typical epithelial islets in culture, which were absent in untreated cells.
Fig. 8. Actin filaments and E-cadherin-positive intercellular contacts in control and SkQ1-treated (40 nM for 7 days) HCT116/p53-/- cell cultures. Bar, 20 µm. a) Immunostaining with specific antibodies. b) See color insert.
Also we have studied several epithelioid cell lines originating from carcinoma of uterine cervix (CaSki, SiHa, C4I, HeLa). These cells contain wild-type p53 but show deregulated p53-dependent signaling due to infection with papilloma viruses HPV16 or -18 and interaction of viral E6 oncoprotein with p53. One of these cell lines, SiHa (contains integrated HPV16), was studied in detail (Fig. 9). These cells showed disorganized system of actin filaments and the absence of organized intercellular E-cadherin-positive contacts. Treatment with SkQ1 increased the total amount of E-cadherin (Fig. 9b) and restored actin bundles and well-organized intercellular contacts of SiHa cells. Morphology of these cells and their islets became almost indistinguishable from normal keratinocytes (Fig. 9a; see also color insert, Fig. 9c). The morphology of non-transformed keratinocyte line HaCaT was not significantly affected by SkQ1 (Fig. 9d; see color insert). Phenotype reversion (normal epithelial-like morphology restoration) by SkQ1 treatment was observed also with other carcinoma cell lines (CaSki, C4I, HeLa; data not shown). Of note, well known carcinoma HeLa cells which are strongly dedifferentiated and lost almost all epithelial features, became organized in a more epithelial fashion after treatment with SkQ1. Our data are in line with previously published results on NAC-induced apical-basolateral differentiation of colon and ovary carcinoma cell lines [50].Fig. 8b. Actin filaments and E-cadherin-positive intercellular contacts in control and SkQ1-treated (40 nM for 7 days) HCT116/p53-/- cell cultures. Colocalization of actin and E-cadherin. Bar, 20 µm.
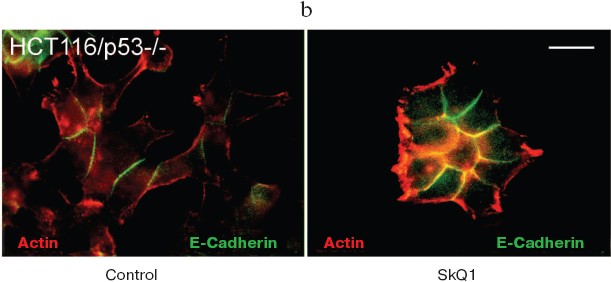
Fig. 9. Effect of incubation with SkQ1 (40 nM for 7 days) on morphology and cytoskeleton of SiHa cell culture. a) Restoration of epithelial morphology and epithelial-like islets after incubation with SkQ1. Phase contrast microscopy; immunostaining of actin filaments, beta-catenin, and E-cadherin-positive intercellular contacts. Bar, 20 µm. b) Effect of incubation with SkQ1 on E-cadherin protein content as revealed by Western analysis. c, d) See color insert.
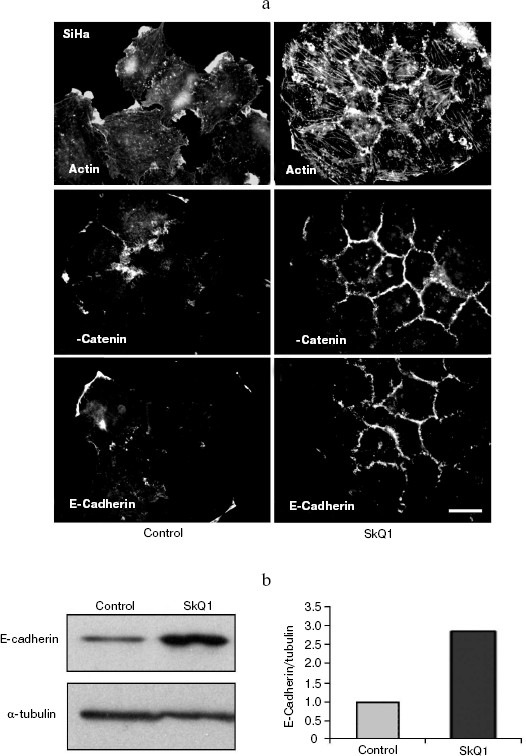
SkQ1 reverses morphological transformation of Ras- and SV40-transformed p53-/- fibroblasts. The effect of SkQ1 on transformed fibroblasts was studied in mouse fibroblast cell line 10(3) with deleted p53 [35]. These cells can be considered as minimally transformed cells. They are non-tumorigenic and retain basic morphological features of normal fibroblasts. The cells were dramatically changed upon transformation by oncogene N-RasAsp13. Transformation was accompanied by strong decrease in cell area (Fig. 10). A major feature of cell transformation during malignization consists in the loss and/or reorganization of actin structures, which leads to enhanced cell motility and invasion [51]. Ras induced dissipation of actin stress fibers and related vinculin-positive focal adhesions in 10(3) fibroblasts (Fig. 10a; see also color insert, Fig. 10b). This effect of Ras is caused, at least in part, by inactivation of the Rho-ROCK-LIMK kinase signaling mediating cofilin phosphorylation and stress fiber formation [52, 53]. Recently we have shown that ROS up-regulation was critical for Ras-induced modulation of cofilin activities responsible for reorganization of cell architecture and actin cytoskeleton [9]. Treatment of the Ras-transformed cells with SkQ1 caused restoration of normal fibroblastic morphology, i.e. re-appearance of the stress fibers and focal adhesion contacts, and enlargement of cell area (Fig. 10, c-e). Of note, original 10(3) fibroblasts, which did not expressed activated Ras, were also affected by SkQ1. Their area was increased and stress fibers as well as focal contacts became more pronounced (Fig. 10, d and e). The changes of the actin system in the SkQ1-treated cells were accompanied with an increase in the level of phosphorylated cofilin (not shown). Similar phenotype reversion was observed in SkQ1-treated SV40-transformed human fibroblasts MRC5-V2 (Fig. 11).Fig. 9 (c, d). Effect of incubation with SkQ1 (40 nM for 7 days) on morphology and cytoskeleton of SiHa cells (c) and non-transformed epithelial cells HaCaT (d). Bar, 20 µm.
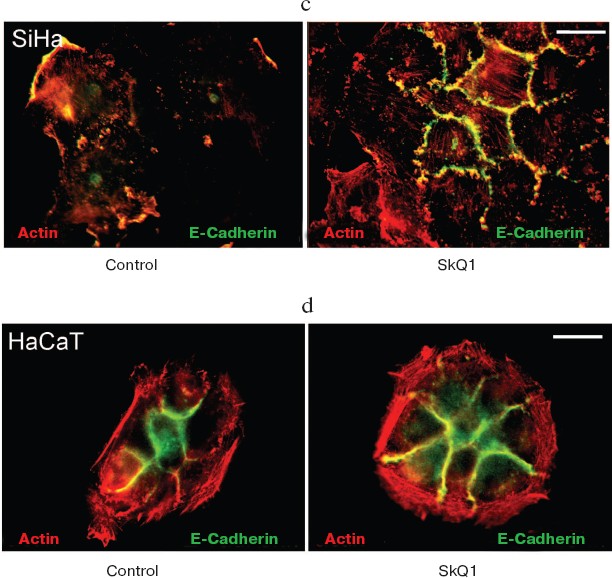
Fig. 10. Reversal of the Ras-induced morphological transformation in 10/3 mouse fibroblasts incubated with SkQ1. The cells were incubated with 20 nM SkQ1 for 7 days. a) Actin filaments and vinculin-positive focal contacts in the control and SkQ1-treated cells. Bar, 20 µm. b) See color insert. c) Number of focal contacts per cell. d) Effect of SkQ on cell shape. e) Measurements of cell area.
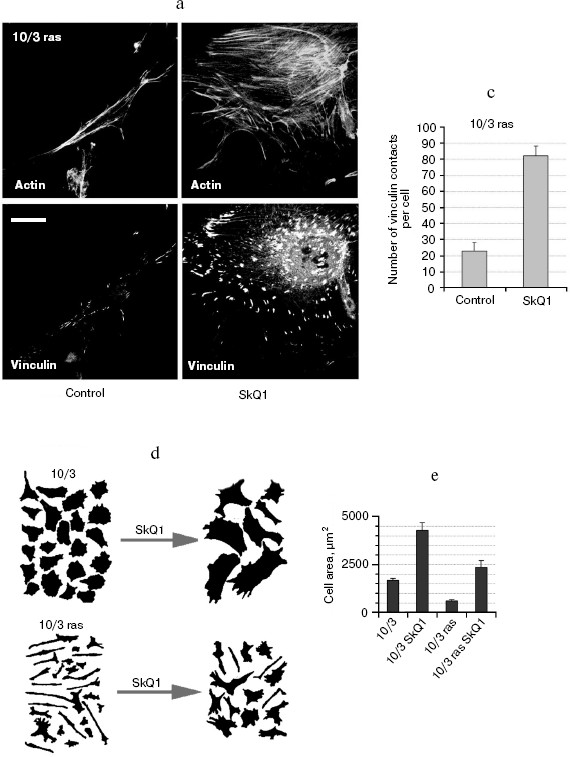
Fig. 10b. Reversal of Ras-induced morphological transformation in 10/3 mouse fibroblasts incubated with SkQ1. The cells were incubated with 20 nM SkQ1 for 7 days. Colocalization of actin and vinculin. Bar, 20 µm.
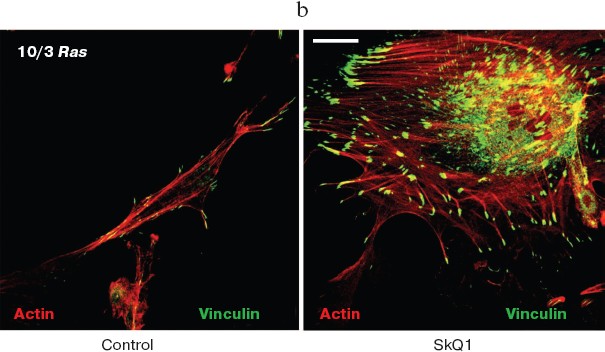
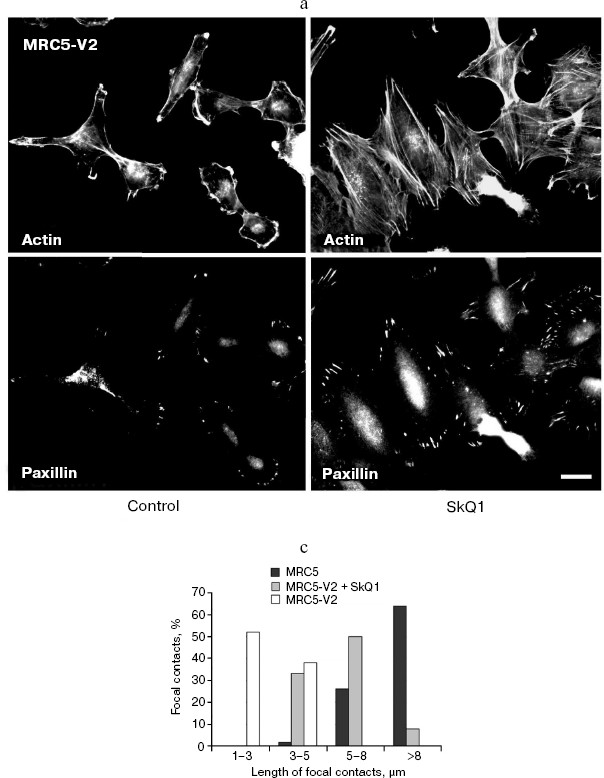
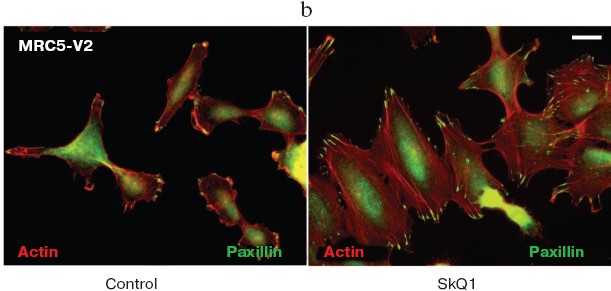
Fig. 11. Reversal of morphological transformation in SkQ1-treated SV-40-transformed MRC5-V2 human fibroblasts. Cells were treated with 20 nM SkQ1 for 7 days. a) Actin filaments and paxillin-positive focal contacts of the control and SkQ1-treated cells. Bar, 20 µm. b) See color insert. c) Distribution of focal contacts of different length.
Accumulation of SkQ1 in tumor cells is dependent on p53-regulated activity of P-glycoprotein pump. To reveal the factors determining differential response of p53-negative and mutant p53-positive HCT116 xenografts to treatment with SkQ1, we studied intracellular accumulation of the drug in different cell cultures. For this purpose, we used SkQR1 [1, 24], a fluorescent analog of SkQ1, bearing rhodamine-19 cationic group instead triphenylphosphonium cation in SkQ1. It was found that alterations of p53 expression were accompanied by some changes in the accumulation of the drug. The pattern of these changes, in particular some decrease of the drug accumulation in derivatives of HCT116 and HCT116/p53-/- cells expressing exogenous mutant R248W p53 protein (Fig. 12a), was consistent with the idea that transport of SkQR1 is carried out by the P-glycoprotein pump, the product of the MDR1 gene whose hyperfunction leads to effective extrusion from cells of a wide variety of hydrophobic positively charged molecules and, as a result, to development of so-called multiple drug resistance (MDR) [54, 55]. In fact, it was reported that oncogenic mutant p53 proteins can up-regulate MDR1 gene expression [56, 57].Fig. 11b. Reversal of morphological transformation in SkQ1-treated SV-40-transformed MRC5-V2 human fibroblasts. Cells were treated with 20 nM SkQ1 for 7 days. Colocalization of actin and paxillin. Bar, 20 µm.
To prove the suggestion that activity of P-glycoprotein decreases intracellular accumulation of SkQs, we studied K-562 erythroleucemia cell line and its multidrug-resistant derivative K-562-MDR selected for resistance to doxorubicin [34]. Over-expression of the MDR1 gene encoding P-glycoprotein was confirmed by Western analysis (N. S. Melik-Nubarov and coworkers, unpublished). It was found that accumulation of SkQR1 in the K-562-MDR cells was decreased approximately 10 times compared with the original K-562 cells (Fig. 12b). Treatment with inhibitors of P-glycoprotein (verapamil and pluronic L61 [58]) increased SkQR1 accumulation in K-562-MDR cells, but not in the parental K-562 cells (Fig. 12b), which is consistent with participation of P-glycoprotein in extrusion of SkQs.Fig. 12. Dependence of intracellular accumulation of SkQR1 on functioning of the P-glycoprotein pump. a) HCT116 and HCT116/p53-/- cell derivatives expressing exogenous R248W mutant p53 protein were loaded with 50 nM SkQR1 for 2 h, and after washing they were incubated for 3 h. Fluorescence of 104 cells is shown. In each case, the average data of two experiments are presented. b) K-562 and K-562-MDR cells were loaded with 50 nM SkQR1 for 1.5 h, and after washing they were incubated for 0.5 h. Inhibitors of P-glycoprotein verapamil (Ver, 50 µM) or pluronic L61 (60 µg/ml) were added to the incubation mixture simultaneously with SkQR1 and then to the washing medium. The data from five experiments are presented. c) K-562 and K-562-MDR cells were treated for 20 h with 100 and 300 µM H2O2, respectively, to reach similar viability. Where indicated, SkQR1 (10 nM) and/or pluronic L61 (5 µg/ml) were preincubated with cells for 6 days.
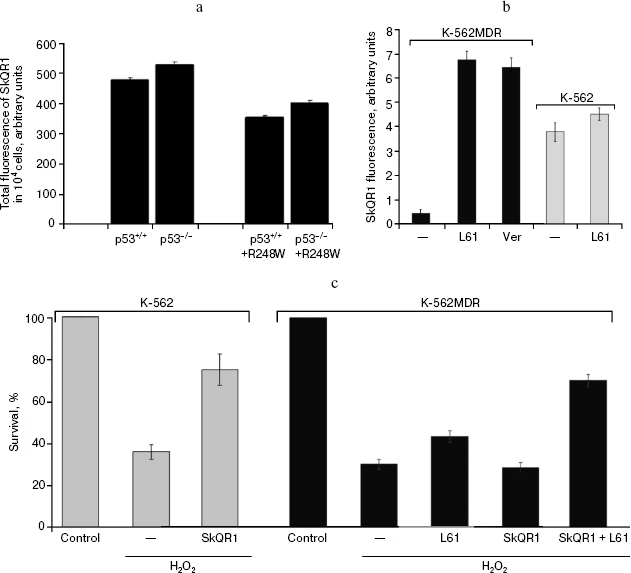
Finally, we have shown that P-glycoprotein activity modulated the protective antioxidant activity of SkQR1, which depends on its accumulation in mitochondria. Apoptosis of K-562 cells induced by H2O2 (which is not a substrate of the MDR pump) was blocked by low concentration of SkQR1. The same SkQR1 concentration did not protect K-562-MDR cells until inhibitors of P-glycoprotein were added (Fig. 12c). A similar effect was observed with SkQ1, indicating that this cationic antioxidant was also a good substrate of P-glycoprotein (not shown).
Concluding remarks. Discovery of a strong antiapoptotic effect of SkQs, described in the first paper of the series [24], brings up an important question whether such compounds possess carcinogenic activity. This question seems relevant since certain types of apoptosis are known to operate as anticancer defense mechanisms. The above results clearly show that SkQs are not carcinogens. Even more, SkQ1 showed an anticancer effect at least in cells deficient in p53, i.e. in a situation typical for many kinds of carcinogenesis. In fact, we found that SkQ1 suppressed spontaneous tumorigenesis in p53-/- mice (Fig. 1) as well as HCT116/p53-/- tumor xenograft growth in athymic mice (Figs. 2 and 3). Apparently, mitochondrial ROS-induced apoptosis, which is specifically inhibited by SkQs, does not participate in the anticancer defense. Moreover, mitochondrial ROS are most probably carcinogenic, so their scavenging by SkQ1 can account for the anticancer effect of our compound.
Our findings suggest that there are several aspects in physiology of tumors that critically depend on mitochondrial ROS production.
First, scavenging of mitochondrial ROS with SkQ1 inhibited proliferation of tumor cells (Fig. 6). This effect was not connected with activation of p53 function and/or up-regulation of expression of p21Cip1/Waf1 and p27Kip1, inhibitors of cyclin-dependent kinases (Fig. 7). It was probably mediated by suppression of some mitogen-activated pathways. In fact, an important role of ROS in transduction of various mitogenic signals and in activation of MAP kinases in particular, is well established [4, 8]. Now we can conclude that mitochondria are important players in this scenario.
Second, SkQ1 initiated differentiation processes in various tumor cells. The coordinated changes in morphology, cytoskeleton, organization of intercellular contacts and focal adhesion contacts were observed in epithelial tumor cells as well as in Ras-transformed fibroblasts (Figs. 8-10). Mitochondrial ROS hyperproduction probably stimulates the Ras-dependent signaling promoting transformed phenotype. Increase in differentiation state of tumor cells by SkQ1 could be related to decrease in their invasiveness and in the general aggressiveness of the tumor. This effect could be one possible explanation why survival of tumor-bearing animals was increased by SkQ1 even in the case when the rate of tumor growth was not affected (Fig. 3h). Another explanation might be that SkQ1 somehow increases resistance of the organism to the factors responsible for the death of athymic mice due to opportunistic infections, intoxication, etc. Within the framework of such an assumption, we should take into account that SkQs seem to be competent in interrupting execution of program of the “biochemical suicide” (phenoptosis) initiated in critical situations [1, 25, 27, 59].
Finally, mitochondrial ROS could be important for tumor angiogenesis. Strong suppression of angiogenesis by SkQ1 was observed in Matrigel implants (Fig. 4) but was less pronounced in tumor xenografts (Fig. 5). Angiogenesis in Matrigel implants probably depended on hypoxia-induced stabilization of HIF-1alpha that was mediated by mitochondrial ROS [41] and prevented by SkQ1. In a tumor, xenograft maturation of the vessels was also partially suppressed by SkQ1 (Fig. 5). A smaller effect of SkQ1 in this case might be explained by lower SkQ1 concentrations used in the xenograft experiments compared with those in Matrigel.
The antitumor activity of SkQ1 (and related positively charged antioxidants) could be limited in the tumors with high level of P-glycoprotein multidrug resistance pump, as was found here in the case of HCT116 xenografts bearing mutant forms of p53. However, it can be proposed that the extrusion of SkQ1 from the tumor cells could be used for the sake of anticancer therapy. For example, mitochondria-targeted antioxidants could protect normal cells (with low level of MDR) against killing by X-ray, photodynamic, or chemotherapy mediated by severe oxidative stress, while killing of tumor cells (with high level of MDR) will not be affected.
Similar effects on tumor growth and tumor cells phenotype were observed with the traditional antioxidant NAC [9, 16], but the efficient NAC dose (6 mmol/kg per day) was more than million times higher than efficient SkQ1 dose (5 nmol/kg per day). Such a high NAC concentration can hardly be recommended for any therapeutic purpose due to toxic side effects like NAC-induced S-glutathionylation and inactivation of p65-NFkappaB [60, 61]. Extremely high efficiency of SkQ1 could be explained by specific targeting of SkQ1 to mitochondria, its accumulation in membranes rather than in aqueous phase, and rechargeable nature of its antioxidant functioning [1, 24]. Moreover, the effect of SkQ1 can be understood only if mitochondrial ROS production is critical for the studied tumorigenesis. This conclusion is in good agreement with the general concept of mitochondrial malfunctioning in rapidly growing highly glycolytic tumors.
We are very grateful to Professor V. A. Sadovnichii, Rector of Moscow State University, for his interest in the project and encouragement. We thank M. L. Domninskaya, D. S. Izyumov, and K. G. Lyamzaev for participation in certain experiments. Generous support of Mr. O. V. Deripaska, which in fact made possible this study, is greatly appreciated.
Supported by Mitotechnology LLC, Lomonosov Moscow State University, the Vol'noe Delo Foundation (grant No. 99F-06), Russian Foundation for Basic Research (grant Nos. 07-04-00335, 06-04-49214, and 08-04-01165), Russian Ministry of Education and Science (grant “Leading Scientific Schools” No. 5762.2008.4).
REFERENCES
1.Skulachev, V. P. (2007) Biochemistry
(Moscow), 72, 1385-1396.
2.Hussain, S. P., Hofseth, L. J., and Harris, C. C.
(2003) Nat. Rev. Cancer, 3, 276-285.
3.Wallace, D. C. (2005) Cold Spring Harbor Symp.
Quant. Biol., 70, 363-374.
4.Wu, W. S. (2006) Cancer Metastasis Rev.,
25, 695-705.
5.Blanchetot, C., and Boonstra, J. (2008) Crit.
Rev. Eukaryot. Gene Expr., 18, 35-45.
6.Kopnin, B. P. (2007) Mol. Biol. (Moscow),
41, 369-380.
7.Giles, G. I. (2006) Curr. Pharm. Des.,
12, 4427-4443.
8.Nagai, H., Noguchi, T., Takeda, K., and Ichijo, H.
(2007) J. Biochem. Mol. Biol., 40, 1-6.
9.Alexandrova, A. Y., Kopnin, P. B., Vasiliev, J. M.,
and Kopnin, B. P. (2006) Exp. Cell Res., 312,
2066-2073.
10.Sablina, A. A., Budanov, A. V., Ilyinskaya, G.
V., Agapova, L. S., Kravchenko, J. E., and Chumakov, P. M. (2005)
Nat. Med., 11, 1306-1313.
11.Kopnin, P. B., Agapova, L. S., Kopnin, B. P., and
Chumakov, P. M. (2007) Cancer Res., 67, 4571-4578.
12.Vafa, O., Wade, M., Kern, S.,
Beeche, M., Pandita, T. K., Hampton, G. M., and Wahl,
G. M. (2002) Mol. Cell., 9, 1031-1044.
13.Irani, K., Xia, Y., Zweier, J. L., Sollott, S.
J., Der, C. J., Fearon, E. R., Sundaresan, M., Finkel, T., and
Goldschmidt-Clermont, P. J. (1997) Science, 275,
1649-1652.
14.Gerald, D., Berra, E., Frapart, Y. M., Chan, D.
A., Giaccia, A. J., Mansuy, D., Pouyssegur, J., Yaniv, M., and
Mechta-Grigoriou, F. (2004) Cell, 118, 781-794.
15.Khromova, N. V., Kopnin, P. B., Stepanova, E. V.,
Agapova, L. S., and Kopnin, B. P. (2008) Cancer Lett., submitted
(Ms#CAN-S-08-00360).
16.Xia, C., Meng, Q., Liu, L. Z., Rojanasakul, Y.,
Wang, X. R., and Jiang, B. H. (2007) Cancer Res., 67,
10823-10830.
17.Kopnin, B. P., Khromova, N. V., Kopnin, P. B.,
and Agapova, L. S. (2008) Clin. Oncohemat., 1, 3-11.
18.Carcamo, J. M., and Golde, D. W. (2006) Mutat.
Res., 593, 64-79.
19.Skulachev, V. P. (1996) FEBS Lett.,
397, 7-10.
20.Skulachev, V. P. (1996) Quart. Rev.
Biophys., 29, 169-202.
21.Skulachev, V. P. (2003) in Topics in Current
Genetics, Vol. 3 (Nystrom, T., and Osiewac, H. D., eds.)
Springer-Verlag, Berlin-Heidelberg, pp. 191-238.
22.Zhang, D. X., and Gutterman, D. D. (2007) Am.
J. Physiol. Heart Circ. Physiol., 292, H2023-2031.
23.Scherz-Shouval, R., and Elazar, Z. (2007)
Trends Cell Biol., 17, 422-427.
24.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L.
E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Yu.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Yu., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Yu., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1273-1287.
25.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. V., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashook, E. V., Studneva, I. M., Tskitishvili, O. V., Vasileva,
A. K., Victorov, I. V., Zorov, D. B., and Skulachev, V. P. (2008)
Biochemistry (Moscow), 73, 1288-1299.
26.Neroev, V. V., Archipova, M. M., Bakeeva, L. E.,
Fursova, A. Zh., Grigorian, E. N., Grishanova, A. Yu., Iomdina, E. N.,
Ivashchenko, Zh. N., Katargina, L. A., Khoroshilova-Maslova, I. P.,
Kilina, O. V., Kolosova, N. G., Kopenkin, E. P., Korshunov, S. S.,
Kovaleva, N. A., Novikova, Yu. P., Philippov, P. P., Pilipenko, D. I.,
Robustova, O. V., Saprunova, V. B., Senin, I. I., Skulachev, M. V.,
Sotnikova, L. F., Stefanova N. A., Tikhomirova, N. K., Tsapenko, I. V.,
Schipanova, A. I., Zinovkin, R. A., and Skulachev, V. P. (2008)
Biochemistry (Moscow), 73, 1317-1328.
27.Anisimov, V. N., Bakeeva, L. E., Egormin, P. A.,
Filenko, O. F., Isakova, E. F., Manskikh, V. N., Mikhelson, V. M.,
Panteleeva, A. A., Pasyukova, E. G., Pilipenko, D. I., Piskunova, T.
S., Popovich, I. G., Roshchina, N. V., Rybina, O. Yu., Samoylova, T.
A., Saprunova, V. B., Semenchenko, A. V., Skulachev, M. V., Spivak, I.
M., Tsybul'ko, E. A., Tyndyk, M. L., Vyssokikh, M. Yu., Yurova, M. N.,
Zabezhinsky, M. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1329-1342.
28.Donehower, L. A., Harvey, M., Slagle, B. L.,
McArthur, M. J., Montgomery, C. A., Jr., Butel, J. S., and Bradley, A.
(1992) Nature, 356, 215-221.
29.Jacks, T., Remington, L., Williams, B. O.,
Schmitt, E. M., Halachmi, S., Bronson, R. T., and Weinberg, R. A.
(1994) Curr. Biol., 4, 1-7.
30.Soussi, T. (2005) in 25 Years of p53
Research (Hainaut, P., and Wiman, K. G., eds.) Springer,
Dordrecht-Berlin-Heidelberg-New York, pp. 255-292.
31.Shi, H., Calvez, F. L., Olivier, M., and Hainaut,
P. (2005) in 25 Years of p53 Research (Hainaut, P., and Wiman,
K. G., eds.) Springer, Dordrecht-Berlin-Heidelberg-New York, pp.
293-320.
32.Chumakov, P. M. (2007) Biochemistry
(Moscow), 72, 1399-1421.
33.Bunz, F., Dutriaux, A., Lengauer, C., Waldman,
T., Zhou, S., Brown, J. P., Sedivy, J. M., Kinzler, K. W., and
Vogelstein, B. (1998) Science, 282, 1497-1500.
34.Kalinina, E. V., Chernov, N. N., Saprin, A.
N., Kotova, Y. N., Andreev, Y. A., Solomka, V. S., and Scherbak, N. P.
(2006) Biochemistry (Moscow), 71, 1200-1206.
35.Kopnin, B. P., Stromskaya, T. P., Kondratov, R.
V., Ossovskaya, V. S., Pugacheva, E. N., Rybalkina, E. Y., Khokhlova,
O. A., and Chumakov, P. M. (1995) Oncol. Res., 7,
299-306.
36.Huschtscha, L. I., and Holliday, R. (1983) J.
Cell Sci., 63, 77-99.
37.Kopnin, P. B., Kravchenko, I. V., Furalyov, V.
A., Pylev, L. N., and Kopnin, B. P. (2004) Oncogene, 23,
8834-8840.
38.Sablina, A. A., Chumakov, P. M., and Kopnin, B.
P. (2003) J. Biol. Chem., 278, 27362-27371.
39.Logunov, D. Y., Ilyinskaya, G. V., Cherenova, L.
V., Verhovskaya, L. V., Shmarov, M. M., Chumakov, P. M., Kopnin, B. P.,
and Naroditsky, B. S. (2004) Gene Ther., 11, 79-84.
40.Hirota, K., and Semenza, G. L. (2006) Crit.
Rev. Oncol. Hematol., 59, 15-26.
41.Bell, E. L., Klimova, T. A., Eisenbart, J.,
Moraes, C. T., Murphy, M. P., Budinger, G. R., and Chande, N. S. (2007)
J. Cell Biol., 177, 1029-1036.
42.Sherr, C. J., and Roberts, J. M. (1999) Genes
Dev., 13, 1501-1512.
43.Mainprize, T. G., Taylor, M. D., Rutka, J. T.,
and Dirks, P. B. (2001) J. Neurooncol., 51, 205-218.
44.Levine, A. J., Hu, W., and Feng, Z. (2006)
Cell Death Differ., 13, 1027-1036.
45.Harms, K., Nozell, S., and Chen, X. (2004)
Cell Mol. Life Sci., 61, 822-842.
46.Pugacheva, E. N., Roegiers, F., and Golemis, E.
A. (2006) Curr. Opin. Cell Biol., 18, 507-515.
47.Brembeck, F. H., Rosario, M., and Birchmeier, W.
(2006) Curr. Opin. Genet. Dev., 16, 51-59.
48.Zhang, J., Betson, M., Erasmus, J., Zeikos, K.,
Bailly, M., Cramer, L. P., and Braga, V. M. M. (2005) J. Cell
Sci., 118, 5549-5562.
49.Lozano, E., Betson, M., and Braga, V. M. M.
(2003) BioEssays, 25, 452-463.
50.Parasassi, T., Brunelli, R., Bracci-Laudiero, L.,
Greco, G., Gustafsson, A. C., Krasnowska, E. K., Lundeberg, J.,
Lundeberg, T., Pittaluga, E., Romano, M. C., and Serafino, A. (2005)
Cell Death Differ., 12, 1285-1296.
51.Campbell, P. M., and Der, C. J. (2004) Semin.
Cancer Biol., 14, 105-114.
52.Lee, S., and Helfman, D. M. (2004) J. Biol.
Chem., 279, 1885-1891.
53.DesMarais, V., Ghosh, M., Eddy, R., and
Condeelis, J. (2005) J. Cell Sci., 118, 19-26.
54.Stavrovskaya, A. A. (2000) Biochemistry
(Moscow), 65, 95-106.
55.Glavinas, H., Krajcsi, P., Cserepes, J., and
Sarkadi, B. (2004) Curr. Drug Deliv., 1, 27-42.
56.Thottassery, J. V., Zambetti, G. P., Arimori, K.,
Schuetz, E. G., and Schuetz, J. D. (1997) Proc. Natl. Acad. Sci.
USA, 94, 11037-11042.
57.Sampath, J., Sun, D., Kidd, V. J., Grenet, J.,
Gandhi, A., Shapiro, L. H., Wang, Q., Zambetti, G. P., and Schuetz, J.
D. (2001) J. Biol. Chem., 276, 39359-29367.
58.Kabanov, A. V., Batrakova, E. V., and
Alakhov, V. Y. (2002) Adv. Drug Deliv. Rev., 54,
759-779.
59.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
60.Qanungo, S., Wang, M., and Nieminen, A. L. (2004)
J. Biol. Chem., 279, 50455-50464.
61.Qanungo, S., Starke, D. W., Pai, H. V., Mieyal,
J. J., and Nieminen, A. L. (2007) J. Biol. Chem., 282,
18427-18436.