REVIEW: Nanocolonies: Detection, Cloning, and Analysis of Individual Molecules
H. V. Chetverina* and A. B. Chetverin
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: helena@vega.protres.ru* To whom correspondence should be addressed.
Received March 23, 2008; Revision received May 5, 2008
Nanocolonies (other names molecular colonies or polonies) are formed upon template nanomolecule (DNA or RNA) amplification in immobilized medium with efficient pore size in the nanometer range. This work deals with the principle, invention, development, and diverse nanocolony applications based on their unique abilities to compartmentalize amplification and expression of individual DNA and RNA molecules, including studying reactions between single molecules, digital molecular diagnostics, in vitro gene cloning and expression, as well as identification of the molecular cis-elements including DNA sequencing, analysis of single-nucleotide polymorphism, and alternative splicing investigation.
KEY WORDS: molecular colonies, polonies, RNA recombination, RNA self-recombination, molecular cloning, gene expression, molecular diagnostics, DNA sequencing, single-nucleotide polymorphism, alternative splicing, fluorescence detection, detection in real timeDOI: 10.1134/S0006297908130014
Abbreviations: AML1-ETO, chimeric sequence consisting of parts of the AML1 and ETO genes; 3D-pol, gene of poliovirus RNA polymerase; FRET, fluorescent (Forster's) resonance energy transfer; GFP, green fluorescent protein; HBV, hepatitis B virus; HIV, human immune deficiency virus; PCR, polymerase chain reaction; Qβ replicase, RNA-dependent RNA polymerase of Qβ bacteriophages; RCA, rolling circle amplification; RQ RNA, a special class of RNA exponentially amplified by Qβ replicase; RT-PCR, reverse transcription followed by polymerase chain reaction; SNP, single nucleotide polymorphism; Taq DNA polymerase, DNA-dependent DNA polymerase of Thermus aquaticus; Tth DNA polymerase, DNA-dependent DNA polymerase of Thermus thermophilus.
Molecular colonies are formed when template nucleic acids (DNA and RNA)
are amplified using template-dependent polymerases not in a liquid
medium as usual but in an immobilized medium like a gel, etc.
Dimensions of polynucleotide molecules that form colonies are in the
nanometer range, i.e. RNA and DNA are nanomolecules. Moreover, the gel
matrix forms a three-dimensional network with the pore size also in the
nanometer range. These two facts allow us to call molecular colonies
nanomolecular colonies or nanocolonies.
The use of an immobilized medium is the key element because the gel matrix prevents convection of the medium and retards diffusion of nanomolecules. As a result, the progeny of each initial nanomolecule is not spread over the whole reaction volume and concentrates in a limited zone around the parental molecule, i.e., it forms a colony. On the other side, the template network does not influence diffusion of smaller molecules--the reaction substrates (nucleoside triphosphates, oligonucleotide primers) and buffer components. Owing to this, reaction within the gel does not suppress amplification of nucleic acids.
In the case of a thin layer gel, nanocolonies are arranged in one focal plane, which facilitates their observation and screening. The number of molecules in a colony depends on the efficiency of the amplification enzyme system and reaction time (the number of amplification cycles), whereas the colony size and density of nanomolecule packing in it are defined by the size of amplified molecules and gel density (pore size). Regulation of these parameters always makes possible to amplify an individual template nanomolecule to amounts detectable by the used recording device. Large nanocolonies become visible by a naked eye after special treatment. Small nanocolonies enable screening of numerous template nanomolecules on a small gel surface, but the use of a microscope is necessary in this case.
Since practically each nanocolony originates from a single template molecule, the use of this method makes possible detection, counting, and identification of single DNA or RNA molecules. Besides, since nanocolonies are descendants of individual template molecules (molecular clones) they make possible cloning pure genetic material and direct clone screening. The amplified DNA or RNA may serve as templates for synthesis of other nanomolecules, proteins, whose properties can be investigated directly in colonies that, unlike cells, are not surrounded by the membrane.
The method of obtaining nanocolonies was first published in 1991 in our article on the nature of “spontaneous” RNA synthesis [1]. We showed that it is possible to grow RNA colonies and used them for detection in the air of single replicating RNA molecules. Simultaneously, we suggested growing DNA colonies and the use of nucleic acid colonies for studying chemical reactions between single molecules, molecular diagnostics, and in vitro gene cloning and expression. These trends are described in our patents for nanocolonies, which were obtained in Russia and the USA [2-7].
In subsequent works, we named our method MCT (Molecular Colony Techniques).
For 10 years, beginning from 1989, this method was developed owing to efforts of members of our laboratory. The situation changed when G. Church of Harvard University (Boston, USA) began to use it in his laboratory. In 1999, R. Mitra and G. Church published it without reference to our works as a method of “polonies” (i.e. polymerase colonies), where PCR (polymerase chain reaction) in DNA colonies [10], almost word for word, reproduced our method described in patent pendings [2-7] applied for as early as 1992. They introduced just a single insignificant modification using an acrydite group to immobilize a primer on polyacrylamide matrix. Now molecular colonies are used under the name of “polonies” at Harvard and other universities of USA for analysis of gene expression and single-nucleotide polymorphism (SNP), highly efficient DNA sequencing, investigation of alternative splicing, determination of allelic gene variations, gene mapping, and determination of their copy number.
Until recently, our method appeared under the name “molecular colonies” in most of our articles. The new name “nanocolonies” more precisely accounts for the nature of this event, because the question concerns not any molecules, but just nanomolecules.
Results of our and foreign laboratories obtained using nanocolonies are summarized in this review.
INVENTION OF THE NANOCOLONY TECHNIQUE
The principle of obtaining nanocolonies is the same as that proposed by Robert Koch in 1881 for growing bacterial colonies [11]: amplification is carried out in an immobilized medium owing to which the progeny (bacteria in works by Koch, template nanomolecules in our case) is localized in a certain limited space.
Bacterial colonies are grown on a solid nutrient medium prepared from agar or gelatin. Viral colonies, forming lysed zones (“plaques”) in a confluent cell monolayer, are obtained in a similar way. Each colony represents the progeny of a unique bacterial cell or virus particle localized in the place where this parental cell or particle was “seeded”. Koch's method allows one to identify the pathogen by the appearance and properties of formed colonies and also makes possible titer determination by direct counting the pathogen colonies; all this makes microbiological analysis simpler, quicker, and cheaper, and the result is more precise. In the case of sufficient dilution of seeded material, colonies are spatially separated and individual cells or virus particles do not interfere with propagation of each other. Owing to this, it is possible to detect the pathogen and determine its precise titer even during investigation of complex mixtures of microorganisms in which the sought pathogen is present in trace amounts or is non-competitive towards the rapidly growing species.
To obtain nanocolonies, individual DNA or RNA molecules are multiplied in a solid hydrated medium (like gel). Unlike cells, nucleic acids multiply not on the surface, but rather within (in a liquid phase) a gel containing an appropriate polymerase, nucleoside triphosphates, and, if necessary, primers. Therefore, propagation is carried out in a thin gel layer to dispose nanocolonies in the same plane.
In principle, any system of nucleic acid amplification can be used to grow nanocolonies. To make a nanocolony visible to the naked eye, it should contain up to one billion copies of the initial molecule. Such number of copies can be obtained in a reasonable period of time only using an exponential amplification system that enables doubling the amount of template nanomolecules in each cycle. The examples of exponential systems are PCR [12], 3SR (or SSSR, self-sustained sequence replication) [13], NASBA (nucleic acid sequence-based amplification) [14], SDA (strand displacement amplification) [15], LCR (ligase chain reaction) [16, 17], and RCA (rolling circle amplification) [18].
We obtained the first nanocolonies (colonies of replicating RNA) in January 1989 using Qβ replicase (RNA-dependent RNA polymerase of bacteriophage Qβ), a unique enzyme able to amplify RNA in vitro at a record rate, producing up to 1010 copies of template RNA in a 10 min incubation [19, 20]. RNA colonies were obtained in an agarose layer containing Qβ replicase, ribonucleoside triphosphates (rNTP), and an appropriate buffer. After incubation for 1 h at 37°C, the agarose was stained with ethidium bromide. Since RNA forms an intensively fluorescing complex with ethidium bromide, bright fluorescent spots visible under UV irradiation appeared at the locations of RNA colonies [21].
First Application of Nanocolonies
Not all RNA can be amplified by Qβ replicase, but only genomic RNA of Qβ bacteriophages and a very limited class of relatively short (from several tens to several hundreds of nucleotides) templates named RQ RNA (from Replicable by Qβ replicase) [19, 20]. In nature, RQ RNA are satellites of Qβ phage [1]. The probability that the randomly chosen RNA of 50-70 nucleotides (nt) will be RQ RNA is about 10-12 [22].
In 1975, M. Eigen et al. published a sensational report on the possibility of “spontaneous” generation of RQ RNA (in the absence of added template) in an incubation mixture containing a highly purified Qβ replicase and all four rNTP [23]. This experiment served as the basis for a concept concerning the RQ RNA self-generation, i.e. by “de novo” synthesis due to disordered nucleotide condensation and evolution of replication-capable polyribonucleotides to efficiently replicating molecules [24, 25]. The authors of the concept considered spontaneous synthesis of RQ RNA by Qβ replicase as a “basic” experimental model for investigation of emergence of genetic information by evolution at the molecular level [26]. This concept dominated until we used nanocolonies to discover the real source of replicating RNA.
We paid attention to the fact that the set of the spontaneous synthesis products was defined by the room used for the experiment. This suggested that synthesis could be caused by RQ RNA molecules caught by reaction mixture from the air. To check this possibility, the Qβ-replicase reaction was carried out as previously in agarose poured into Petri dishes. However, two layers of agarose were used to keep track of RNA molecule coming from the air. The lower layer contained rNTP, while the upper, poured after solidification of the lower one, contained Qβ replicase (Fig. 1a). Due to separation of rNTP and polymerase, RNA amplification could take place only after pouring the enzyme layer at the interface between layers. Before that, dishes with the substrate layer were kept for 1 h on the table, and one dish was closed while the other was open. After pouring the upper enzyme layer dishes were incubated on ice till agarose hardening and then for 1 h at 25°C. The agarose staining by ethidium bromide showed that more RNA colonies are formed in the dish that was open. The least number of colonies appeared in the case when all procedures were carried out in a room in which no work with RQ RNA was done before [1, 27]. These experiments have shown that RQ RNA molecules are present in the laboratory air and can infect reaction mixture, thus giving the impression of spontaneous synthesis. Thus, it became unnecessary to take advantage of the “de novo” synthesis concept; a number of “mystic” features of spontaneous synthesis such as almost complete identity of RQ RNA produced in different “spontaneous” reactions got a rational explanation; finally, the central dogma of molecular biology managed to stand this [28], because it was not necessary any more to postulate “creation of genetic information by protein enzyme [29]”.
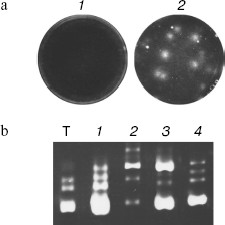
Main Properties of NanocoloniesFig. 1. Replication of RQ RNA in immobilized medium (a) and in liquid (b). a) Progeny of each molecule, amplified in immobilized medium (agarose), forms a separate colony and therefore does not compete with progeny of other molecules. Agarose gels containing a complete set of reaction components are stained by ethidium bromide either before (1) or after (2) incubation for 1 h at 25°C. b) Competition of RQ RNA, observed upon their amplification in liquid, results in non-reproducibility of synthesis products: 1-4) RQ RNA synthesized by Qβ replicase in four different tubes using the same RQ RNA preparation as template (T). An electrophoregram of synthesis products is shown; the gel was stained with ethidium bromide.
The RQ RNA colonies grown in agarose are quite diffuse. It was possible to overcome this problem by replacement of the agarose substrate layer by a nylon membrane filter, moistened with rNTP solution, which was applied on agarose containing Qβ replicase [8]. The nylon membrane reversibly binds nucleic acids and thus prevents their diffusion. Besides, since some of the RNA molecules bind the membrane during amplification, it can be used for detection of nanocolonies by incorporation of labeled substrates or by hybridization with labeled probes. At the same time, since a significant part of RNA remains in agarose, replicas of nanocolonies can be prepared using new membranes.
When the α-32P-labeled rNTP are used, RNA colonies can be registered by the label incorporation within first 10 min of incubation at room temperature. Based on the substrate specific radioactivities, time of X-ray film exposure, and the RQ RNA size, it can be concluded that, on the average, about 0.02 pmol or 1010 molecules of RQ RNA are formed in each colony. Since almost every nanocolony originates from a single molecule, this means that about 30 replication cycles are carried out in 10 min and as a result, the time of RQ RNA duplication is about 20 sec. Thus, the rate of RQ RNA amplification in agarose is not lower than in liquid media, where duplication time at the exponential growth stage was estimated as 30 sec [30, 31].
If known amounts of replicating RNA are seeded on agarose, the number of RNA colonies increases over spontaneous level and in this case the increment is proportional and close to the number of seeded molecules [8]. The proportion of nanocolonies to the number of seeded RQ RNA molecules is comparable with the fraction of viable (plaque-forming) particles (10%) in preparations of the wild-type phage Qβ [32].
Seeding a mixture containing various species of RQ RNA and following colony analysis by hybridization with appropriate labeled probes have shown that different colonies contain different RQ RNA [8]. Thus, RNA colonies are identical to seeded RQ RNA molecules. This showed that each colony represents the progeny of a single RQ RNA molecule, i.e. a molecular clone.
STUDYING RNA RECOMBINATION
The next application of nanocolonies was the investigation of RNA recombinations, rare events resulting in emergence of new molecules consisting of covalently bound fragments of parental RNAs.
For the first time RNA recombination was reported at the beginning of 1960s by Hirst and Ledinko, who studied the exchange by genetic markers between related strains of RNA-containing poliomyelitis virus (poliovirus) [33, 34]. In 1980 it was shown in the laboratory of V. I. Agol that recombination in poliovirus results in formation of a chimeric polyprotein, i.e. it is really associated with rearrangement of primary structure of the viral genomic RNA; in 1982 formation of recombinant viral RNA was detected by direct sequencing in the laboratory of A. King [35-37]. It was assumed that RNA recombination is the prerogative of viruses of higher organisms, animals and plants, until we isolated from bacterial lysates two types of recombinant RQ RNA (RQ120 and RQ135) (the number shows the length of nucleotide sequence) consisting of fragments of known sequences [38, 39].
However, since all studied recombinant RNA emerged in living cells or in crude cell lysates, in no case the possibility of reverse transcription was excluded, and as a result there always existed the probability that recombination involves not RNA molecules, but their DNA copies, while recombinant RNA are the result of transcription of recombinant cDNA [9, 40, 41].
The situation developed by the beginning of our investigations of RNA recombination can be characterized as follows. Recombinant RNAs were found in practically all RNA-containing viruses. Recombinant RNA could be both homologous and not homologous to parental molecules at crossover site. In this case, homologous recombination was considered as a rule (“legal”) and non-homologous was considered as an exclusion (“illegal”) [42]. The template switch mechanism was considered as the only possible and proved one [43, 44]. However, all evidence for both the mechanism and occurrence of recombination at the level of RNA were indirect. Therefore, there was the urgent need for a pure cell-free system of RNA recombination [44].
RNA Recombination in a Cell-Free System
Direct recombination between RNA molecules is possible. Nanocolonies obtained using Qβ replicase made possible investigation of reaction between RNA molecules in a pure system, free of DNA, dNTP, reverse transcriptases, DNA-dependent RNA polymerases, and enzymes responsible for DNA recombination, and as a result the possibility of recombinant RNA formation via DNA intermediates is excluded. We used as recombination substrates mutually supplementing 5′ and 3′ fragments of the RQ135 RNA minus strand [RQ135(-) RNA] supplied with foreign extensions (Fig. 2a) [9]. Since there predominated the idea that recombinant RNA always results from template switch [43, 44], foreign extensions were purposely made homologous in order to increase the probability of such an event. Separate fragments of RQ135 RNA are incapable of replication, but their combination in a covalently continuous molecule with restoration of correct mutual orientation could result in formation of recombinant RQ RNA replicable by Qβ replicase. This provided for positive selection of recombinant molecules, as it happens with selective multiplication of viable recombinant viruses.
Recombinant molecules were detected by growing RQ RNA colonies [8]. The mixture of fragments was seeded on the Qβ replicase-containing agarose layer which was then covered by a nylon membrane, moistened by rNTP solution, incubated at 22°C for 60 min, after which the membrane was hybridized with the 32P-labeled probe. The RQ135(-) RNA 5′ fragment or an oligodeoxyribonucleotide homologous to the foreign extensions was used as a probe. Each such probe is able to hybridize with (+)-strand of an expected recombination product, which is synthesized during exponential amplification along with (-)-strand. The emergence of recombinants was judged by formation of RQ RNA colonies that hybridized with the probe, and the number of such colonies was indicative of recombination frequency.Fig. 2. RNA recombination in vitro. a) A scheme of recombination between non-replicable RQ135(-) RNA fragments. The result of recombination is formation of RNA replicated by Qβ replicase. b) Recombination between RQ135(-) RNA fragments carried out by Qβ replicase. RNA colonies grown after seeding onto the Qβ replicase containing gel of an annealed mixture containing 5′ and 3′ fragment, 106 molecules of each. c) Spontaneous RNA recombination. Prior to seeding onto the gel, RNA was incubated for 64 h at 37°C without or in the presence of 9 mM Mg2+. Intermolecular recombination in a mixture containing 5′ and 3′ fragments of RQ135(-) RNA, 1010 molecules of each, is shown to the left; intramolecular recombination (the sample contained 1010 molecules of non-replicated RQ mRNA) is shown to the right.
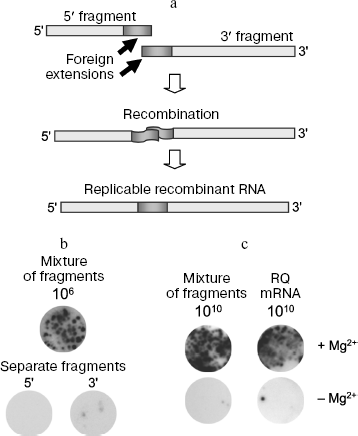
It appeared that the RNA colonies are really formed, and then and only then, when a mixture of 5′ and 3′ fragments, but not each fragment was seeded on agarose separately (Fig. 2b). This was the first evidence showing the possibility of direct recombination between RNA molecules. The recombination frequency is very low but it increases if fragments are not simply incubated but annealed before seeding; in this case, fragments associate, probably due to Watson-Crick interactions between complementary regions [9].
It should be noted that in the absence of nanocolonies it would be practically impossible to detect and isolate recombinant molecules and especially to study the mechanism of recombination due to very low recombination frequency and competition from “spontaneous” synthesis. In the case of amplification in liquid of even natural RQ RNA, selected on the basis of high replication efficiency, the set of products is not reproduced: molecules of different types dominate in different tubes (Fig. 1b). The situation is even much worse with amplification of recombinant RNA. Even if recombination results in emergence of replicating molecules, most of them will be faulty (only if recombination did not result in exact restoration of a natural sequence) and will not be able to compete with contaminant natural RQ RNA. On the contrary, in the case of amplification in gel different RQ RNA molecules are in different nanocolonies and do not interfere with amplification of each other (Fig. 1a). This makes possible amplification and counting of even poorly replicating molecules against the background of efficiently replicating RNA.
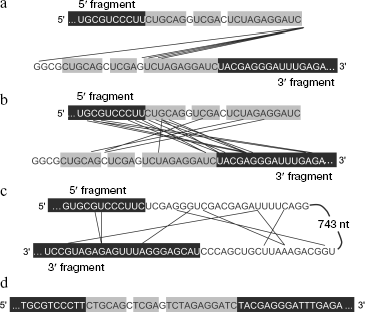
Mechanism of RNA recombination may differ from template switch. Recombinant RQ RNA extracted from colonies was cloned as cDNA within a plasmid vector and sequenced (Fig. 3a). All recombinants contained a complete 5′ fragment and a complete or shortened from 5′ end 3′ fragment. Despite the presence of homologous extensions, in all cases recombination of fragments involved non-homologous regions, i.e. all recombinants were non-homologous.
In accordance with the hypothesis of template switch, which at that time was the only discussed model of RNA recombination, at the beginning a complementary copy (or a part of it) of the first template (in our case it is 3′ fragment) is synthesized, which then serves as a primer for copying the second template (5′ fragment). The existence of such a mechanism was proved for the retroviral RNA-dependent DNA polymerases [45]. In fact, incubation of the same mixture of RNA fragments in the presence of reverse transcriptase of avian myeloblastosis virus (AMV) and its substrates (dNTP) resulted in formation of a homologous recombinant (Fig. 3d) [9]. This means that fragments used in our experiments are able to be substrates for homologous recombination, but Qβ replicase does not use this possibility. Moreover, frequency of recombination in the presence of Qβ replicase did not change if extensions of 5′ and 3′ fragments were changed for non-homologous ones. Moreover, all recombinants were structurally identical to those originated from the fragments with homologous extensions, i.e. they contained the whole 5′ fragment and a part of the 3′ fragment [46].
Conditions (pH, temperature, salt and Mg2+ concentrations) at which recombination is observed fully correspond to intracellular ones, therefore similar intracellular recombination can be also expected. Extrapolation to the Qβ-RNA concentration in infected E. coli cells (10-5 M according to Weissmann [47]) suggests that the similar intracellular process should have the frequency of 10-4-10-5 per nucleotide per hour (which is approximately equal to the duration of infection cycle). This agrees with the observed frequency of non-homologous recombination in RNA-containing bacteriophages in vivo and shows indirectly that within cells recombinant molecules are formed in reactions between RNA molecules without involvement of DNA intermediates [21].Fig. 3. Nucleotide sequences of recombination products. The RQ135 RNA sequence is shown in part (white letters against black background). Foreign extensions (a, b, d) or an insert (c) are designated by black letters: homologous sites are shown against gray background. Straight lines join nucleotides that appeared to be neighbors in recombinant molecules. a) Recombination between 5′ and 3′ fragments of RQ135(-) RNA carried out by Qβ replicase. b) Spontaneous recombination between 5′ and 3′ fragments of RQ135(-) RNA. c) Intramolecular spontaneous recombination resulting in deletion of the mRNA insert from RQ mRNA encoding chloramphenicol acetyltransferase. d) A homologous recombinant formed during reverse transcription of a mixture of the RQ135(-) RNA 5′ and 3′ fragments (reaction product is recombinant cDNA).
Role of 3′-terminal hydroxyls in RNA recombination. Taking into account that the whole 5′ fragment is incorporated in the recombinant, we have supposed that recombinants can be formed in a trans-esterification reaction in which free hydroxyl of 3′ end of 5′ fragment attacks internal phosphate of the 3′ fragment.
Further investigations have shown that the observed recombination really requires the presence of free hydroxyl groups at the 3′ end of the 5′ fragment: it is completely inhibited if the 5′ fragment is oxidized by periodate, which results in oxidation of 2′,3′-cis-glycol group at the RNA 3′ end with the ribose ring rupture and dialdehyde formation, or if its terminal 3′ hydroxyl is blocked by a phosphate group. The removal of this phosphate group using alkaline phosphatases restores recombination up to the initial level. A similar modification of 3′ end of 3′ fragment does not significantly influence the recombination efficiency [9].
Spontaneous RNA Recombination
Separation of stages of recombination and recombinant amplification. The use of 5′ fragment oxidation for recombination inhibition in the presence of Qβ replicase made it possible to separate recombination by itself (formation of recombinants) from amplification of recombination products in the form of nanocolonies. To achieve this, the mixture of RNA fragments was first incubated under the chosen conditions, then, before seeding on agarose, it was oxidized by periodate and melted after preliminary chelation of all free Mg2+ by EDTA [48]. Melting helped in lowering the level of residual recombination, caused by incomplete RNA oxidation, due to destruction of noncovalent interfragment complexes. Such treatment practically completely prevented further recombination and at the same time it did not interfere with colony growth, if a ready recombinant was present in the sample. Therefore, RNA colonies could be formed only if recombination happened before oxidation and melting of the fragment mixture.
Such an approach, i.e. recombination suppression during colony growth, makes it possible to determine whether recombination is possible in the absence of Qβ replicase, and it also allows investigation of the role of replicase and other enzymes in RNA recombination.
Intermolecular recombination. RNA fragments are also capable of recombination in the absence of Qβ replicase and rNTP due to formation of replicating molecules. The unique but obligatory condition is the presence of bivalent magnesium cations (Fig. 2c, on the left). However, frequency of such “spontaneous” recombination is several orders of magnitude below that in the presence of replicase: it is possible to detect spontaneous recombination only after incubation of the fragment mixture in the presence of Mg2+ at 37°C for 16-64 h. The mechanism of spontaneous recombination is also different: it does not require the presence of free hydroxyl groups, because it is equally efficient with any modification of 3′ end of 5′ fragment [48].
Sequencing the RNA isolated from colonies has shown that recombination involves internal regions of fragments with partial loss of the sequence of both the 3′ and 5′ fragments (Fig. 3b). Recombination does not require sequence homologies because the recombination efficiency is the same for fragments with homologous and non-homologous extensions and all recombinants are non-homologous. Finally, recombination sites are more or less randomly distributed along the nucleotide sequence of the 5′ and 3′ fragments within ±10-20 nucleotides from the stacking sites of RQ135 RNA and foreign extension sequences [48]. (Evidently, the absence of recombination beyond these sites is the result of selection: Qβ replicase poorly replicates both the recombinants with badly damaged initial structure of RQ135 RNA and recombinants carrying inserts that are too long.) This suggests that neither proteins nor any ribozyme-like structures are responsible for the observed recombination. In other words, the capability of recombinations is an integral feature of polyribonucleotides themselves.
Intramolecular recombination. Similar experiments already not with fragments but with long derivatives of RQ RNA (650-930 nt) with preliminarily inserted different mRNA sequences have shown that intramolecular spontaneous recombination is also possible. Such RQ mRNA cannot be replicated by Qβ replicase [49], but “in cis” recombinations take place during incubation in the presence of Mg2+, which result, as shown by sequencing, in deletion of inserts with formation of replicating molecules revealed by nanocolonies (Figs. 2c and 3c).
Spontaneous recombination probably follows the mechanism including non-hydrolytic rupture of the RNA sugar-phosphate backbone with formation of termini carrying 2′,3′-cyclophosphate and 5′ hydroxyl with following reversion of reaction with involvement of termini of different molecules (cross-ligation). Another possible mechanism is an intermolecular attack of internucleotide phosphate by 2′ hydroxyl resulting in a lariat (branched molecule) formation. If replicase can copy such structures, then the lariat reading may result in formation of a linear recombinant RNA. Since both these mechanisms require the presence of 2′ hydroxyls, spontaneous recombination is most likely a feature specific exclusively of RNA and it should not take place between DNA molecules [40].
Characteristics of spontaneous recombination. The use of nanocolonies allowed qualitative and quantitative characterization of features of RNA spontaneous recombination [48]. It was found that the number of recombinant molecules increased linearly: by 64 h of incubation, their number exceeded that by 16 h by 4.0 ± 1.8 times (n = 10). The RNA spontaneous recombination is a reaction of pseudo-first order, i.e. it is independent of the recombining fragment concentrations within the studied interval of concentrations (5-500 nM). Both linearity of kinetics and recombination independence of fragment concentration are caused by the fact that the rate of noncovalent association of RNA fragments under reaction conditions significantly exceeds the rate of the chemical reaction. The rate of recombination increases about three times upon temperature increase by 10°C. Such value of the Van't Hoff coefficient is characteristic of non-catalyzed (non-enzymatic) chemical reactions. The reaction rate constant will be about 10-9 h-1 per phosphodiester bond at 37°C.
Possible biological role of spontaneous RNA recombination. Since spontaneous recombination requires nothing but RNA and Mg2+, it must everywhere and continually happen in nature. Although in a “pure” (all components of which are known, unlike lysates whose composition cannot be controlled) cell-free system the rate of spontaneous recombination is far below that of non-homologous recombination in viruses, it is high enough to play a certain evolutionary role both in the prebiotic “world of RNA” and in the present time. At first glance, the rate of spontaneous recombination is extremely low. In the absence of nanocolonies it is practically impossible to register products of this reaction, especially against the background of tremendous excess of fragments not involved in reaction. Nevertheless, spontaneous rearrangements of RNA molecules may play an important role in evolution of RNA- and DNA-genomes. Even if spontaneous recombination is not promoted by cellular RNA-binding proteins, it should result in emergence of a novel recombinant RNA in a single eukaryotic cell once a minute. This means that, for example, up to 1020 such events should happen during the life span of a human.
It is quite probable that enzymes encoded by genomic mobile elements [50, 51] are able to copy recombinant RNA and insert their cDNA copies into the chromosome. This supposition is supported by detection in mammalian genomes of pseudogenes that are chimeric DNA copies of cellular RNA [52]. Integration into the genome even of a very small fraction of formed recombinant sequences may result in its significant alteration. Both the enzymic apparatus of retrotransposons [52] and cellular DNA-dependent DNA polymerases are able, to one extent or another, to use RNA as template [53].
With the account for the above-mentioned possibilities, self-recombination should be considered as an important factor influencing genome variability and probability of malignant transformation [54]. It is also necessary to keep in mind that some cellular proteins, especially RNA-binding ones, could nonspecifically stimulate spontaneous RNA recombination similarly to stimulation of activity of some ribozymes [55, 56].
Also, spontaneous RNA recombination could play an important role in the formation and development of the RNA world [21, 54, 57-59]. It should be also added that the ability of self-recombination is another argument in favor of the idea, first suggested by A. N. Belozersky in 1957 (see [58]), concerning the priority of RNA relative to DNA.
Variety of mechanisms of non-replicative RNA recombination. Spontaneous recombination is non-replicative because it does not require RNA synthesis (replication) and happens in the absence of replicase. After publication of our first results on in vitro recombination between RQ RNA fragments [9, 48] (Fig. 2), similar experiments with fragments of viral RNA and their different modifications were carried out in vivo. For this aim, RNA fragments obtained by in vitro transcription were transfected into cells. It was shown in the laboratory of V. I. Agol using fragments of poliovirus genome and in the laboratory of P. Becher using cattle diarrhea virus that intracellular RNA recombination can be also non-replicative (in the absence of active replicase) [60-62]. Sequencing of the formed viable viruses revealed non-homologous and homologous recombinants. Relatively high content of homologous recombinants in this case (compared to in vitro experiments) is, probably, the result of selection due to their higher viability.
The frequency of intracellular non-replicative recombination appeared to be several orders of magnitude higher than that of spontaneous recombination catalyzed by Mg2+ [41], which points to possible involvement of cellular enzymes. In fact, hydrolytic enzymes, cleaving RNA with formation of 5′-hydroxyl and 2′,3′-cyclophosphate, as well as RNA ligases, joining fragments with such groups at their ends, are present in practically all cell types [63]. Moreover, one and the same enzyme can both fragment RNA and ligate fragments, as it was shown for T1 ribonuclease [64]. A 2′,3′-cyclophosphate, necessary for ligation, can be formed from a 3′ terminal phosphate group using cyclase [63]. The existence of other mechanisms of RNA recombination is also quite probable when it is also considered that there are many intracellular enzymes capable of the nonspecific cleavage and ligation of RNA [65].
Mechanisms of Replicative Recombination
An experimental approach that made possible the demonstration of spontaneous recombinations (i.e. separation of recombinant formation and amplification stages) was also used to study the effect on recombination of viral RNA-dependent RNA polymerases [66]. In this case, before growing nanocolonies, incubation mixture was not only oxidized, but was phenol extracted to remove proteins.
Recombination in the presence of Qβ replicase is replicative. Experiments on incubation of the 5′ and 3′ fragment mixture with addition of the replication mixture components in various combinations have shown that it is possible to increase significantly the rate of recombination compared to the spontaneous level only after addition of all components of the replication system, i.e. Qβ replicase, Mg2+, and all four rNTP (ATP, CTP, GTP, and UTP). In this case, the mechanism of recombination is changed: it is inhibited by oxidation of the 5′ fragment [66]. This suggested that RNA recombination detected by us in the first work [9] was carried out by Qβ replicase itself and, evidently, RNA synthesis is necessary for recombination.
In experiments using cordycepin triphosphate (3′-deoxy-ATP, terminator of RNA synthesis), it was shown that not simply RNA synthesis is necessary for recombination, but synthesis of rather extended regions. Even when 3′-deoxy-ATP was incorporated into the growing chain along with ATP in a ratio of 1 : 5, the recombination was significantly inhibited [66]. In other words, recombination carried out by Qβ replicase is replicative.
Variety of replicative RNA recombination mechanisms. Poliovirus is a classical object for observation of in vivo RNA recombination. To explain homologous recombination just in this virus, in 1974 Cooper postulated for the first time the mechanism of template switch by viral replicase (RNA-dependent RNA polymerase) [67]. After that, the template switch hypothesis was applied to explain all cases of RNA recombination, although, unlike template switch in reverse transcription, it was not proved experimentally. Moreover, the terms “template switch” and “replicative recombination” were used as synonyms.
After publication of our work on in vitro RNA recombination in the presence of Qβ replicase, which expressed our doubt concerning the template switch mechanism, Perez Bercoff et al. [68] carried out the in vitro experiment on cells, transfected with plasmid, encoding the full-sized poliovirus genome. The plasmid transcription produced RNA that governed synthesis of active replicase but was incapable of replication due to two point mutations in 5′ non-translated region [68]. In the case of combined transfection with the poliovirus RNA fragment carrying the intact 5′ non-translated region, infectious virus was formed due to recombination. At the same time, no recombination took place if the RNA fragment carrying 5′ non-translated region had cordycepin (3′-deoxyadenosine) at the 3′ end and thus had no free 3′ hydroxyl. Thus, like in our in vitro experiments [9], recombination in vivo also required the presence of 3′ hydroxyl on the 5′ substrate.
Since all recombinants were homologous and formed in the presence of active replicase, the authors proposed a mechanism of “primer alignment and extension” which is in essence a component of the mechanism of replicative template switch. According to the authors, real recombination substrates are the RNAs of opposite polarity, in the case of our experimental scheme these are the 5′ fragment and a complementary copy of the 3′ fragment. These fragments are hybridized by complementary segments, and then one fragment or both serve as primers for elongation by the polymerase that uses the other fragment as template [68]. Naturally, in this case a homologous recombinant is obtained. Such recombination frequency should depend on the hybrid stability and, as a result, on the length of the primer and template complementary sites.
Since our investigations have shown that RNA recombination by Qβ replicase is not homologous, though replicative, it was important to compare directly the effect of Qβ replicase and poliovirus replicase (protein 3D-pol) on RNA recombination under identical conditions. The same recombination substrates and optimal reaction conditions, that appeared to be similar, were presented to both replicases. The same system of recombinant detection, i.e. the Qβ replicase version of nanocolonies, was used.
To make easier formation of recombinants by the mechanism of “primer alignment and extension”, pairs of RQ135 RNA fragments of opposite polarity including 5′ fragment and the complement of 3′ fragment (“3′C-fragment”) with extensions of different length were used as recombination substrates. Extensions are complementary within each pair (in pairs of the same polarity fragments they are homologous), but since extensions have different length, the lengths of complementary regions are different (Fig. 4, on the left).
Recombination between different polarity fragments, carried out by poliovirus replicase, appeared to be very efficient and completely correlated with the “primer alignment and extension” mechanism: pairs of fragments with longer complementary regions recombine at higher frequency (Fig. 4, column “3D-pol”) and all recombinants are homologous [66]. At the same time, recombination between the same fragments carried out by Qβ replicase is 3-4 orders of magnitude less efficient, there is no correlation between recombination frequency and the length of complementary regions (Fig. 4, column “Qβ-rep”), and all recombinants are non-homologous [66].Fig. 4. Comparative efficiencies of recombinations carried out by poliovirus polymerase (3D-pol) and Qβ replicase (Qβ-rep). The nucleotide sequence of recombination substrates is shown to the left. The RQ135 RNA sequence is shown in part (white letters against black background). Foreign extensions are designated by black letters: homologous segments (fragments of the same polarity) or complementary segments (fragments of different polarity) are against gray background. Complementary pairs are shown by dots. 3′C is the complement of the 3′-fragment. To the right there are RNA colonies grown after seeding of the annealed mixture of fragments onto the Qβ replicase-containing gel, either without incubations and treatments (recombination in the presence of Qβ replicase (Qβ-rep)) or incubated for 30 min in conditions, optimal for poliovirus polymerase, after treatment suppressing recombination during colony growth (recombination in the presence of poliovirus polymerase - 3D-pol). Figures show the number of molecules of each pair of fragments in the seeded mixture.
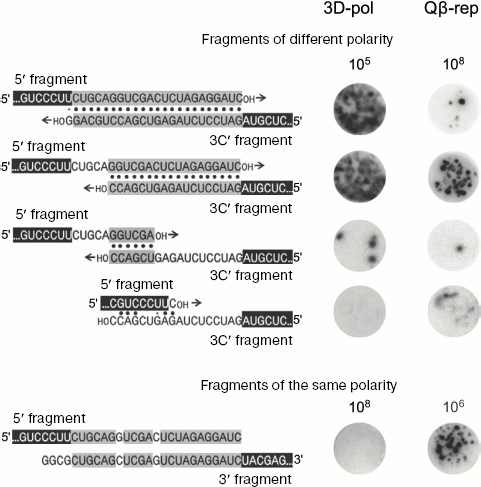
Contrary to the “primer alignment and extension” mechanism, recombination by Qβ replicase between different polarity fragments is 2-3 orders of magnitude less efficient compared to that between fragments of the same polarity (Fig. 4). Oxidation of the 5′ fragment is not enough for its inhibition (as in reaction between fragments of the same polarity), but in this case oxidation of both fragments is necessary [66]. Unlike Qβ replicase, poliovirus RNA polymerase does not produce detectable amounts of recombinants in reaction between fragments of the same polarity (Fig. 4).
So, nanocolonies helped us to determine that different RNA-dependent RNA polymerases use different mechanisms for RNA recombination: poliovirus RNA polymerase uses the “primer alignment and extension” mechanism, while Qβ replicase rejects it.
Possible mechanisms of RNA recombination by Qβ replicase. Recombinants obtained by recombination between pairs of fragments, one of which was oxidized by periodate, were sequenced. Almost all recombinants contained both whole fragments with additional different length inserts between fragments. The inserted sequences depended on the oxidized fragment and were complementary in part to segments of foreign extensions. Based on these observations and taking into account that Qβ replicase can elongate RNA from 3′ end, thus forming hairpins [69, 70], we supposed that the inserts are formed by elongation of the fragment 3′ terminus and partial copying the same fragment. Then, two ways of further event development are possible: either 3′ hydroxyl of the elongated 3′C fragment attacks internucleotide 5′ phosphate on the 5′ fragment copy (trans-esterification reaction), or 3′C-fragment is further elongated using the 5′ fragment as template. In other words, the second mechanism suggests elongation of a primer not hybridized to the template. In accordance with a number of arguments, the first mechanism seems more likely [66].
It is still not clear what the role of RNA synthesis in recombination of the same polarity fragments is when both substrates are ready for recombination from the very beginning. One possibility is that the trans-esterification reaction requires Qβ replicase in active conformation that can be acquired only during synthesis. It should be noted in this connection that the supposed trans-esterification reaction, consisting in the attack of internucleotide phosphate of one RNA by the 3′-terminal hydroxyl of the other, is chemically analogous to the attack of α-phosphate of the nucleotide to be joined by the 3′-hydroxyl of growing chain, i.e. it is analogous to the reaction that is the main function of all polymerases [53].
Application of Nanocolonies in Chemistry of Single Molecules
The use of a pure cell-free system of RNA recombination, created on the basis of nanocolonies, produced the following main results. It is proved that recombination takes place at the level of RNA without involvement of DNA intermediates. The RNA recombination is significantly more diverse concerning molecular mechanisms than it seemed previously. It can be both replicative and non-replicative. RNA is capable of self-recombination under physiological conditions at a biologically significant rate, without involvement of proteins and ribozymes. Different polymerases employ different mechanisms of RNA recombination. Contrary to previous concepts, RNA recombination is possible without template switch.
The experiments have shown that nanocolonies can be used to study chemical reactions between single RNA molecules. In principle, nanocolonies can also be used in investigation of different rare chemical reactions or rare reaction intermediates. It is only necessary that the reaction products or intermediates have a sufficiently complex surface to form at least two high affinity-binding regions, as discussed in the section “Detection of Molecules of Non-nucleic Nature”.
DEVELOPMENT OF THE NANOCOLONY TECHNIQUE
Limitations of the Qβ Replicase Nanocolony Version
The demonstration of the ability of Qβ replicase to amplify foreign sequences within RQ RNA [31, 71] gave rise to the hope that it will be possible to use RNA colonies for diagnostics as well as for cloning foreign genetic material such as mRNA by its incorporation into a natural replicating molecule.
This became the basis for schemes proposed in the laboratory of F. Kramer for a diagnostic use of Qβ replicase. In the original variant [72], a nucleotide sequence complementary to the region of DNA or RNA to be assayed (target) was incorporated into RQ RNA and such artificial recombinant RNA was mixed with the analyzed sample. Then the sample was treated with magnetic particles with immobilized oligonucleotide probe complementary to another region of the target. If the sample contained target molecules, they were captured by the magnetic particles and carried along with the recombinant RNA that was then amplified by Qβ replicase. The presence and amount of the target in the sample was determined from the kinetics of RNA synthesis: the more target molecules there were in the sample, the earlier synthesis was registered. However, due to unspecific sorption, up to 10,000 recombinant RNA molecules were caught by magnetic particles even in the absence of a target, which made the method extremely insensitive.
In the improved scheme [73] the whole recombinant RNA was replaced by its supplementary fragments extended at the truncated ends with nucleotide sequences complementary to adjacent regions of the target, and the sample was additionally incubated with phage T4 DNA ligase that is able of end-to-end joining the hybridized RNA fragments. Thus, the replicating recombinant RNA was formed directly in the sample, depending on the presence of the target in the latter. According to the authors, in this case sensitivity of the method was ~40 target molecules.
We have tried to use RNA colonies for diagnostics of viral infections by the above-described scheme. Unfortunately, it was found that the applicability of this approach is quite limited. First, foreign inserts, even short ones, often inhibit replication of natural replicase templates [20, 74] and as a result such approach is not universal. Second, RNA recombination, especially in the presence of replicase (see above), results in emergence of replicating molecules and subsequent appearance of numerous nanocolonies independently of the presence in the sample of the target under study.
The use of this approach for mRNA cloning was fully hopeless, because RQ mRNA are incapable of exponential amplification [49], and spontaneous deletions of mRNA inserts result in formation of efficiently replicating RQ RNA [48].
DNA Colonies
Unlike Qβ replicase, PCR is more suitable for diagnostics and cloning because it allows amplification of any sequence using a pair of oligonucleotide primers. However, PCR cannot be carried out in agarose gel, because the reaction medium requires periodic heating up to ~100°C.
We have elaborated a method for PCR in a thermostable polyacrylamide gel. This method allows one to obtain DNA colonies [2-7, 75, 76]. RNA may also serve as initial template that should be first converted to the cDNA form using RNA-dependent DNA polymerase (reverse transcriptase). The gel is prepared in a suitable container such as a well on a microscopic slide.
Initially the gel was polymerized in the presence of all PCR components including DNA polymerase [2-7], and this method was reproduced in the laboratory of G. Church at Harvard University [10]. However, it appeared that another approach gave better results: the gel is first polymerized, then it is soaked in water to remove all soluble substances, autoclaved and dried, and immediately before experiment it is impregnated with a complete reaction mixture including dNTP, oligonucleotide primers, thermostable DNA-dependent DNA polymerase, and the sample. In this case, DNA polymerase completely retains its activity, while DNA molecules that could get into the gel during its preparation are eliminated. During impregnation by reaction medium the gel completely regains its initial volume and mechanical properties. The volume of the gel prepared in a well 14 mm in diameter and 0.4 mm in depth is 65 µl (i.e. consumption of reagents is almost the same as in a standard PCR of 50 µl).
For PCR the gel is sealed and placed in the DNA amplifier with a flat heating element (such DNA amplifier is usually used for in situ PCR). Then the reaction is carried out in approximately the same temperature conditions as in standard PCR in tubes, with only annealing time increase to 10-20 sec.
Methods of Nanocolony Detection
Nanocolonies grown in the gel are invisible to a naked eye, but they can be detected in different ways. For example, when amplification is over, the gel can be stained with intercalating dyes like ethidium bromide [1, 8] or SYBR Green I [10]. However, in this case both specific nanocolonies (products of the analyzed target amplification) and nonspecific ones, formed by primer “dimers” (products of primer elongation due to hybridization with each other) are stained, as well as those formed by products of foreign DNA amplification due to erroneous primer hybridization with foreign templates.
Specific nanocolonies can be revealed by hybridization to a probe complementary to the internal part of the amplified target, after transferring them onto a membrane filter by blotting the gel. In the first experiments, we used radiolabeled probes for detection of nanocolonies [8, 9, 48]. Disadvantages of such probes are the necessity of using harmful radiolabeled substances (32P, 33P, or 35S), which in addition have a short half-life, and duration of the procedure of obtaining an autoradiograph often takes more than 24 h.
In another known method, using target-specific probes, nanocolonies are hybridized in situ (in the gel where they were grown) with fluorescent probes or with unlabeled oligonucleotide probes that are then labeled by terminal incorporation of fluorescent nucleotides [77]. Owing to the use of fluorescent labels, it becomes unnecessary to use radioactive materials and the time necessary to obtain a picture is reduced to several minutes. However, gel hybridization makes the procedure significantly more complicated and expensive, because covalent immobilization of amplified nucleic acids on the gel matrix is necessary (to achieve this, one of the oligonucleotide primers used for amplification is immobilized). Moreover, it is necessary to remove from the gel one of the amplified target complementary strands using a reversibly crosslinked gel. Amplification is followed by partial uncrosslinking the gel to make diffusion of nucleic acids easier, and then electrophoresis is used to increase the rate of removal of the non-immobilized strand. Besides, upon hybridization in the gel targets appear to be less accessible for probes, and removal of unbound probes from the gel proceeds at a lower rate compared to that in the case of hybridization on the membrane.
Detection of nanocolonies by hybridization with fluorescent probes on the membranes. Hybridization with fluorescent probes on nylon membranes could become a solution of the problem, but it followed from the literature that this is unlikely due to the extremely low content of nucleic acids in a colony and too high level of the membrane background fluorescence. As it has been shown in different laboratories, about 108 DNA molecules are present in a single colony of 0.2-0.5 mm in diameter formed by PCR in gel [10, 75]. About 10% is transferred onto the membrane by blotting [78], i.e. 107 molecules or ~10 amol DNA. It has been repeatedly stated that it is impossible to detect such amounts of nucleic acids immobilized on nylon membrane by hybridization with fluorescent probes due to the high intrinsic fluorescence of nylon [79-83].
Nevertheless, we managed to develop nanocolony hybridization with fluorescent probes on a membrane, total duration of which (from colony fixation to obtaining scanning result) takes less than 1 h [84, 85].
Nanocolony hybridization with fluorescent probes on a membrane allows quick detection of single molecules and determination of target DNA and RNA titers. Since the number of colonies coincides within the limit of statistical data scattering with the number of DNA molecules introduced into the gel before beginning PCR, it can be concluded that all DNA colonies are detected. Moreover, the membrane can be hybridized with a mixture of fluorescent probes and information concerning hybridization of each probe can be obtained separately if spectral characteristics of the labels differ enough to provide for selective recording of fluorescence of each of them, like that observed for fluorophores Cy3 and Cy5. A mixture of probes labeled by these fluorophores was used for detection of different parts of chimeric cDNA molecule AML1-ETO consisting of parts of the AML1 and ETO genes (chimeric mRNA AML1-ETO is a marker of a frequent form of leukemia [86]): the Cy5-labeled probe is specific to the AML part, whereas the Cy3-labeled probe is specific to the ETO part of the AML1-ETO sequence (Fig. 5, a and c). Hybridization of both probes with the same nanocolony shows that the latter is formed by chimeric molecules. This makes it possible to enhance the specificity of diagnostics by discrimination against nanocolonies formed by non-chimeric molecules present in normal leucocytes.
Simultaneous hybridization with several fluorescent probes of different base composition and fluorophores can be used in other aspects, such as separate determination of several targets at once or for determination of sequence variants in the same target, in particular, for determination of so-called single-nucleotide polymorphism (SNP).Fig. 5. Detection of nanocolonies using fluorescent probes. a, b) Sites of fluorescent probe hybridization with the amplified fragment of chimeric cDNA AML1-ETO: a) probes used for hybridization on the membrane; b) a pair of FRET probes. Probes are complementary to the strand synthesized during reverse transcription. The Cy5-AML probe and the donor of the FRET probe pair are hybridized with the AML1 part of cDNA, while the Cy3-ETO and acceptor of the FRET probe pair are hybridized with its ETO part. A short distance (2 nucleotides) between donor (FAM) and acceptor (Cy5) provides for efficient resonance energy transfer between them when these probes are hybridized with the product of the chimeric cDNA amplification. c) The AML1-ETO cDNA colonies revealed by hybridization with fluorescent probes on the membrane. After PCR in gel, DNA was transferred by blotting onto nylon membrane that was hybridized with a mixture of oligodeoxynucleotides Cy5-AML and Cy3-ETO, and the Cy5 and Cy3 fluorescence was registered, respectively. d) Detection of molecular colonies in real time. The AML1-ETO cDNA colonies were obtained by asymmetrical PCR in the presence of FRET probes (FAM/Cy5). Figures show the number of the PCR cycle after which the gel was scanned using a blue laser (488 nm) and a red emission filter (670 nm). The amplified PCR product was 260 bp long (upper row) or 395 bp (lower row).
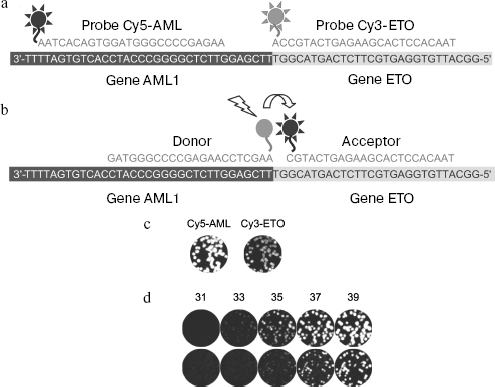
Detection of nanocolonies under real time conditions. We have shown that growing nanocolonies can be observed under real time conditions [87, 88] using different homogeneous detection systems (allowing DNA detection without removal of unbound fluorophore) using intercalating dyes, hybridization probes, or their various combinations. To observe growing DNA colonies, PCR was carried out as usual in the same polyacrylamide gel prepared in the slide wells and covered by coverslip. To provide for the gel fluorescence recording, the coverslip was sealed by sticky foil with a hole equal to the well size. The use of homogeneous systems allows detection of nanocolonies without opening the gel and colony transfer onto the membrane and does not require a special hybridization stage when PCR is over.
It appeared that any presently known principle of homogeneous fluorescent detection [89, 90] can be used for detection of nanocolonies. The highest detection sensitivity was obtained using a pair of adjacent hybridized probes having at their proximal termini fluorophores fluorescein (FAM) and cyanine-5 (Cy5), between which FRET (fluorescent resonance energy transfer) is possible (Fig. 5, b and d). Amplification of longer fragment results in a later appearance of colonies, but they become smaller, which makes it possible to increase the resolution (Fig. 5d).
Nanocolonies can be also revealed using a “molecular beacon” (an oligonucleotide probe with the hairpin structure carrying on the closely adjacent termini a fluorophore and a group that quenches the fluorescence when the probe is unbound, but stops quenching when the probe is extended due to hybridization with the target) or a combination of an intercalating fluorescent dye and a specific probe carrying a fluorescent group whose excitation maximum coincides with that of the dye emission [87, 88].
Although detected nanocolonies are also formed under conditions of usual (symmetrical) PCR, the detection sensitivity can be increased using conditions of asymmetrical PCR at the primer concentrations favorable for synthesis of strands complementary to fluorescent probes. In the case of asymmetrical PCR, the intensity of colony fluorescence is significantly higher because some DNA nanocolonies remain single-stranded and thus are more accessible for hybridization with probes [87, 88].
In the above-mentioned examples, fluorescent pictures were obtained at constant (room) temperature after finishing the elongation stage. Evidently, images can be registered at different temperatures and at different stages of the PCR cycle. Since hybridization temperature falls to 4-8°C in the case of even a single alteration of the target and probe complementarity, analysis of hybridization (melting) curve makes possible detection of point mutations in the target if they emerged in the site of probe hybridization [91]. Temperature variations at which colony image appears or disappears can be used as an instrument of SNP analysis.
It is probably possible to adapt methods using probe degradation for fluorophore removal from its quencher (the principle was used in the TaqMan probe) for detection of nanocolonies by immobilization of the probe fluorescent group on the gel matrix in order to prevent its diffusion in the gel. In such situation the gel matrix will fluoresce when hydrolysis of the probe in the point, previously occupied by the colony, will be over, even when amplification products diffused or were washed away from the gel; therefore, such gels can be stored and used as a documentary evidence of analysis [92].
MOLECULAR DIAGNOSTICS
The discovery of replicating RNA in the air, which demonstrated that the “spontaneous” synthesis was template-directed [1], actually became the first case of nanocolonies application to diagnostics. These experiments have shown that “infectious” molecules can be detected as nanocolonies and their number can be determined. Carrying out PCR in nanocolonies and development of non-radioactive methods for their detection has transferred the application of nanocolonies for diagnostics to the practical level.
Diagnostic Procedure
In addition to the nanocolony growing and detection, the diagnostic procedure includes a number of consecutive operations, such as clinical material conservation, isolation of nucleic acids, and reverse transcription (if RNA is the target).
We have elaborated methods providing for isolation of nucleic acids from clinical samples including unfractionated whole blood, with almost 100% yield: a method of DNA isolation [75] and a universal technique providing for simultaneous isolation of RNA and DNA [93]. It was also shown that in the case of storage of whole blood samples as lysates containing 4 M guanidine thiocyanate, high-molecular-weight RNA and DNA are preserved for 3 days at room temperature, at least during two weeks at 4°C, and for more than one year at -20°C [94, 95]. This method of conservation provides for sample storage and transport at the environmental temperature and is completely compatible with the procedure of subsequent isolation of nucleic acids.
We have found in the course of reverse transcription optimization that nucleic acids in high concentrations inhibit the reverse transcriptase activity of Tth DNA polymerase, often used for RNA detection [96], and we have shown that in the case of analysis of samples with a low titer of RNA targets it is preferable to use for cDNA synthesis reverse transcriptase of mouse leukemia virus (M-MLV) lacking RNase H activity [93].
Diagnostic Potential of Nanocolonies
Sensitivity of DNA and RNA detection. In the first experiments we used the universal reaction mixture with DNA polymerase of Thermus thermophilus (Tth DNA polymerase) in order to have the possibility of detecting both DNA and RNA molecules. In the presence of Mn2+, this polymerase is able to use both DNA and RNA as template and synthesizes cDNA [96]. Prior to PCR, the gel impregnated by such reaction mixture was incubated for 30 min at ~60°C providing for reverse transcription. In such system the sensitivity of the target molecule detection was 98 ± 21% for HBV DNA, 13 ± 3% for HIV RNA, and 15 ± 3% for Qβ RNA [75]. The standard deviation in this case is indicative of the error in serial dilutions and the statistics of random distribution of a low number of molecules between samples rather than the error of the nanocolony technique. DNA molecules are detected with the same efficiency upon amplification using Taq DNA polymerase in the presence of Mg2+.
Thus, nanocolonies enable detection of about 100% of DNA molecules. The lower percentage of RNA molecule detection shows the yield of the stage of reverse transcription. Nevertheless, the number of nanocolonies was always proportional to that of seeded RNA molecules, and as a result, in this case the method allows determination of exact titer of the target. In the next experiments, we increased the yield of reverse transcription up to 50% using the M-MLV reverse transcriptase [93], which made possible detection of each second RNA molecule.
Absence of competition between simultaneously amplified molecules. Simultaneous detection of several targets (multiplex PCR) is often used in clinical investigations. In order to determine to what extent nanocolonies make it possible to escape competition between amplified molecules, an experiment was carried out in which 300 molecules of HIV-1 RNA were amplified simultaneously with HBV DNA, the initial number of which was changed from 0 to 1 billion molecules (Fig. 6a) [75]. After reaction, the membrane was first hybridized with the probe specific to the amplified fragment of the HIV-1 genome. Then this probe was washed and the same membrane was hybridized with a probe specific to amplified fragment of HBV DNA. It is seen in the figure that the number and size of colonies of one target (HIV-1) are independent of simultaneous growth in the same gel of several million times more abundant colonies of another target (HBV).
Although these targets use different primers, they use the same DNA polymerase in an amount lower than that of primers, which is a factor limiting DNA amplification. The complete absence of competition between targets shows that polyacrylamide gel has a very high resolving ability. In the terms of “digital PCR” [97], the gel of 14 mm in diameter used in this work is equivalent in its resolution power to a PCR plate with one billion wells.Fig. 6. Diagnostic potential of nanocolonies. a) Absence of competition between targets amplified in the same gel. DNA colonies grown as a result of RT-PCR in polyacrylamide gel containing, in addition to everything else, MnCl2 and Tth DNA polymerase (able in the presence of Mn2+ to carry out both reverse transcription and PCR), 300 molecules of the RNA fragment of human immune deficiency virus (HIV-1), the indicated number of molecules of the hepatitis B virus (HBV) DNA fragment, as well as HIV-1- and HBV-specific primers. When PCR was over, membranes were first hybridized with radiolabeled HIV-1-specific probe (upper row) and then with the HBV-specific probe (lower row). b) Simultaneous detection of HIV-1 RNA and HBV DNA in human whole blood. Total nucleic acids were isolated from a sample (100 µl) of donor blood to which the target was not added, or 150 molecules of HIV-1 RNA and 50 molecules of HBV DNA were added at the moment of lysis. The total preparation was used for cDNA synthesis by M-MLV SuperScript™ II reverse transcriptase and following PCR in gel using Taq DNA polymerase. Nanocolonies were revealed by membrane hybridization with radiolabeled probes, first with the HIV-1-specific (upper row) and then with HBV-specific (lower row); 78 ± 18 HIV-1 RNA molecules and 53 ± 11 HBV DNA molecules were revealed in the form of nanocolonies in a series of seven experiments.
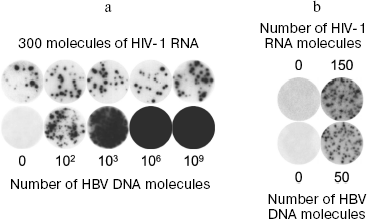
Absence of interference from nonspecific synthesis. Since in addition to the assayed target a clinical sample usually contains a large number of different nucleic acids, in the case of PCR in liquid there is a high probability of interference from nonspecific (not caused by the target) DNA synthesis. Nonspecific synthesis is the result of the limited specificity of hybridization of oligonucleotides used for target amplification, because at a certain probability the oligonucleotide hybridizes, instead of with target, with a not quite complementary sequence--another oligonucleotide or foreign DNA. Although the probability of a single event of such kind is low, it is almost always realized during analysis of biological samples because the concentration of oligonucleotides and foreign nucleic acids in the analyzed sample is much higher than the concentration of the target. Thus, in the case of amplification in liquid of 1000 molecules of HBV DNA, the amount of specific product decreased and that of unspecific product increased in the presence of 0.4 µg nucleic acids (isolated from 15 µl whole blood), which corresponds to 109-fold excess of human nucleic acids over the target of 392 bp [98]. At the same time, neither brightness nor the number of specific DNA colonies were changed upon amplification in gel of 50 molecules of the same target in the presence of 5 µg nucleic acids (isolated from 100 µl blood) [75], which corresponds to 200 times higher (2⋅1011-fold) excess of human nucleic acids over the target.
Detection of single molecules of viral DNA and RNA in whole blood. Model experiments simulating clinical diagnostics in which HBV DNA was added to 100 µl of uninfected blood and then total DNA isolated from the blood was analyzed have shown that the number of HBV-specific nanocolonies was equal (within the limits of statistical data scattering) to the number of target molecules added into the blood [75]. Thus, nanocolonies make it possible to lower the detection limit of DNA-containing viruses in blood to an absolute minimum of one molecule, which is 500 times higher than the current sensitivity of PCR diagnostics [98].
A similar result was obtained in the case of simultaneous detection of HIV-1 RNA and HBV DNA in human whole blood (Fig. 6b). In this case, 106 ± 20% DNA molecules and 50 ± 12% RNA molecules are detected in 100 µl of human blood (such sensitivity of RNA detection reflects the yield of the stage of reverse transcription using M-MLV reverse transcriptase) [93]. In other words, nanocolonies enable detection of one DNA molecule and two RNA molecules. These results not only demonstrate previously unattainable sensitivity of diagnostics, but they show that our methods of nucleic acid isolation provide for the yield close to 100% and practically complete elimination of contaminants that inhibit reverse transcription and PCR.
Determination of marker RNA in oncological diseases. We have applied nanocolonies to detection of the α-fetoprotein mRNA [99], a marker of hepatocellular carcinoma and some tumors of the reproductive system [100] as well as to revealing a chimeric transcript AML1-ETO [84], a marker of the leukemia associated with translocation t(8;21)(q22;q22) [86].
The developed diagnostic procedure was tested by determination of absolute titer of the AML1-ETO mRNA in clinical samples obtained from patients with this type of leukemia at different stages of the disease [101]. It appeared that nanocolonies made possible detection of a tumor-specific marker in the blood and bone marrow of patients at the stage of remission, several months prior to its titer outburst, which can be used for the timely repeated course of chemotherapy.
The results obtained using nanocolonies were compared with those for standard (liquid) PCR in real time. At a high titer of RNA target in the blood or bone marrow of patients, a satisfactory coincidence of results obtained by these two methods was observed. However, at the stage of remission when the titer of RNA target is low, the standard PCR sensitivity is not always enough for detection of minimal residual disease. (The “minimal residual disease” is called the state of a patient during remission when a small number of leukemia cells still exist in the body. The appearance of RNA marker or increase in its amount during hematological remission is called the “molecular relapse” and is the precedent of clinical relapse.)
An additional resource for increase in specificity is the ability of our method to discriminate between DNA colonies formed by chimeric RNA and non-chimeric precursors present in normal cells. Chimeric colonies have to hybridize with every probe specific of genes composing the chimera (Fig. 5), whereas non-chimeric colonies can hybridize with only one of them. A similar effect could be achieved using probes that hybridize with constituent portions of the chimeric template close to the junction point and carry fluorophores capable of the resonance energy transfer (FRET). Since FRET is possible only when fluorophores are in the immediate vicinity of each other [102], the FRET-mediated fluorescence of nanocolonies proves that they consist of chimeric sequences able to hybridize with both probes (Fig. 5).
Thus, the developed procedure for detection of minimal residual disease in the case of leukemia by revealing the RNA marker using nanocolonies far exceeds by sensitivity and reliability the existing methods, including real time PCR.
Overcoming Problems of Liquid PCR Diagnostics
PCR is now the most sensitive instrument of diagnostics. However, for some reasons such as competition from another amplified target and/or nonspecific synthesis, deviation of the reaction kinetics from calculated curves, and the possibility of false emergence of a target as a result of recombination, this reaction in liquid can give wrong results [76, 98]. The main problems of liquid PCR, the competition between amplicons and the difficulty of quantitative determination, are in principle the same as those that in his time faced R. Koch: how to overcome the competition between different microorganisms and how to estimate their exact titer. Robert Koch solved this problem by elaboration of a solid nutrient medium.
Nanocolonies allow one to overcome all problems of liquid PCR (like of other liquid systems of exponential amplification) and to make molecular diagnostics the highly sensitive, quantitative, and reliable one. The observation of growing nanocolonies in real time enables performing of a whole list of diagnostic applications developed for the PCR in real time [103] with the difference that in this case much higher sensitivity and accuracy of the results are ensured.
The use of nanocolonies in quite a number of parameters exceeds methods based on nucleic acid amplification in solution. Spatial separation of nanocolonies significantly decreases the competition between targets upon simultaneous detection of several of them, and at the same time interference from unspecific synthesis caused by the primer mishybridization with foreign DNA and RNA also becomes significantly lower. Nanocolonies make possible detection of nucleic acids with absolute sensitivity: they reveal one molecule of target DNA even against the background of enormous excess of non-target nucleic acids. Counting the number of nanocolonies makes possible direct determination of the number of target molecules, thus turning molecular diagnostics to a digital technique, significantly enhancing its reliability. The plateau effect, nonreproducibility of reaction conditions, different rates of different target amplification in multiplex PCR, influencing the size of colonies, do not change their number. For reliable diagnostics, it is simply necessary to register the nanocolony (to obtain answer “yes” or “no”) without measuring the signal intensity.
Amplification in different nanocolonies excludes inter-template recombination that is often observed in liquid PCR [104-107]. This prevents the emergence of artifact sequences and thus enhances the diagnostics reliability. Although there are ways to lower this recombination (sparing denaturation conditions, increased time of elongation [105, 108]), the complete elimination of recombination under the liquid PCR conditions is impossible because recombination may not only be the result of annealing and subsequent elongation incomplete strands [109] (similarly to the “primer alignment and extension” mechanism for RNA recombination), but also the result of template switch at the stage of elongation [110].
It is especially important to consider the possibility of DNA recombination during PCR in situations when the aim is revealing the differences between homologous sequences. Such problem emerges upon analysis of a single gene alleles or mRNA corresponding to these alleles [109] as well as during analysis of partially degraded DNA (like DNA from archeological specimens) [106, 111]. Moreover, amplification of partially fragmented DNA results in insertions of single nucleotides [106], which causes a shift in the reading frame in coding sequences and makes impossible reconstruction of amino acid sequence and following synthesis of a correct protein upon analysis of gene expression.
The nanocolony technique is just a little more complicated than standard PCR: the only difference is that reaction mixture is placed not in a tube but in a well with a film of dry polyacrylamide gel. It is also important that PCR in nanocolonies is not more expensive than standard PCR: the gel volume in the described experiments was only 65 µl, i.e. consumption of reagents is almost the same as in standard PCR of 50 µl. In fact, the nanocolony technology is even cheaper, because it allows many times decrease in the number of samples necessary for analysis.
Distinctions from Solid-Phase Amplification
It is stated in a series of articles that DNA colonies can be obtained by so-called “solid-phase amplification” (SPA) [112-114]. These “colonies” differ in principle from nanocolonies, because in the case of SPA the PCR components are not in an immobilized medium, but in liquid, and only primers are immobilized by covalent bonding to the slide surface. Although in SPA technique both primers and, as a result, the PCR products are covalently bound to the glass surface, a significant fraction of amplification products migrate from parental template due to instability of this bond. And the template itself also migrates. Despite significant enhancement of the bond stability compared to the original variant of the technique, the “leakage” of primers during PCR still reaches up to 60% [115]. Authors of the SPA technique note that the bond instability results in lowering the PCR yield and mixing the colony content (as a result, these colonies are already not molecular clones) [115]. After all, the method is deprived of advantages over liquid PCR. In particular, it becomes impossible to estimate the target titer, because due to migration of templates and amplification products, the number of colonies increases during development without any control.
CELL-FREE GENE CLONING AND EXPRESSION
As mentioned above, each nanocolony is a molecular clone, i.e. it is a genetically homogeneous progeny of a single parental molecule. Recently we have used PCR and realized in nanocolonies the idea of gene cloning (more exactly, cloning of protein-encoding DNA sequences surrounded by all elements necessary for transcription and translation), their expression in nanocolonies, and in situ clone screening by the encoded protein functions [78, 116, 117]. In other words, it was possible to carry out real molecular cloning, that unlike traditional gene cloning is called “molecular cloning” but in reality it is not cloning of molecules, but cloning of cells or viruses propagated in these cells.
Genes of bioluminescent proteins were used as models for cloning and expression in nanocolonies. The advantage of these proteins is their easy detection on the basis of functional activity: by chemiluminescence (like obelin from hydroid polyp Obelia longissima [118] and luciferase of the firefly Luciola mingrelica [119]) or by fluorescence (GFP, green fluorescent protein from the medusa Aequorea victoria) [120].
Gene Amplification in the Form of Nanocolonies
Although amplification of a relatively short fragment is sufficient for detection of a specific sequence, as in diagnostics of different diseases, gene cloning requires amplification of long sequences. It was logical to suppose (and it was really so) that in a less dense medium PCR and gene expression reactions (transcription and translation) will be more efficient. At the same time, the gel should exhibit sufficient density in order the colonies remain compact. A 5% acrylamide with the acrylamide/methylene-bis-acrylamide ratio 100 : 1 appeared to be optimal [78]. The use in PCR of thermostable DNA polymerases of Thermus aquaticus (Taq DNA polymerase) and of the hyperthermophile Pyrococcus woesei (Pwo DNA polymerase) also resulted in a noticeable increase of the product in colonies. Carrying out PCR during gene amplification in the presence of Pwo DNA polymerase is also useful due to the presence in this polymerase of 3′→5′-correcting activity: gene amplification appears to be more exact, with fewer mistakes [121].
As a result, nanocolonies containing up to 108 and more gene copies were obtained. The number of colonies in this case coincides, within the limits of statistical scattering, with the number of seeded DNA molecules (Fig. 7a), which allows cloning and testing up to 100% elements of a genetic library (compared to 0.0001-0.01% in intracellular cloning [122]).
Cell-Free Cloning of cDNA from Biological MaterialFig. 7. Gene cloning and expression in nanocolonies. a) Amplification of the GFP gene (the 1570 bp long DNA includes the protein-encoding sequence and the elements necessary for gene expression) in the form of nanocolonies. DNA colonies grown during PCR in polyacrylamide gel were revealed by hybridization on a membrane with radiolabeled probe. b) GFP gene expression within nanocolonies. When PCR was over, the gel was washed successively with ethanol-saline solution and 50% ethanol to remove PCR components, dried, impregnated with components of the cell-free transcription-translation system, and incubated for 30 min at 25°C. “GFP” are colonies of green fluorescent protein. Protein fluorescence was registered directly in the gel using a blue laser (488 nm) and a green emission filter (508 nm). The “GFP gene” are DNA colonies with GFP gene in the same gel. An autoradiograph of the membrane hybridized with the labeled probe is shown.
The applicability of the method for cloning cDNA up to 2⋅103 bp in length from natural biological material was shown on an example of mRNA for luciferase of Luciola mingrelica. In this case it could be isolated without preliminary amplification of genetic material from a portion of total RNA corresponding to a single cell, which is practically impossible with traditional cloning techniques.
The clones obtained were expressed in situ. To carry out transcription, the PCR gel was simply dried and impregnated with solution containing transcription buffer, DNA-dependent RNA polymerase, and rNTP. Incubation of the gel for 2 h at 37°C resulted in synthesis in colonies of at least 10 RNA copies up to 1700 nt per each DNA copy [78]. This variant enables cloning and direct selection of RNA molecules as ribozymes and aptamers.
Gene Expression in Nanocolonies
It was necessary to modify the procedure for protein synthesis in nanocolonies because it appeared that PCR components (especially buffer solution with relatively high pH) inhibit translation. The problem could be overcome by soaking the gel in the alcohol-saline solution that was first used by us for extraction of the reverse transcriptase and PCR inhibitors from alcohol precipitates during nucleic acid isolation from clinical samples [75]. Such treatment resulted in washing out of translation inhibitors from the gel, but DNA colonies were completely retained in this case. Then the gel was dried and impregnated with a mixture containing all components for combined transcription-translation.
Expression in nanocolonies of the green fluorescent protein (GFP) gene of Aequorea victoria resulted in synthesis of a functionally active (and thus correctly folded) polypeptide (Fig. 7b, on the left). Fluorescence of GFP synthesized in nanocolonies can be observed in real time. The colonies become detectable in 30 min and their fluorescence reaches maximum in 2 h after start of the combined transcription-translation. The rate of the fluorescent GFP accumulation is defined both by the rate of polypeptide synthesis and by the rate of protein maturation, including the chromophore formation [123]. It follows from comparison of the rates of these processes in liquid and in gel that the immobilized medium does not inhibit either translation or protein maturation [78].
The number of fluorescent spots correlates with the number of GFP gene molecules introduces into the gel before PCR. Moreover, the arrangement of fluorescent spots practically coincides with the arrangement of GFP-specific DNA colonies detected in the same gel by hybridization with the radiolabeled probe (Fig. 7b, on the right). On the average, about 109 protein molecules are synthesized, which is enough for its detection in situ, including that by binding to antibodies or ligands as well as by enzyme activity.
Advantages of Cloning in Nanocolonies
A classic and generally accepted method of gene cloning is cloning in vivo, including gene insertion into a vector molecule, transformation of bacterial or eukaryotic cells by the obtained genetic construct (like a plasmid), and growing the transformed cell colonies (clones) on a solid medium (like agar in a Petri dish) [121]. Besides the gene under study, such clones contain cell components that make necessary isolation of plasmid DNA. Moreover, in the course of cloning the gene under study inevitably undergoes natural selection. And finally, one of the most significant limitations is the efficiency of gene insertion into the vector and transformation. Only 0.0001-0.01% of the initial DNA preparation (the gene library content) give rise to individual clones [122].
Gene cloning in nanocolonies has a number of advantages over cloning in vivo. First, gene cloning and expression in nanocolonies are possible without natural selection and in the presence of unnatural nucleotides and amino acids. Second, nanocolonies make possible direct gene screening by properties of their expression products, because there are no cell walls and membranes. Third, conditions for analysis of properties of the nanocolony expression products may differ from conditions of transcription-translation: the composition of reaction medium can be easily changed by gel impregnation with an appropriate solution. Fourth, a nanocolony is genetically pure DNA that can be immediately used for genetic manipulations, omitting the stage of isolation. Finally, nanocolonies link the gene to the product of expression and thus provides for molecular display. Unlike the phage and other forms of genetic display, in this case linking of a protein to its gene does not require protein modification by the tag-sequence or “fusion” with another protein, which may influence the structure and function of the sought protein. This means that cloning in nanocolonies is able to do the same as traditional cloning and even much more.
Gene cloning in nanocolonies has a number of advantages over cell-free cloning in liquid [21]. A variety of methods have been proposed for molecule cloning by DNA amplification in liquid, which in principle could be helpful in obtaining individual clones. These approaches are based on exhausting dilution of template solutions so that just single molecules could get into the amplification reaction. A tube or a plate well [97, 124], a microchip element [125], microsphere [126], or a drop of water-oil emulsion [127, 128] can be used as reaction compartments.
Although each of these methods may result in clone isolation, the clone purity of obtained preparation is not assured and should be determined by independent techniques. It follows from the Poisson distribution that if the template is diluted so that on the average there is one molecule per one reaction compartment (like a well), then 36.8% wells will be empty, 36.8% will contain one molecule each, and 26.4% wells will contain two and more molecules [21]. The acceptable probability of the pure clone isolation (99.5%) is expected in the case of mean well population no higher than 0.01 (i.e. at template dilution to one molecule per 100 wells).
In the case of nanocolonies there are no discrete compartments. Of course, if more than a single molecule is seeded onto a gel, some colonies may overlap. With colony diameter ~0.5 mm and gel diameter 14 mm (Fig. 7) the probability that the isolated DNA is not a clone is equal to 0.02% when two molecules and 0.5% when 25 molecules are seeded [21]. In this case it should be kept in mind that the increase in the gel density and the length of amplified fragment may allow reducing the diameter of nanocolonies to 10 µm and less [10]. This allows resolution of tens of thousands clones on a gel 14 mm in diameter.
STUDIES ON GENES AND THEIR EXPRESSION
Sequencing in Nanocolonies
Sequencing by synthesis. In 2003 G. Church et al. [129] demonstrated the possibility of direct DNA sequencing in nanocolonies. To this end, they created a library of short DNA fragments flanked by the primer-binding sites and amplified these fragments in nanocolonies using PCR. One primer was covalently “cross-linked” to the gel, owing to which its elongation resulted in immobilization on the gel matrix of one strand of synthesized DNA. After PCR, DNA was denatured in formamide and the not cross-linked complementary strand was removed by electrophoresis.
Sequencing was carried out simultaneously in all colonies using “sequencing by synthesis” elaborated previously by Affimetrix company for detection of single nucleotide substitutions [130]. The immobilized DNA was hybridized with a primer, complementary to the flanking region of a known sequence, and then the primer was elongated by DNA polymerase using a fluorescently labeled nucleotide. After that, the gel was washed free of the non-incorporated nucleotide and scanned. This made it possible to identify a colony with the newly-incorporated nucleotide and to determine the template nucleotide opposite to the incorporated one. Nucleotides with free 3′-hydroxyl group were used for strand elongation (“reading through” more than one base). In order that the fluorescent label from the preceding cycle did not interfere with detection of nucleotides introduced during following cycles, the fluorophore was cross-linked to the nucleotide by a cleavable bond, disulfide [131] or photolabile [132], and scanning the fluorophore was removed by mercaptoethanol treatment or UV radiation. Then the elongation-scanning-cleavage cycle was repeated with another fluorescently-labeled nucleotide.
In theory, the cycles can be repeated as long as necessary, to complete detection of the immobilized DNA sequence upstream from the hybridized primer in each colony. The single cycle yield is 99.8%, which allows reading up to 150 nucleotides. In practice, the procedure was limited to 10-14 cycles due to the detachment of the gel from the glass. However, this is just a technical problem that can be solved in different ways. Thus, sequencing is possible on nylon membrane after the nanocolony content is transferred onto it [84]. Since single-stranded DNA after denaturing (in alkali) can be immobilized on the membrane, it is not necessary to immobilize one of primers on the gel matrix and to remove not immobilized DNA strands from the gel by electrophoresis.
Another problem is associated with homonucleotide tracts, reading of which may result in primer elongation for several nucleotides during one cycle. Measurement of the colony fluorescence intensity does not allow exact determination of the number of simultaneously incorporated nucleotides. A solution of this problem is the use of fluorescent nucleotides with reversibly blocked 3′-hydroxyl functioning as elongation terminator [133]. In this case, independently of the length of homonucleotide tract, only a single nucleotide can be added in each cycle.
Sequencing of nanocolonies both in a gel and on a membrane can be enhanced by primer elongation by a mixture of deoxynucleotides where individual fluorophore color corresponds to each base type (Fig. 8). Such method is already elaborated for sequencing on DNA chips [134], and there are no contraindications to its application to nanocolony sequencing.
The transfer of the nanocolony material into a capillary by electrophoresis and following sequencing using traditional capillary electrophoresis can be an alternative approach [135].
Sequencing by oligonucleotide ligation. The adjacent template sequence can be determined by primer ligation with all possible fluorescently labeled complementary oligonucleotides instead of multiple successive elongations of immobilized primer. Recently [136] the feasibility of such approach has been shown on an example of sequencing DNA fragments amplified by the RCA technique and immobilized on microgranules introduced into the layer of polyacrylamide gel. The primer was ligated with a set of fluorescently labeled up to 9-nt long oligonucleotides, degenerate in one of nine positions. In each mixture, the fluorophore color corresponded to one of four possible bases in the degenerate position.Fig. 8. Principle of sequencing by synthesis. Gel pictures with nanocolonies are shown after the first four cycles. A mixture of all four rNTP labeled by different color fluorophores are used for primer elongation (letters against the gray background). The colony color shows what nucleotide was incorporated into the growing strand by DNA polymerase. The nucleotide incorporated during the given cycle into the colony, marked by an arrow, is underlined under each gel picture on the scheme; the read-through template sequence is shown in bold.
One sequencing cycle included hybridization of DNA strands, immobilized on microgranules, with one of nine mixtures of labeled oligonucleotides, ligation, washing off not incorporated labeled oligonucleotides, gel scanning at four wavelengths, and removal of ligation products. The next cycle was carried out with a mixture of labeled oligonucleotides degenerate in another position. It appeared that the template can be so read through at a distance of 6-7 nucleotides from the site of ligation with primer.
The E. coli genome was re-sequenced using the described method. Since sequenced fragments were obtained by random cleavage of the E. coli genome, these fragments were overlapped, which made it possible to reconstruct the complete genome. Several distinctions from the E. coli genome sequence kept in GeneBank were found.
Since tens of thousands of nanocolonies can be resolved in a single gel [10, 137], this approach is potentially able to enhance significantly the sequencing output and lower its cost. Since relatively short sequences are read through, sequencing in nanocolonies is the best suited for re-sequencing of already known genomes for improvement and detection of individual differences.
Analysis of Single-Nucleotide Polymorphism
Individual peculiarities of different human genomes, like of any other biological species, often are single-nucleotide substitutions (SNP). The SNP-mapping is important for identification of genes associated with genetic diseases, for detection of diagnostic markers, and prediction of drug resistance and infectious pathogens [130].
Haplotype determination. Recent investigations have shown that in many cases SNP exhibit a cis-effect, i.e. they influence gene expression or function of encoded protein only if a certain combination of several SNP is present in one and the same DNA molecule [138]. The set of SNP in one chromosome is called haplotype. As a rule, SNP are inherited in large haplotypic blocks of over 100,000 bp (the mean gene size), and a limited number of each block variants circulate in the population [139-141]. It was found that the haplotypic block structure is conserved among mammals [142]. The haplotype determination has diagnostic and prognostic significance, but it is a complicated technical problem, first of all due to the difficulties in separation of sister chromosomes of the diploid genome. The first human haplotypic map with detailed SNP structure of ten 500,000 bp regions has been recently designed by efforts of a large international consortium [143]. The cost of this project was 100 million US dollars [144].
G. Church et al. were the first who demonstrated that the use of nanocolonies enables easy and quick haplotype determination without separation of sister chromosomes [77]. This is possible because amplification in gel of cis-elements (of two or more segments of the same DNA molecule, for example, loci of the same chromosome) results in emergence of a mixed nanocolony. At the same time, separate nanocolonies are formed upon amplification of sites of different molecules (for example, of different chromosomes). Belonging of two or more SNP to the same colony can be detected by sequencing by synthesis or by ligation (see above), or by colony hybridization with oligonucleotide probes under conditions when a single mutation results in significant hybrid destabilization.
Recently G. Church et al. have mapped SNP on a 153⋅106 bp long segment of the seventh human chromosome [145]. To do this, they introduced condensed chromosomes (smaller than 1 µm) into polyacrylamide gel, then proteins were removed by proteinase K and chromosomal DNA was degraded to relatively short fragments by freezing-thawing of the gel. DNA fragmentation resulted in increased yield of PCR product in colonies, but in this case connection between SNP belonging to the same chromosome was not broken, because fragments were immobilized in the gel. The use of nanocolonies in the same gel allowed distinguishing the SNP groups belonging to 414 different chromosomes, and exact haplotype identification during analysis of a heterogeneous mixture of chromosomes of several tens of people.
The authors state that it is possible to determine in one gel the 14-locus haplotypes of 50 humans, which replace 2600 allele-specific liquid PCR, thus increasing the efficiency of analysis by three orders of magnitude. According to the authors, such remote haplotyping makes possible detection and characterization of recombinations occurring in single cells at meiosis, and even more rare recombinations occurring at mitosis, as well as chromosomal translocations resulting in malignant transformation of normal cells.
Determination of viral drug resistance. Another example of nanocolony applications is the search for SNP causing drug resistance in a population of human immune deficiency virus (HIV) in a particular patient [137]. The work was carried out at Duke University (USA). It was shown that the nanocolony single-step sequencing by synthesis (see above) enables detection of minor variants of the virus exhibiting multiple drug resistance even if they make up 0.01% of the total population. This exceeds by three orders of magnitude the sensitivity achieved by direct sequencing of viral RNA (20%). Revealing the drug resistant viral variants makes it possible to choose the most adequate scheme of therapy for the particular patient. Among advantages of such approach over other existing methods, the authors point to the absence of false-positive results due to recombinations in liquid PCR.
A similar approach was used for quantitative determination of known drug resistance mutations in the fused (chimeric) gene of tyrosine kinase BCR-ABL that is expressed in a type of chronic myeloid leukemia [146]. Nine mutations in the kinase domain of chimeric gene BCR-ABL define over 90% cases of resistance to the kinase inhibitor imanitib that is widely used for chemotherapy [147]. Nanocolonies enable detection of a single imanitib-resistant cell among 104 sensitive ones [148].
Nanocolonies obtained using isothermal DNA amplification by the RCA technique were used for quantitative analysis of gene P16 methylation in stomach tumor cells [149]. Preliminary treatment of the DNA with bisulfite results in the conversion of unmodified cytosine to uracil without affecting methylated cytosines. The C→U substitutions formed after such treatment were detected by nanocolony hybridization with fluorescent probes.
Studying Alternative Splicing
Similarly to detection of SNP cis-combinations on the same chromosome, the search and quantitative estimation of different splicing variants are possible, because exons belonging to one mRNA molecule will be present in one nanocolony.
There can be many alternative splicing variants of the same transcript. If the presence or absence of one exon produces only two variants of mRNA, 10 exons are able to form up to 210 or 1024 combinations either containing the given exon or not. In standard analysis of alternative splicing using liquid RT-PCR, minor variants can remain undetected and the labor-consuming analysis is required to reveal all variants and estimate their proportion. Besides, in the case of analysis in liquid the emergence of false splicing variants is possible due to recombinations during reverse transcription and PCR.
G. Church et al. [150] have shown that nanocolonies can be used to overcome these problems (Fig. 9). After reverse transcription and following PCR in gel with a pair of primers, corresponding to the terminal constant exons and allowing amplification of all mRNA variants, nanocolonies were detected by hybridization with exon-specific probes. As a result, it was found for each colony which exons were present in it and which were absent. This made it possible to detect in one gel all variants of splicing and estimate exact number of copies of each variant. In this work, to identify exon in nanocolonies, the latter were hybridized with unlabeled oligonucleotide probes that were then labeled in situ by a single fluorescent nucleotide (see above). However, the same information could be obtained easier and more rapidly by nanocolony hybridization with preliminarily labeled fluorescent probes on nylon membrane. Such membrane can be repeatedly used for successive hybridization with different probes (or with different sets of probes) [84].
The efficiency of nanocolonies in investigation of alternative splicing was demonstrated with the example of the primary gene transcript of mouse CD44 [150], a glycoprotein exposed on the cell surface and involved in intercellular interactions. All 10 exons encoding the extracellular part of this protein are variable. Analysis of ~9000 nanocolonies revealed 69 mRNA species with different exon composition that more than twice exceeded the number of isoforms found previously. Comparison of exon composition of mRNA in normal and transformed cells has shown that malignant transformation results in extremely reliable alterations in splicing profile (P < 10-10), which could serve as a diagnostic feature. Changes in the content of minor forms and just by several times are observed. It would be impossible to detect such changes by non-digital methods of analysis such as liquid RT-PCR. These results confirm that isoforms of alternative splicing can serve as markers of oncological diseases, but only if a digital method is used for their quantitative determination.Fig. 9. Detection and quantitative determination of alternative splicing variants using nanocolonies. a) Possible exon combinations. The RNA sequence beyond variable exons is shown by a black line; three variable exons are designated by black, gray, and white rectangles; arrows point to the annealing sites of PCR primers. b) The nanocolony hybridization with the probes, marked by the same colors as exons, means that all these exons are present in the molecule that gave rise to this nanocolony.
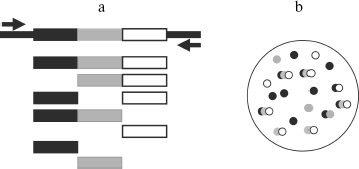
Another group of researchers applied nanocolonies for determination of a relative level of two products of K-ras oncogene alternative splicing, K-RAS2A and K-RAS2B [151]. Nanocolonies made possible the reliable detection of alterations in exon composition of mRNA of τ protein, playing an important role in neuron morphology and associated with Alzheimer's disease, although these alterations are not revealed in liquid PCR [152]. Nanocolonies also showed the linkage of a certain SNP cis-combination with a variant of alternative splicing of the survival motor neuron (SMN) gene mRNA in spinal muscular atrophy [150].
Determination of Allele and mRNA Copy Numbers
Since direct counting nanocolonies is possible, they enable exact determination of individual DNA and RNA copy numbers using a procedure elaborated by us for determination of virus and tumor-specific marker titers. In this case, any convenient method, including hybridization on membrane or in gel (using homogeneous detection systems) as well as primer elongation by a labeled nucleotide can be used for detection of nanocolonies.
In one of the first works of this kind, nanocolonies were used for determination of the ratio of individual yeast strains upon their competitive growth. In this experiment, eight strains carrying plasmids encoding different phosphoglycerate kinase mutants were mixed and grown together. The change in the ratio of individual strains during their combined growth was determined by counting the number of colonies grown upon seeding onto a gel of total plasmid DNA and containing appropriate SNP [153]. The same approach was used later for quantitative analysis of competition between 54 yeast strains producing different mutant forms of human glucose-6-phosphate dehydrogenase [154].
In another work, nanocolonies were used to detect deletions resulting in the loss of heterozygosity (LOH) [155]. If LOH occurs within a gene-suppressor of tumor growth and the remaining gene copy is inactive (for example, due to point mutation), the cell may undergo malignant transformation. Owing to this, LOH of cancer gene suppressors are used as a diagnostic feature. The authors used nanocolonies for analysis of a pancreatic cell line (Panc-1) and found that the number of the K-ras2 colonies was almost doubled compared to the number of gene p53 colonies, which is indicative of the role of LOH relative gene p53.
One promising application of nanocolonies is digital monitoring of gene expression by counting the transcript copy number in individual cells or in a group of cells at a given moment. Such approach makes it possible to detect with a high sensitivity low but statistically reliable and biologically significant alterations of gene expression. The potential of nanocolonies was demonstrated in measurement of expression of 14 genes involved in galactose metabolism in yeast Saccharomyces cerevisiae cells grown in a medium minimal in glucose or galactose [156]. The authors concluded that nanocolonies present unlimited possibilities for functional genomics, because such method of the gene expression estimation is digital in its nature and therefore is the most precise among all existing techniques. They have also noted that the method of nanocolonies has a wider dynamic range compared to other methods of studying gene expression profiles: it allows measurement with identical accuracy of expression levels of the least and most active genes.
In a recent work, copy numbers of some mRNA have been measured using nanocolonies in a limited number of embryonic stem cells [157]. The amount of RNA isolated from 1/5 of a single blastocyte appeared to be enough for statistically reliable determination in one gel of titers of three different mRNA, the content of which differed by one order of magnitude.
Finally, nanocolonies enable exact measurement of relative expression levels of different alleles of the same gene. Allele variants of mRNA were detected as in the SNP analysis: by nanocolony hybridization with an unlabeled primer, labeled in situ by the fluorescent nucleotide incorporation. The ratio of allele activities was determined precisely by simple counting of the colonies labeled by different fluorophores. This approach was tested on measurements of a relative level of proteinase D2 expression in eight heterozygous patients with SNP 4208G/A [158]. The ratio of alleles determined to be 1 : 1 served as a control. The authors point out that nanocolonies allow estimation of absolute levels of allele expression, whereas in other methods the results are normalized to the averaged ratio of allele expression which is assumed to be 1 : 1.
PERSPECTIVES OF NANOCOLONIES
The use of nanocolonies makes it possible to determine the level of gene expression in single cells, to determine the amounts of individual DNA and RNA in many cells simultaneously, to carry out gene selection according to various properties of encoded RNA or proteins, to study functional interactions of proteins, and to detect nanomolecules of non-nucleic nature.
Studying gene expression in single cells. The sensitive and quantitative assays with nanocolony technique make it possible to analyze with high accuracy the contents of a single cell and, moreover, to analyze nucleic acids upon taking material by a manipulator from different single cell compartments (nucleus, organelles, different zones of the cytoplasm). Such approach should allow one to study gene expression and distribution of template and other RNA within cells during differentiation, at different stages of the cell cycle, in various pathologies, and in response to external factors.
Quantitative in situ assay. We recently proposed a quantitative in situ assay (QISA) [88, 92] using nanocolonies and allowing quantitative determination of the nucleic acid content in each of numerous cells embedded within a single gel.
When colony growth is monitored in real time, virtually all colonies become visible simultaneously (Fig. 4). This happens because each nanocolony is produced by a single molecule. However, if a nanocolony is produced by a cluster of molecules, it becomes visible at earlier PCR cycles [88, 92]. Strictly speaking, such a colony is not a molecular clone, because it represents a mixed progeny of different molecules. The initial number of molecules in each colony can be determined similarly to how it is done in a usual quantitative PCR in liquid for determination of the target amount in the tube, by noting the number of the cycle in which the fluorescent signal reaches a certain threshold level.
For QISA, individual cells or cell groups (like tissue slices) are introduced into the gel during its preparation. The cells are lysed also in gel (for example, by soaking the gel with solution of SDS or guanidine thiocyanate), then lysing reagents and most of cell components are washed away with ethanol-saline mixture, causing precipitation of high-molecular-weight RNA and DNA at the location occupied by the cell before lysis. After salt removal with ethanol and drying, PCR is carried out in the gel. In the case of RNA determination, it is preceded by reverse transcription [88, 92].
At first glance, QISA resembles the well-known method of in situ PCR [159, 160], but these two methods differ in several principal ways. In traditional in situ PCR amplification is carried out in liquid, cells in this case are either suspended in solution, or attached to a solid surface. The main problem of this method is how to deliver reaction components (dNTP, primers, polymerase) into a cell and to prevent the leakage of nucleic acids (DNA- or RNA-target and PCR products) from the cells. In practice, this problem has no satisfactory solution. The treatment of the cell makes its membrane permeable for added PCR components, but the same treatment results in leakage of nucleic acids from the cell and their migration into other cells. Therefore, neither precise localization of the target under study nor its quantitative determination is possible.
QISA includes full lysis of cells immobilized in the gel, which provides for complete accessibility of cellular polynucleotides to the components of the amplification reaction. Owing to the medium immobilization, both cellular polynucleotides and their amplification products remain at the location where the cell was present, and the monitoring nanocolonies in real time allows simultaneous measurement of the target amount in each cell.
QISA is applicable for detection and determination of titers of microbial pathogens in clinical samples such as blood or feces. QISA can be used for cancer diagnostics, for example, for detection of cells among normal leukocytes that produce chimeric AML1-ETO mRNA [86], and for estimation of the chimeric gene expression level in individual leukemia cells. It is possible to use QISA for determination of mRNA localization and local concentration in tissue slices. Analysis of a series of successive slices would allow reconstruction of a three-dimensional map of expression of a certain gene or a group of genes in a tissue, organ, and even in a whole organism such as an embryo.
Gene selection by functions of encoded RNA and proteins. Nanocolonies are a kind of genetic display, because they combine in the space the gene and its expression product. This makes it possible to isolate from genetic library those genes or their variants whose expression products (RNA and protein) exhibit required functions.
Transcription of nanocolonies can be used for direct selection of RNA molecules with desired properties, such as ribozymes or aptamers. Since a nanocolony already contains a DNA clone encoding the selected RNA, reverse transcription of this RNA and cloning its cDNA in vivo become unnecessary.
We have shown by the example of GFP that a functionally active protein can be obtained upon gene expression in nanocolonies. In the same way it is possible to carry out screening of the library of GFP and other fluorescent protein variants, selecting nanocolonies by the fluorescence color and intensity. Nanocolonies can also be selected by the ability of produced proteins or peptides to bind particular ligands, antibodies, and cellular receptors or to catalyze certain chemical reactions. In the last case, it is convenient to carry out screening if an intensely stained or fluorescent product is formed.
The use of translation systems composed of purified components opens additional possibilities. Such a system has been recently designed in the laboratory of T. Ueda from translation components of Escherichia coli. It includes purified ribosomes, translation factors, all aminoacyl-tRNA-synthases, and all tRNAs [161]. The advantages of a pure cell-free translation system are its higher efficiency with PCR product as template (in particular, due to the absence of nucleases) and the possibility to control the composition of the reaction mixture. The use of chemically modified aminoacyl-tRNAs allows one to introduce amino acid derivatives into the translation product, to carry out translation in the absence of some components (like termination factors, which makes it possible to fix the synthesized protein on the template), or on the contrary, in the presence of some additional substances.
The use of a pure cell-free transcription-translation system will make it possible to expand the set of features suitable for selection of a protein synthesized in nanocolonies. Screening of nanocolonies is impossible with usual transcription-translation systems prepared on the basis of a crude cell extracts if the extract itself contains the enzyme or any other activity used for selection. The use of cell extract can also preclude nanocolony screening by binding to certain ligands or antibodies due to high unspecific background. The pure cell-free expression system is free of these disadvantages.
Investigation of protein functional interactions. Another possibility for the use of gene expression in nanocolonies is the study of functional interactions of proteins. In fact, expression of nanocolonies enables production of protein microchips. The treatment of a gel or its replica (membrane) with the labeled protein, polynucleotide, or a low-molecular-weight ligand will also label nanocolonies if they synthesize proteins exhibiting affinity to the labeled substance. To prevent diffusion of the synthesized protein, it is possible to immobilize it on the gel matrix, for example, by furnishing the synthesized proteins with oligohistidine tags and gel matrix with the groups chelating zinc or nickel ions.
Detection of molecules of non-nucleic nature. Nanocolonies can be potentially used for detection of single molecules of substances other than DNA and RNA. It is possible to detect a protein capable of simultaneous binding to two ligands like two different antibodies [162]. To this end, ligands are linked to two DNA fragments. As a result of ligand binding to the protein, these fragments come to close proximity, which makes possible their combination in one molecule with the help of DNA ligase. The amplification in gel of a combined DNA molecule, a substitute of the original target, will result in formation of a nanocolony. In addition to antibodies, DNA- or RNA-aptamers as well as such ligands as substrate analogs or enzyme cofactors can play the role of protein-specific reagents. Evidently, such approach can be used for detection of single molecules of any substance having sufficiently complex surface to form two or more high-affinity binding sites. Such approach can be used both for diagnostics (as for detection of single drug or doping molecules) and for detection of rare products or intermediates of chemical reactions.
Various applications of nanocolonies are based on their unique ability to separate in space (compartmentalize) the amplification and expression of individual DNA or RNA molecules. Three groups of applications, already used now, immediately follow from this basic ability: (i) cloning template nanomolecules; (ii) detection and quantitative determination of individual molecules; (iii) identification of cis-elements of complex molecules, i.e. the elements covalently bound to each other as the result of belonging to the same molecule.
Cloning is the most natural application of nanocolonies, because, by its origin, each individual colony is a molecular clone (i.e. copies of a single DNA or RNA molecule). Cloning in nanocolonies is incomparably more efficient than methods based on exhausting template dilution. In the case of combination with expression in situ, cloning in nanocolonies can be used for detection of interrelationship between gene and encoded function (carried out by a protein and/or RNA), for identification of gene groups encoding interacting proteins and/or RNA, for revealing regulatory networks, as well as a universal instrument for creation of new proteins, design of aptamers, and drug discovery.
The ability to detect and quantify individual molecules makes nanocolonies a powerful tool for a variety of analytical applications. The examples include detection of the rare chemical reaction products, absolute quantitative determination of individual gene copies, their alleles, any RNAs, including those in single cells, and undoubtedly, any kind of quantitative molecular diagnostics (diagnostics of infectious and oncological diseases, environmental monitoring, solution of forensic problems, and detection of trace amounts of genetically modified organisms).
Unlike standard methods based on nucleic acid amplification in liquid, the diagnostics using nanocolonies does not require measurement of signal intensity, only the presence or absence of a detectable colony is important independently of the number of constituent molecules. This means that the method of nanocolonies is digital by nature and thus more reliable compared to the analog methods based on signal intensity measurements.
Another important peculiarity of nanocolonies is spatial separation of amplification of individual DNA or RNA molecules present in the sample. This practically excludes the competition between simultaneously amplifying templates in multiplex analysis as well as interference from nonspecific synthesis, which, in the case of amplification in liquid, is a factor limiting the sensitivity of target detection in clinical samples. As a result, the method of nanocolonies exhibits enormous sensitivity allowing detection of one DNA and two RNA molecules in the presence of trillion-fold excess of foreign nucleic acids.
Finally, the ability to identify cis-elements forming mixed colonies makes nanocolonies a unique instrument for gene mapping and haplotype determination, elucidation of the RNA exon composition, and sequencing in situ.
Thus, the use of nanocolonies can be quite useful for different fields of fundamental science, biotechnology, and medicine, such as chemistry of single molecules, cell-free gene cloning and screening, sequencing, and molecular diagnostics. In each of these directions, nanocolonies exhibit outstanding results. This shows that the method has become a developed technology with unique abilities and a high potential for solution of scientific and applied problems.
The authors are grateful to our colleagues participated in development of nanocolony technologies in Russia--T. R. Samatov, M. V. Falaleeva, V. I. Ugarov, and A. V. Kravchenko.
This work was supported by the “Molecular and Cellular Biology” Program of the Presidium of the Russian Academy of Sciences, Russian Foundation for Basic Research, Federal Targeted Scientific-Technological Program “Investigations and Developments in the Priority Directions of Science and Technology”, Howard Hughes Medical Institute, INTAS European Agency, and International Research Foundation.
REFERENCES
1.Chetverin, A. B., Chetverina, H. V., and Munishkin,
A. V. (1991) J. Mol. Biol., 222, 3-9.
2.Chetverin, A. B., and Chetverina, H. V. (1995) The
RF Patent No. 2048522.
3.Chetverin, A. B., and Chetverina, H. V. (1998) The
RF Patent No. 2114175.
4.Chetverin, A. B., and Chetverina, H. V. (1998) The
RF Patent No. 2114915.
5.Chetverin, A. B., and Chetverina, H. V. (1997) US
Patent 5,616,478.
6.Chetverin, A. B., and Chetverina, H. V. (1999) US
Patent 6,001,568.
7.Chetverin, A. B., and Chetverina, H. V. (1999) US
Patent 5,958,698.
8.Chetverina, H. V., and Chetverin, A. B. (1993)
Nucleic Acids Res., 21, 2349-2353.
9.Chetverin, A. B., Chetverina, H. V., Demidenko, A.
A., and Ugarov, V. I. (1997) Cell, 88, 503-513.
10.Mitra, R. D., and Church, G. M. (1999) Nucleic
Acids Res., 27, e34.
11.Koch, R. (1881) Mittheilungen aus dem
Kaiserlichen Gesundheitsamte., 1, 1-48.
12.Saiki, R. K., Scharf, S., Faloona, F., Mullis, K.
B., Horn, G. T., Erlich, H. A., and Arnheim, N. (1985) Science,
230, 1350-1354.
13.Guatelli, J. C., Whitfield, K. M., Kwoh, D. Y.,
Barringer, K. J., Richman, D. D., and Gingeras, T. R. (1990) Proc.
Natl. Acad. Sci. USA, 87, 1874-1878.
14.Compton, J. (1991) Nature, 350,
91-92.
15.Walker, G. T., Little, M. C., Nadeau, J. G., and
Shank, D. D. (1992) Proc. Natl. Acad. Sci. USA, 89,
392-396.
16.Wu, D. Y., and Wallace, R. B. (1989)
Genomics, 4, 560-569.
17.Barany, F. (1991) Proc. Natl. Acad. Sci.
USA, 88, 189-193.
18.Fire, A., and Xu, S.-Q. (1995) Proc. Natl.
Acad. Sci. USA, 92, 4641-4645.
19.Chetverin, A. B. (1998) Uspekhi Biol.
Khim., 38, 3-75.
20.Chetverin, A. B., and Spirin, A. S. (1995)
Progr. Nucleic Acid Res. Mol. Biol., 51, 225-270.
21.Chetverin, A. B., and Chetverina, H. V. (2007)
Mol. Biol. (Moscow), 41, 284-296.
22.Brown, D., and Gold, L. (1995)
Biochemistry, 34, 14775-14782.
23.Sumper, M., and Luce, R. (1975) Proc. Natl.
Acad. Sci. USA, 72, 162-166.
24.Biebricher, C. K., Eigen, M., and Luce, R. (1981)
J. Mol. Biol., 148, 369-390.
25.Biebricher, C. K., Eigen, M., and Luce, R. (1986)
Nature, 321, 89-91.
26.Biebricher, C. K., Eigen, M., and McCaskil, J.
S. (1993) J. Mol. Biol., 231, 175-179.
27.Chetverin, A. B. (1996) Mol. Biol.
(Moscow), 30, 981-990.
28.Crick, F. H. (1958) Symp. Soc. Exp. Biol.,
12, 138-163.
29.McCaskill, J. S., and Bauer, G. J. (1993)
Proc. Natl. Acad. Sci. USA, 90, 4191-4195.
30.Biebricher, C. K., Eigen, M., and Luce, R. (1981)
J. Mol. Biol., 148, 391-410.
31.Lizardi, P. M., Guerra, C. E., Lomeli, H.,
Tussie-Luna, I., and Kramer, F. R. (1988) BioTechnology,
6, 1197-1202.
32.Paranchych, W. (1975) RNA Phages (Zinder,
N. D., ed.) Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, New York, pp. 85-111.
33.Hirst, G. K. (1962) Cold Spring Harbor Symp.
Quant. Biol., 27, 303-309.
34.Ledinko, N. (1963) Virology, 180,
107-119.
35.Romanova, L. I., Tolskaya, E. A.,
Kolesnikova, M. S., and Agol, V. I. (1980) FEBS Lett.,
118, 109-112.
36.King, A. M. Q., McCahon, D., Slade, W. R., and
Newman, J. W. I. (1982) Cell, 29, 921-928.
37.Tolskaya, E. A., Romanova, L. I.,
Kolesnikova, M. S., and Agol, V. I. (1983) Virology,
124, 121-132.
38.Munishkin, A. V., Voronin, L. A., and
Chetverin, A. B. (1988) Nature, 333, 473-475.
39.Munishkin, A. V., Voronin, L. A., Ugarov, V.
I., Bondareva, L. A., Chetverina, H. V., and Chetverin, A. B. (1991)
J. Mol. Biol., 221, 463-472.
40.Chetverin, A. B. (1999) Mol. Biol.
(Moscow), 33, 985-996.
41.Chetverin, A. B. (1999) FEBS Lett.,
460, 1-5.
42.Lai, M. M. C. (1992) Microbiol. Rev.,
56, 61-79.
43.Kirkegaard, K., and Baltimore, D. (1986)
Cell, 47, 433-443.
44.Agol, V. I. (1997) Semin. Virol.,
8, 77-84.
45.Diaz, L., and DeStefano, J. J. (1996)
Nucleic Acids Res., 24, 3086-3092.
46.Chetverina, H. V. (2007) Molecular
Colonies: Doctoral dissertation in Biological Sciences [in
Russian], Moscow State University, Moscow.
47.Weissmann, C. (1974) FEBS Lett.,
40, S10-S18.
48.Chetverina, H. V., Demidenko, A. A., Ugarov, V.
I., and Chetverin, A. B. (1999) FEBS Lett., 450,
89-94.
49.Morozov, I. Yu., Ugarov, V. I., Chetverin, A.
B., and Spirin, A. S. (1993) Proc. Natl. Acad. Sci. USA,
90, 9325-9329.
50.Gvozdev, V. A. (1993) Mol. Biol. (Moscow),
27, 1205-1217.
51.Pasyukova, E. G., Nuzhdin, S. V., Filatov, D. A.,
and Gvozdev, V. A. (1999) Mol. Biol. (Moscow), 33,
26-37.
52.Gogvadze, E. V., and Buzdin, A. A. (2005) Mol.
Biol. (Moscow), 39, 364-373.
53.Vlasov, V. A., Dymshits, G. M., and Lavrik, O. I.
(1998) Mol. Biol. (Moscow), 32, 5-18.
54.Chetverin, A. B. (2004) FEBS Lett.,
567, 35-41.
55.Herschlag, D., Khosla, M., Tsuchihashi, Z., and
Karpel, R. L. (1994) EMBO J., 13, 2913-2924.
56.Coetzee, T., Herschlag, D., and Belfort, M.
(1994) Genes Dev., 8, 1575-1588.
57.Spirin, A. S. (2002) FEBS Lett.,
530, 4-8.
58.Spirin, A. S. (2005) Mol. Biol. (Moscow),
39, 550-556.
59.Spirin, A. S. (2005) Paleontol. Zh.,
39, 25-32.
60.Gmyl, A. P., Belousov, E. V., Maslova, S. V.,
Khitrina, E. V., Chetverin, A. B., and Agol, V. I. (1999) J.
Virol., 73, 8958-8965.
61.Gmyl, A. P., Korshenko, S. A., Belousov, E. V.,
Khitrina, E. V., and Agol, V. I. (2003) RNA, 9,
1221-1223.
62.Gallei, A., Pankraz, A., Thiel, H.-J., and
Becher, P. (2004) J. Virol., 78, 6271-6281.
63.Filipowicz, W., Billy, E., Drabikowski, K., and
Genschik, P. (1998) Acta Biochim. Pol., 45, 895-906.
64.Tsagris, M., Tabler, M., and Sanger, H. L.
(1991) Nucleic Acids Res., 19, 1605-1612.
65.Gmyl, A. P., and Agol, V. I. (2005) Mol. Biol.
(Moscow), 39, 618-632.
66.Chetverin, A. B., Kopein, D. S., Chetverina, H.
V., Demidenko, A. A., and Ugarov, V. I. (2005) J. Biol. Chem.,
280, 8748-8755.
67.Cooper, P. D., Steiner-Pryor, S., Scotti, P. D.,
and Delong, D. (1974) J. Gen. Virol., 23, 41-49.
68.Pierangeli, A., Bucci, M., Forzan, M., Pagnotti,
P., Equestre, M., and Perez Bercoff, R. (1999) J. Gen. Virol.,
80, 1889-1897.
69.Biebricher, C. K., and Luce, R. (1992) EMBO
J., 11, 5129-5135.
70.Ugarov, V. I., Demidenko, A. A., and Chetverin,
A. B. (2003) J. Biol. Chem., 278, 44139-44146.
71.Miele, E. A., Mills, D. R., and Kramer, F. R.
(1983) J. Mol. Biol., 71, 281-295.
72.Lomeli, H., Tyagi, S., Pritchard, C. G.,
Lizardi, P. M., and Kramer, F. R. (1989) Clin. Chem.,
35, 1826-1831.
73.Tyagi, S., Landegren, U., Tazi, M., Lizardi, P.,
and Kramer, F. R. (1996) Proc. Natl. Acad. Sci. USA, 93,
5395-5400.
74.Axelrod, V. D., Brown, E., Priano, C., and Mills,
D. R. (1991) Virology, 184, 595-608.
75.Chetverina, H. V., Samatov, T. R., Ugarov, V. I.,
and Chetverin, A. B. (2002) BioTechniques, 33,
150-156.
76.Chetverin, A. B., and Chetverina, H. V. (2002)
Mol. Biol. (Moscow), 36, 320-327.
77.Mitra, R. D., Butty, V. L., Shendure, J.,
Williams, B. R., Housman, D. E., and Church, G. M. (2003) Proc.
Natl. Acad. Sci. USA, 100, 5926-5931.
78.Samatov, T. R., Chetverina, H. V., and Chetverin,
A. B. (2005) Nucleic Acids Res., 33, e145.
79.Caldwell, K. D., Chu, T.-J., and Pitt, W. G.
(1992) US Patent 5,112,736.
80.Weiss, R. B., Kimball, A. W., Gesteland, R. F.,
Ferguson, F. M., Dunn, D. M., Di Sera, L. J., and Cherry, J. L. (1995)
US Patent 5,470,710.
81.Guerasimova, A., Ivanov, I., and Lehrach, H.
(1999) Nucleic Acids Res., 27, 703-705.
82.Guerasimova, A., Nyarsik, L., Girnus, I.,
Steinfath, M., Wruck, W., Griffiths, H., Herwig, R., Wierling, C.,
O'Brien, J., Eickhoff, H., Lehrach, H., and Radelof, U. (2001)
Biotechniques, 31, 490-495.
83.Andreoli, R., Amin, M., Meyering, M.,
Chesterson, R., and Ostreicher, E. (2004) US Patent 6,734,012.
84.Chetverina, H. V., Kravchenko, A. V., Falaleeva,
M. V., and Chetverin, A. B. (2007) Bioorg. Khim., 33,
456-463.
85.Chetverina, H. V., Chetverin, A. B., and
Kravchenko, A. V. (2006) Application for RF Patent
No. 2006108053.
86.Miyoshi, H., Kozu, T., Shimizu, K., Enomoto,
K., Maseki, N., Kaneko, Y., Kamada, N., and Ohki, M. (1993) EMBO
J., 12, 2715-2721.
87.Samatov, T. R., Chetverina, H. V., and
Chetverin, A. B. (2006) Analyt. Biochem., 356,
300-302.
88.Chetverin, A. B., Samatov, T. R., and
Chetverina, H. V. (2007) PCT Publication WO2007111639.
89.Mackay, I. M., Arden, K. E., and Nitsche, A.
(2002) Nucleic Acids Res., 30, 1292-1305.
90.Wittwer, C. T., Herrmann, M. G., Moss, A. A., and
Rasmunssen, R. P. (1997) BioTechniques, 22, 130-138.
91.Bernard, P. S., Ajioka, R. S., Kushner, J. P.,
and Wittwer, C. T. (1998) Am. J. Pathol., 153,
1055-1061.
92.Chetverin, A. B., Samatov, T. R., and
Chetverina, H. V. (2006) Application for RF Patent
No. 2006109271.
93.Chetverina, H. V., Falaleeva, M. V., and
Chetverin, A. B. (2004) Analyt. Biochem., 334,
376-381.
94.Chetverina, H. V., Chetverin, A. B., and
Kravchenko, A. V. (2005) Application for RF Patent
No. 2005138583.
95.Kravchenko, A. V., Chetverina, H. V., and
Chetverin, A. B. (2006) Bioorg. Khim., 32, 609-614.
96.Myers, T. W., and Gelfand, D. H. (1991)
Biochemistry, 30, 7661-7666.
97.Vogelstein, B., and Kinzler, K. W. (1999)
Proc. Natl. Acad. Sci. USA, 96, 9236-9241.
98.Chetverina, H. V., and Chetverin, A. B. (2003)
Mol. Med., 2, 30-40.
99.Rusinova, A. V., Chetverina, H. V., and
Chetverin, A. B. (2005) in Abst. 9th Int. School/Conf. for Young
Scientists in Pushchino “Biology Is a Science of XXI
Century”, Pushchino, p. 168.
100.Abelev, G. I. (1994) Immunology,
3, 4-9.
101.Falaleeva, M. V., Chetverina, H. V.,
Kravchenko, A. V., Zabolotneva, Yu. A., Samatov, T. R., Bobrynina, V.
O., Maschan, M. A., and Chetverin, A. B. (2006) in Materials of
Sci.-Pract. Conf. “Live Systems” within Framework of
Federal Sci.-Techn. Program “Research and Applications in
Priority Directions of Development in Science and Technology in
2002-2006”, pp. 238-243.
102.Cardullo, R. A., Agrawal, S., Flores, C.,
Zamecnik, P. C., and Wolf, D. E. (1988) Proc. Natl. Acad. Sci.
USA, 85, 8790-8794.
103.Klein, D. (2002) Trends Mol. Med.,
8, 257-260.
104.Saiki, R. K., Gelfand, D. H., Stoffel, S.,
Scharf, S. J., Higuchi, R., Horn, G. T., Mullis, K. B., and Erlich, H.
A. (1988) Science, 239, 487-491.
105.Meyerhans, A., Vartanian, J. P., and
Wain-Hobson, S. (1990) Nucleic Acids Res., 18,
1687-1691.
106.Paabo, S., Irwin, D. M., and Wilson, A. C.
(1990) J. Biol. Chem., 265, 4718-4721.
107.Zaphiropoulos, P. G. (1998) Nucleic Acids
Res., 26, 2843-2848.
108.Yu, W., Rusterholtz, K. J., Krummel, A. T.,
and Lehman, N. (2006) Biotechniques, 40, 499-507.
109.Jansen, R., and Ledley, F. D. (1990)
Nucleic Acids Res., 18, 5153-5156.
110.Odelberg, S. J., Weiss, R. B., Hata, A., and
White, R. (1995) Nucleic Acids Res., 23, 2049-2057.
111.Marton, A., Delbecchi, L., and Bourgaux, P.
(1991) Nucleic Acids Res., 19, 2423-2426.
112.Adessi, C., Matton, G., Ayala, G., Turcatti,
G., Mermod, J. J., Mayer, P., and Kawashima, E. (2000) Nucleic Acids
Res., 28, e87.
113.Mercier, J. F., and Slater, G. W. (2005)
Biophys. J., 89, 32-42.
114.Turcatti, G., Romieu, A., Fedurco, M., and
Tairi, A. P. (2008) Nucleic Acids Res., 36, e25.
115.Fedurco, M., Romieu, A., Williams, S.,
Lawrence, I., and Turcatti, G. (2006) Nucleic Acids Res.,
34, e22.
116.Samatov, T. R., Chetverina, H. V., and
Chetverin, A. B. (2006) Vestnik Biotekhnol., 2,
45-49.
117.Chetverin, A. B., Samatov, T. R., and
Chetverina, H. V. (2008) Cell-Free Protein Synthesis (Spirin, A.
S., and Swartz, J. R., eds.) Wiley-VCH, Weinheim, pp. 191-206.
118.Vysotskii, E. S., Markova, S. V., and Frank, L.
A. (2006) Mol. Biol. (Moscow), 40, 404-417.
119.Ugarova, N. N., Maloshenok, L. G., Uporov, I.
V., and Koksharov, M. I. (2005) Biochemistry (Moscow),
70, 1262-1267.
120.Zubova, N. N., Bulavina, A. Yu., and Savitskii,
A. P. (2003) Uspekhi Biol. Khim., 3, 163-224.
121.Sambrook, J., and Russell, D. W. (2001)
Molecular Cloning, 3rd Edn., CSHL Press, Cold Spring Harbor.
122.Roberts, R. W., and Ja, W. W. (1999) Curr.
Opin. Struct. Biol., 9, 521-529.
123.Verkhusha, V. V., Akovbyan, N. A., Efremenko,
E. N., Varfolomeeva, S. D., and Vrzheshch, P. V. (2001) Biochemistry
(Moscow), 66, 1342-1351.
124.Lukyanov, K. A., Matz, M. V., Bogdanova, E.
A., Gurskaya, N. G., and Lukyanov, S. A. (1996) Nucleic Acids
Res., 24, 2194-2195.
125.Chetverin, A. B., and Kramer, F. R. (1994)
BioTechnology, 12, 1093-1100.
126.Brenner, S., Williams, S. R., Vermaas, E. H.,
Storck, T., Moon, K., McCollum, C., Mao, J.-I., Luo, S., Kirchner, J.
J., Eletr, S., DuBridge, R. B., Burcham, T., and Albrecht, G. (2000)
Proc. Natl. Acad. Sci. USA, 97, 1665-1670.
127.Tawfik, D. S., and Griffiths, A. D.
(1998) Nat. Biotechnol., 16, 652-656.
128.Dressman, D., Yan, H., Traverso, G., Kinzler,
K. W., and Vogelstein, B. (2003) Proc. Natl. Acad. Sci. USA,
100, 8817-8822.
129.Mitra, R. D., Shendure, J., Olejnik, J.,
Krzymanska-Olejnik, E., and Church, G. M. (2003) Analyt.
Biochem., 320, 55-65.
130.Shapero, M. H., Leuther, K. K., Nguyen, A.,
Scott, M., and Jones, K. W. (2001) Genome Res.,
11, 1926-1934.
131.Shimkus, M. L., Guaglianone, P., and Herman,
T. M. (1986) DNA, 5, 247-255.
132.Olejnik, J., Krzymanska-Olejnik, E., and
Rothschild, K. J. (1998) Meth. Enzymol., 291,
135-154.
133.Welch, M. B., and Burgess, K. (1999)
Nucleosides Nucleotides, 18, 197-201.
134.Ju, J., Kim, D. H., Bi, L., Meng, Q., Bai, X.,
Li, Z., Li, X., Marma, M. S., Shi, S., Wu, J., Edwards, J. R., Romu,
A., and Turro, N. J. (2006) Proc. Natl. Acad. Sci. USA,
103, 19635-19640.
135.Kosobokova, O., Gavrilov, D. N., Khozikov,
V., Stepukhovich, A., Tsupryk, A., Pan'kov, S., Somova, O.,
Abanshin, N., Gudkov, G., Tcherevishnik, M., and Gorfinkel, V. (2007)
Electrophoresis, 28, 3890-3900.
136.Shendure, J., Porreca, G. J., Reppas, N. B.,
Lin, X., McCutcheon, J. P., Rosenbaum, A. M., Wang, M. D., Zhang, K.,
Mitra, R. D., and Church, G. M. (2005) Science, 309,
1728-1732.
137.Cai, F., Chen, H., Hicks, C. B., Bartlett, J.
A., Zhu, J., and Gao, F. (2007) Nat. Meth., 4,
123-125.
138.Davidson, S. (2000) Nat. Biotechnol.,
18, 1134-1135.
139.Daly, M. J., Rioux, J. D., Schaffner, S. F.,
Hudson, T. J., and Lander, E. S. (2001) Nat. Genet., 29,
229-232.
140.Patil, N., Berno, A. J., Hinds, D. A., Barrett,
W. A., Doshi, J. M., Hacker, C. R., Kautzer, C. R., Lee, D. H.,
Marjoribanks, C., McDonough, D. P., Nguyen, B. T., Norris, M. C.,
Sheehan, J. B., Shen, N., Stern, D., Stokowski, R. P., Thomas, D. J.,
Trulson, M. O., Vyas, K. R., Frazer, K. A., Fodor, S. P., and Cox, D.
R. (2001) Science, 294, 1719-1723.
141.Gabriel, S. B., Schaffner, S. F., Nguyen, H.,
Moore, J. M., Roy, J., Blumenstiel, B., Higgins, J., DeFelice, M.,
Lochner, A., Faggart, M., Liu-Cordero, S. N., Rotimi, C., Adeyemo, A.,
Cooper, R., Ward, R., Lander, E. S., Daly, M. J., and Altshuler, D.
(2002) Science, 296, 2225-2229.
142.Guryev, V., Smits, B. M. G., van de Belt, J.,
Verheul, M., Hubner, N., and Cuppen, E. (2006) PLoS Genet.,
2, e121.
143.The International HapMap Consortium
(2005) Nature, 437, 1299-1320.
144.Couzin, J. (2002) Science, 298,
941-942.
145.Zhang, K., Zhu, J., Shendure, J., Porreca, G.
J., Aach, J. D., Mitra, R. D., and Church, G. M. (2006) Nat.
Genet., 38, 382-387.
146.Daley, G. Q., van Etten, R. A., and Baltimore,
D. (1990) Science, 247, 824-830.
147.Hughes, T., Deininger, M., Hochhaus, A.,
Branford, S., Radich, J., Kaeda, J., Baccarani, M., Cortes, J., Cross,
N. C., Druker, B. J., Gabert, J., Grimwade, D., Hehlmann, R.,
Kamel-Reid, S., Lipton, J. H., Longtine, J., Martinelli, G., Saglio,
G., Soverini, S., Stock, W., and Goldman, J. M. (2006) Blood,
108, 28-37.
148.Nardi, V., Raz, T., Cao, X., Wu, C. J.,
Stone, R. M., Cortes, J., Deininger, M. W., Church, G., Zhu, J., and
Daley, G. Q. (2008) Oncogene, 27, 775-782.
149.Zhou, D., Zhang, R., Fang, R., Cheng, L., Xiao,
P., and Lu, Z. (2008) Electrophoresis, 29, 626-633.
150.Zhu, J., Shendure, J., Mitra, R. D., and
Church, G. M. (2003) Science, 301, 836-838.
151.Butz, J. A., Roberts, K. G., and Edwards, J. S.
(2004) Biotechnol. Prog., 20, 1836-1839.
152.Conrad, C., Zhu, J., Conrad, C., Schoenfeld,
D., Fang, Z., Ingelsson, M., Stamm, S., Church, G., and Hyman, B. T.
(2007) J. Neurochem., 103, 1228-1236.
153.Merritt, J., DiTonno, J. R., Mitra, R. D.,
Church, G. M., and Edwards, J. S. (2003) Nucleic Acids
Res., 31, e84.
154.Merritt, J., Butz, J. A., Ogunnaike, B. A., and
Edwards, J. S. (2005) Biotechnol. Bioeng., 92,
519-531.
155.Butz, J., Wickstrom, E., and Edwards, J. S.
(2003) BMC Biotechnol., 3, 11.
156.Mikkilineni, V., Mitra, R. D., Merritt, J.,
DiTonno, J. R., Church, G. M., Ogunnaike, B., and Edwards, J. S.
(2004) Biotechnol. Bioeng., 86, 117-124.
157.Rieger, C., Poppino, R., Sheridan, R., Moley,
K., Mitra, R., and Gottlieb, D. (2007) Nucleic Acids Res.,
35, e151.
158.Butz, J. A., Yan, H., Mikkilineni, V., and
Edwards, J. S. (2004) BMC Genet., 5, 3.
159.Haase, A. T., Retzel, E. F., and Staskus, K. A.
(1990) Proc. Natl. Acad. Sci. USA, 87, 4971-4975.
160.Uhlmann, V., Silva, I., Luttich, K., Picton,
S., and O'Leary, J. J. (1998) Mol. Pathol., 51,
119-130.
161.Shimizu, Y., Inoue, A., Tomari, Y., Suzuki, T.,
Yokogawa, T., Nishikawa, K., and Ueda, T. (2001) Nat.
Biotechnol., 19, 751-755.
162.Gustafsdottir, S. M., Schallmeiner, E.,
Fredriksson, S., Gullberg, M., Soderberg, O., Jarvius, M., Jarvius, J.,
Howell, M., and Landegren, U. (2005) Analyt. Biochem.,
345, 2-9.