REVIEW: Intermediate Vimentin Filaments and Their Role in Intracellular Organelle Distribution
A. A. Minin1* and M. V. Moldaver2
1Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: alexminin@gmail.com2Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received March 15, 2008; Revision received May 21, 2008
Intermediate filaments (IF) represent one of three main cytoskeletal structures in most animal cells. The human IF protein family includes about 70 members divided into five main groups. The characteristic feature of IF is that in various cells and tissues they are formed by proteins of different groups. Structures of all IF proteins follow a unique scheme: a central α-helical part is flanked at the N and C ends by positively charged polypeptide chains devoid of a clear secondary structure. The central part is highly conserved for all proteins in all animals, whereas the N and C termini strongly differ both in size and amino acid composition. This review covers the broad spectrum of recent investigations of IF structure and diverse functions. Special attention is paid to the regulatory mechanisms of IF functions, mainly to phosphorylation by different protein kinases whose role is well studied. The review gives examples of hereditary diseases associated with mutations of some IF proteins, which point to an important physiological role of these cytoskeletal structures.
KEY WORDS: intermediate filaments, vimentin, keratins, cytoskeletonDOI: 10.1134/S0006297908130063
Abbreviations: ABD, actin-binding domain; Bcl-2, oncogene 2 of B-cell lymphoma; BPAG1, bullous pemphigoid antigen 1; CaMKII, Ca-calmodulin-sensitive protein kinase; cdc2, cell division cycle protein kinase; EPR, electron paramagnetic resonance; ER, endoplasmic reticulum; GFAP, glial filament acidic protein; GSK3, glycogen synthase kinase; IF, intermediate filaments; IFAP, intermediate filament-associated proteins; LAMP-2, lysosome-bound membrane protein; MBD, microtubule-binding domain; NF-L, NF-M, and NF-H, light, medium, and heavy neurofilament proteins; PAK, protein kinase activated by p21 protein; PKA, protein kinase A; PKC, protein kinase C; SAXS, small-angle X-ray scattering; SUN and KASH, integral proteins of nuclear membrane.
Intermediate filaments (IF) are among three main cytoskeleton components
in eukaryotic cells. They are called intermediate because their
diameter (8-12 nm) is intermediate between those of microtubules
(25 nm) and actin microfilaments (5-8 nm) [1-4]. IF consisting of different
proteins are expressed at different stages of embryonic development and
differentiation as well as in different cell types. Several different
IF are present simultaneously in different cell types [1, 3, 5].
Altogether, about 70 genes have been found in human genome that encode
different IF proteins, forming one of most numerous protein families
[6].
Unlike actin and tubulin, having highly conserved structures, the homology of IF protein sequences sometimes does not exceed 20% [1, 4]. Nevertheless, five different types of intermediate filaments are distinguished on the basis of biochemical, immunological, and structural similarity [1, 3, 4]. Keratins representing two types of proteins, I and II, comprise the most numerous and compositionally complex group of IF proteins [7]. Acidic keratins belong to type I, all the rest being of type II. Proteins of both types are necessary for assembly of keratin IF--they form heteropolymers [1, 8]. Epidermal keratins, simple epithelial keratins, and those expressed in hair, wool, and nails are known [3]. Four proteins belong to type III IF proteins: desmin, vimentin, peripherin, and GFAP (glial filament acidic protein). Desmin is expressed in all types of muscle cells; vimentin is found in fibroblasts, lymphocytes, endothelial cells, and some other mesenchymal tissues; peripherin is present mainly in peripheral neurons, where it together with type IV proteins is involved in IF assembly; GFAP is expressed in glial cells [3, 9-11]. Unlike keratins, type III IF proteins can form homopolymers, but they are also able to form heteropolymers with other type III proteins and with NF-L protein [1, 12, 13]. Type IV IF proteins are mainly expressed in nerve cells, where they are involved in the radial growth of axons. They include α-internexin and a triplet of neurofilament proteins NF-L, NF-M, and NF-H (light, medium, and heavy neurofilament proteins) [1, 3, 14-16]. Another protein, nestin, was first found in the nerve cell progenitors and is sometimes assigned to the special type VI of IF proteins. However, structural peculiarities of nestin allow its classification as type IV [4]. In addition to the abovementioned proteins that are among cytoplasmic IF and are the main subject of this review, there are two more types, very different from the previous ones, namely type V-forming nuclear lamins, and two type VI proteins detected in the crystalline lens [1, 17, 18].
Five types of IF proteins, distinguished on the basis of their sequence homology, are divided into three groups differing by assembly principles. The first group includes keratins, the second contains type III and IV IF, and the third assembly group is formed by lamins. These three groups may co-exist as three independent IF systems within one and the same cell [19]. Cytoplasmic IF can be assembled in vitro in the absence of auxiliary proteins [1]. Members of the first group are obligatory heteropolymers, i.e. the assembly of keratin filaments requires coupling of type I and II IF. Keratins are not able to form polymers with IF of other types. Proteins of type III IF and NF-L of the second assembly group form homopolymers in vitro but very often they appear as intracellular copolymers. Lamins are not able to form copolymers with proteins of cytoplasmic IF [20].
The main intracellular function of IF, based on their mechanical properties and ability for self-assembly, is maintenance of cell and tissue integrity [1]. Besides this “traditional” function, they play an important role in intracellular distribution of organelles and proteins [3, 21], which, in turn, influences functions of these organelles. Thus, it was shown in a number of works that functions of mitochondria in various cells depend on desmin, vimentin, keratin IF, and neurofilaments [4, 22]. The role of IF in localization of such organelles as Golgi apparatus, endosomes, lysosomes, as well as of nuclei is well documented in many works [3, 21, 23, 24].
Structural peculiarities of different IF, many of which are now well studied, suggest that their interactions with different cell organelles are defined both by structural features common for all IF and by peculiarities of individual members of this large family. Thus, the highly conserved central part of all of IF molecules, namely, α-helix and the filament structural units, can be involved in interactions identical for all IF proteins. At the same time, regions located on the ends of the molecules and sharply distinct in different proteins can be responsible for specific interactions of only these IF.
In this review we consider the results of structural investigations, paying special attention to relationships between IF structure and functions, particularly to interactions with different organelles. A significant place in the review is given to vimentin IF as the best studied ones.
STRUCTURE OF INTERMEDIATE FILAMENTS
Primary Structure of IF Proteins
Analysis of IF protein primary structures has shown that despite great variety, all of them share a common structure (Fig. 1) [1, 25]. All IF proteins have a central α-helical domain flanked by non-helical N-terminal (“head”) and C-terminal (“tail”) domains. The terminal domains of the different type IF strongly differ in size and primary amino acid sequence [1].
On the contrary, the central domain structure is extremely conserved. It contains four helical segments 1A, 1B, 2A, and 2B divided into three points by short non-helical linker regions L1, L1-2, and L2, often containing proline and glycine residues. The amino acid sequence of the central domain α-helical regions consists of (abcdefg)n type heptade repeats where positions “a” and “d” are preferably occupied by small hydrophobic residues leucine, isoleucine, methionine, and valine [25]. Such repeating motif of 7 amino acid residues is characteristic of proteins capable of formation coiled-coil α-helix (or superhelix) consisting of two α-helices [26].Fig. 1. Scheme of some IF protein molecules (based on data published in [136]).
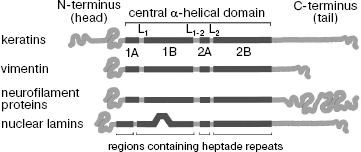
The surface of the α-helical domain of IF proteins is highly charged. For example, 116 (70 acidic and 46 basic) out of 310 amino acids forming central domain of vimentin are charged. So, the excess of negative charge of 24 amino acids falls in each vimentin subunit [20]. Unlike the central domain, the head of many IF proteins is positively charged. The N-terminal domain of vimentin contains 12 arginines, whereas that of B2 lamin has only three Arg residues. In this case, the number of positively charged amino acid residues correlates with the length of the N-terminal domain (it consists of 102 amino acid residues in vimentin and of 25 amino acid residues in lamin B2). Even the acidic glial protein GFAP with a short N-terminal domain of 68 amino acids contains nine basic residues. It is assumed that positively charged amino acid residues of the head interact with negatively charged amino acid residues of the central domain. Besides, these charged clusters on the filament surface can serve as potential binding sites for various cell components [19].
The approximately 45 nm long central domain of cytoplasmic IF consists of 310 amino acids. In nuclear lamins due to the presence of the additional 42 amino acids in the 1B segment, the central domain contains 356 amino acid residues and reaches 53 nm in length. Dimensions of segments forming the α-helical domains of IF are highly conserved: 1A consists of 35 amino acids, 1B of 101 amino acids, 2A of 19 amino acids, and 2B of 115 amino acids [1]. It appeared that IF with different amino acid sequences contain two regions of constant amino acid composition at the ends of the α-helical domain. One of them of 26 amino acid residues comprises the first two thirds of segment 1A (with eight absolutely conserved residues), and the other region of 32 residues is located at the very end of segment 2B, containing 13 absolutely conserved residues [27]. It was shown using the cross-linking technique (this method is described in detail below) that both these regions are involved in interaction of two neighboring IF protein dimers in mature filaments [26, 28]. Another specific structural peculiarity of segment 2B is a slight distortion of heptade structure. An insert of four additional residues is found in all IF proteins after eight complete heptades [29]. This structural distortion is also important for IF assembly. The L1 linker strongly varies in size and sequence, whereas the L2 sequence is highly conserved in all five IF types and consists of eight amino acids [20].
The most pronounced differences in size and amino acid composition are observed in non-helical terminal domains of IF proteins--head and tail. Thus, the tail length in keratin K19 is only nine residues, while the nestin tail consists of 1491 residues [1, 30].
Thus, the central domain of all IF proteins has similar structure and size and carries total negative charge, whereas terminal domains of these proteins are characterized by varied length and amino acid composition and carry total positive charge.
Initial Stages of Assembly: Dimer Formation
Intermediate filaments are polymers exhibiting a unique ability of self-assembly without involvement of additional proteins and, unlike microtubules and actin microfilaments, without additional energy in the form of ATP or GTP. Data concerning IF protein structures suggested a possible mechanism of their assembly that was later largely confirmed by electron microscopy as well as by X-ray analysis and EPR spectroscopy.
IF assembly includes several steps (Fig. 2). First, dimers are formed when central domains of two polypeptide chains are wound around each other forming a twisted helix with a pitch of 14 nm [31-33]. Dimers are formed due to hydrophobic interactions between amino acid residues in positions “a” and “d” of heptade repeats of the interacting protein molecules [34].
The central domain structure, predicted on the basis of its primary sequence, and the interaction of central domains within the dimer were confirmed and studied by X-ray analysis. The large size of IF supramolecular structures makes it difficult to obtain three-dimensional crystals, and owing to this separate small fragments of IF proteins were crystallized [35]. It appeared contrary to expectations that the vimentin 1A segments within the dimer are located in parallel, i.e. they are single α-helices. It is supposed that monomeric helices of segment 1A serve as mobile “couplers” between the back-turned head of one chain and 1A segment of the other chain of the dimer [35].Fig. 2. Scheme of individual steps of IF self-assembly (half of a single protofilament consisting of two octamers is shown).
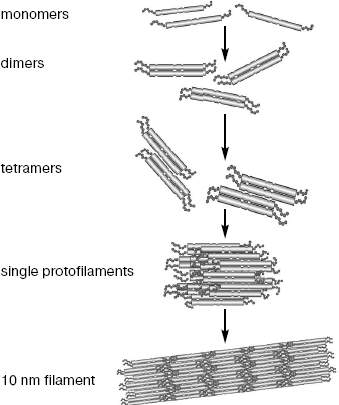
The distortion of heptade structure in the central domain 2B segment was also an important feature for the filament assembly. This distortion results in the local untwisting of a twisted helix by approximately 28°. The result of partial untwisting is a different mutual orientation of functionally important N- and C-terminal groups of the central domain of vimentin. The distortion of this mutual orientation may result in incorrect intramolecular interactions during assembly [35]. Actually, it was shown that although insertion of three amino acid residues for restoration of heptade structure does not hamper formation of filament subunits, it interferes with their assembly into mature filaments [36].
The central domain terminal regions play a special role in IF assembly. Investigations of filament formation in vitro have shown that even a point replacement of a single amino acid at the N-terminus of segment 1A or at the C-terminus of segment 2B result in serious distortions of their structure [37].
Data of X-ray analysis were confirmed by EPR spectroscopy. This method provides information concerning structural elements and assembly mechanism. It is based on the usage of spin labels that are chemical groups containing an unpaired electron (such as nitrogen oxide) covalently bound to certain protein regions. The unpaired electron enables detection of such groups in a magnetic field as those having magnetic moment with characteristic spectrum. The spectral line width is indicative of the presence of a certain secondary structure and the extent of interaction between regions in tertiary structure. The spectral line broadening value is defined by the magnetic interaction of the label and can be estimated in terms of interspin distances that can be used for determination of subunit positions during assembly. Spectra are sensitive to reorientation of molecules, their local structure, and dynamics of the protein molecule side chains containing the EPR label. Over 100 protein variants containing spin label in different positions have been obtained for vimentin. Investigations using EPR spectroscopy have shown that the IF central domain, except for separate regions, is a twisted helix, and they confirmed principles of dimer assembly to tetramers earlier obtained by cross-linking technique [38].
Role of N-Terminal Domain (Head)
When studies of IF structure had just begun, it seemed that only the central domain was necessary for IF assembly, while the terminal domains were involved in interaction with adjacent dimers and other cell components [31]. However, it became clear later that the N-terminal domain plays an important role in IF formation and stabilization. Vimentin and keratin molecules devoid of this domain do not form filaments. Interaction of the positively charged N-terminal domain with the acidic central domain is probably critical for the first phase of assembly [39]. This was studied in detail in experiments with gradual shortening the vimentin polypeptide chain at the N-terminus. The amino acid sequence at the very end of the vimentin head appeared to be the most important. It includes a nonapeptide whose sequence SSYRRIFGG is highly conserved among all type III IF [40]. The deletion or modification of this sequence in vimentin from Xenopus laevis as well as addition of peptides with analogous or identical sequence during in vitro assembly disturbed this process. However, no visible effect was observed after removal of the first four amino acid residues [40].
Analysis of mouse vimentin mutants with different N-terminal deletions revealed residues most essential for vimentin polymerization [41]. The first 24 residues appeared to be important. The removal of these residues as well as of the whole head makes the IF assembly absolutely impossible. The removal of amino acids 25-38 has no effect on the protein polymerization either in vitro or in vivo. At the same time, IF are not assembled in vitro after removal of amino acids 25-63, but they can be formed in vivo. The removal of a fragment that is five residues longer (25-68) results in emergence of a mutant protein incapable of polymerization. Unexpectedly, after a small deletion within this region (44-69 amino acid residues) the protein formed IF in vivo, but not in vitro. It is surprising that further removal of residues beginning from 44 results in a protein that is completely capable of polymerization (Δ44-95) or completely incapable of it (Δ44-103). This indicated that specific residues in the head C-terminal part might play the crucial role in IF assembly, which was confirmed later when it was found that vimentin Δ88-103 was also incapable of assembly (both in vivo and in vitro), while vimentin Δ40-95 was able to form IF [41]. The absolute dimension of the head domain middle deletion is probably less important than the position of the region to be deleted. It follows from these data that amino acid residues 64-69 are necessary for IF assembly in vivo. Besides, except for the sequence at the very N-terminus of the molecule, residues 97-103 are most important for vimentin polymerization.
The middle part of the head is not very important for assembly. It is assumed that this part of the N-terminal sequence extends from the vimentin tetrameric complex, forms a loop, and draws the nonapeptide region SSYRRIFGG nearer to the central domain of the adjacent protein subunit, thus stimulating their association [41].
The involvement of the N-terminus of vimentin in IF assembly was also shown indirectly by another group of researchers who found that numerous regions within the vimentin head domain can be phosphorylated in solution and they become inaccessible for protein kinases if the protein is incorporated in IF [42].
C-Terminal Domain (Tail)
The role of the C-terminal domain of vimentin is not very pronounced. Its removal does not distort IF assembly. However, the diameter of such IF formed by the “tailless” vimentin is for 3 nm greater than that in the case of the wild-type molecules. In other words, the removal of the C-terminal domain alters control of the assembling filament thickness [43].
Tetramer Formation
The next stage of assembly is the joining of dimers into larger complexes. For cytoplasmic IF it is formation of tetramers (Fig. 2), which distinguishes IF from lamins, which follow the “head-to-tail” principle upon assembly and thus forms linear polymers [1, 44]. Tetramer formation involves electrostatic interactions between alternating zones of positive charges, the excess of which exists in the N-terminal domain of vimentin, and negative ones that dominate in the central domain of the molecule [34].
There are several models of dimer joining to tetramers based on the results obtained by the cross-linking technique (Fig. 3). The method involves covalent bonding between certain amino acid residues of adjacent molecules using special reagents containing two reactive groups interacting with these residues. Reagents interacting with lysine or cysteine are used most often. The resulting complexes have been isolated, purified, cleaved by proteinases, and analyzed by chromatography. Comparison of chromatographic profiles of specimens before and after cross-link cleavage by sodium periodate revealed several possible variants of inter- or intramolecular cross-links. Peaks on chromatograms that disappear after treatment by sodium periodate correspond to molecules formed by cross-linking between different molecules or within one protein molecule. So, this method reveals protein region(s) located in the immediate vicinity of each other, i.e. determines mutual arrangement of subunits within filaments. Most peptides obtained by proteolysis of vimentin IF were rather short (less than 10 residues), owing to which amino acid composition allowed unambiguous detection of peptide position within the sequence of vimentin [26]. In some cases, when only a single peptide was found after periodate treatment, this was indicative of cross-link formation within the chain. Many lysine residues formed cross-links in several different reactions.
Sixteen unique cross-links were detected for vimentin, five of which are between two parallel chains included “in register” and forming the double-stranded twisted homodimer vimentin molecule. Eleven cross-links can be ascribed to three possible models of intermolecular interaction shown in Fig. 3. The A11 model (six cross-links) suggests that two antiparallel dimers interact in such a way that 1B segments of their central domains are significantly overlapped [26]. According to another model A22 (three cross-links), two antiparallel dimers are overlapped in the region of 2B segments. In the third model A12 (two cross-links), two dimers are antiparallel and completely overlapped (located “in register”).Fig. 3. Scheme showing three possible models of tetramer formation during IF assembly.
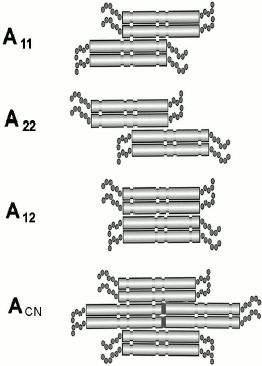
The existence of these three types of dimer interactions was confirmed for keratins. In the case of vimentin, even a fourth model ACN was proposed for interaction between dimer complexes within filaments. If two tetramers formed according to models A11 and A22 are adjacent to each other, then one dimer of each tetramer will follow the head-to-tail principle of interaction. In this case, from 5 to 10 N-terminal residues of 1A segment of one dimer will overlap with 5-10 C-terminal residues of 2B segment of another dimer (Fig. 3) [26]. Similar data were also obtained for the structure of desmin IF [45, 46].
Further Stages of Assembly
Until recently, it was possible to obtain structural information about IF assembly only from electron microscopic analysis of preparations fixed at certain time intervals. Later the small angle X-ray scattering technique (SAXS), used for investigation of macromolecules in solution, was applied to the assembly process [47, 48]. The assembly of vimentin IF in vitro appeared to consist of successive formation of different oligomers including tetramers, octamers, and a single protofilament consisting of two octamers (Fig. 2). Measurements using this method showed that dimers within tetramer are at a distance of 3.4 nm from each other, while the distance between twisted helices is 1.5 nm. This separation might be due to the electrostatically unfavorable direct contact between acidic central domains. It also became clear that the dimer joining in tetramer according the model A11 occurs largely due to the second half of the head domain (35-70 residues), i.e. electrostatic attraction of positively charged head and acidic central domain is probably the driving force [39, 49].
The final IF subunit, the single length (65 nm) protofilament, on average consists of four octamers. Most likely the single length protofilament is rather oval than round in cross section. Measurements using scanning electron microscope have shown that individual protofilaments can significantly differ in the number of vimentin chains forming these protofilaments.
At the last stage of assembly filament subunits are thickened to 10-12 nm. This stage might be also accompanied by intra-filament rearrangement because assembly variants for tetramers A22 and A12 are detected only in mature structures [49].
The existence of vimentin tetramers in vivo is already proved, whereas the presence of two other intermediate components of assembly, such as octamers or single filaments, requires further investigations [49, 50].
The number of protofibrils (octamers) per IF cross-section varies from two to six depending on the IF type and assembly conditions [51]. It is supposed that each protofibril consists of two protofilaments, and each protofilament, in turn, consists of tetramers joined end-to-end. Thus, as shown for vimentin [20], one filament can consist of 24-40 polypeptides (usually 32--eight tetramers) across the width. Thickness may change both from one filament to another and even within a single filament [51].
Intracellular IF
Mature IF and their precursors are dynamic and mobile inside cells. Continuous intracellular assembly, disassembly, and transfer of IF have been described. Formation of intracellular network of different type IF differs in different cells. This may be caused by interactions of IF with different types of molecular motors and other factors [52].
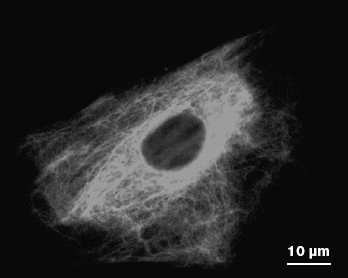
IF subunits and mature polymers are in equilibrium in the cytoplasm and incorporation of a new subunit is possible in any site of a mature filament. However, formation of an IF network begins near the cell nucleus. Not only individual subunits, but mature fibrils move within cells as well. It was shown for vimentin IF that they can move in the cytoplasm both towards the cell surface and to the cell nucleus. The average speed of the long IF transfer is 0.2-0.3 µm/min, while IF subunits move at a higher speed (1-2 µm/sec) mainly along microtubules [53, 54]. Most of them (65-70%) move towards the cell periphery. The ability of vimentin subunits to move along microtubules in different directions is explained by their association with motor proteins kinesin and dynein. Association with kinesin is responsible for movement of vimentin filaments and their subunits to the microtubule plus-ends and to the cell surface [55]. Movement to microtubule minus-ends to the cell nucleus is caused by interaction with the motor complex consisting of dynein and dynactin [56]. These interactions are probably necessary for formation and maintenance of the IF network (Fig. 4). Long vimentin IF bind to the IF-associated proteins (IFAP) such as plectin that forms cross-links between individual IF and other cytoskeleton structures, thus preventing their motility. Such interaction is probably necessary for IF stabilization in special regions of the cytoplasm undergoing mechanical stress or deformation [52].
Unlike vimentin IF, consisting of a single protein, keratin IF are always heteropolymers. Owing to this, the balance between types I and II keratins is necessary to maintain the stable network of keratin filaments, because an excess of any one keratin type results in alteration of the IF network. It is interesting that in cells simultaneously expressing keratin and vimentin, long keratin filaments move at three times lower speed compared to that of vimentin filaments (0.06 µm/min), while the speed of keratin subunits is 15 times slower than of vimentin subunits. Besides, transport of keratin subunits is mainly directed towards the cell nucleus (84%). These distinctions might be caused by different abilities of keratins and vimentin to bind microtubules and motor proteins [57]. Actually, a large part of keratin subunits are associated with the network of actin microfilaments and dynamic properties of keratin IF are caused by interaction with myosin. However, since myosin moves more slowly than the microtubule-associated motor proteins, just this may explain differences in the rate of keratin and vimentin transport within one cell type [52].Fig. 4. Vimentin IF network in a mouse fibroblast as revealed by immune fluorescence.
Formation in neurons of an IF system called neurofilaments is of special interest. The neuronal processes can reach one meter in length. If the speed of IF transport was equal to that of the slow transport of other cytoskeleton structures (0.3-8.0 mm/day), years would be necessary for them to reach distal regions of the process [58]. However, neurofilament subunits move along an axon with the speed of 1.8 µm/sec [59] because they are transported via microtubules with involvement of kinesin and dynein [52, 58]. True, the motility of these structures is interrupted by long breaks, owing to which they move only 27% of the time and in mature sensory axons neurofilaments are at rest for about 99% of the time [58].
It should be said in conclusion that significantly different N- and C-terminal domains are involved in the assembly of different IF types. They also take part in interaction of IF with different proteins such as molecular motors or IFAP that, in turn, help them in distribution and formation of intracellular IF networks. Regulatory mechanisms of IF assembly and their interaction with different proteins will be considered below.
FUNCTIONS OF INTERMEDIATE FILAMENTS
According to current concepts, based on the mechanical properties and ability of self-assembly of IF, their main function is maintenance of cell and tissue integrity [1]. Enhanced interest in their mechanical role is due to information concerning hereditary diseases caused by disturbances of IF structure in tissues subjected to mechanical stress, such as skin, muscle, and blood vessels. However, IF are present in cells of all types, including those not subjected to mechanical stress, but alteration of their networks also results in pathological consequences. Thus, there are serious diseases associated with disturbance of IF functions in the nervous system [60]. This fact as well as dynamic nature of IF and the presence of a large number of IF-associated signal proteins show that IF not only provide for resistance to external factors, but they also have different specialized intracellular functions. Although these functions in different cell types are still not sufficiently studied, it is already known that their significance is high. Non-mechanical functions of IF are probably associated with their participation in intracellular distribution of organelles and proteins, as well as in lipid transport [61].
The functions of IF are defined by their interactions with various cell components. Thus, IF provide for cell mechanical integrity by binding to other cytoskeleton components such as microtubules and microfilaments and to the plasma membrane, interaction with which takes place in special attachment sites like desmosomes and hemidesmosomes of epithelial cells and in the points of fibroblast focal contacts [19]. For interaction with such organelles like mitochondria, Golgi apparatus, endosomes, and lysosomes, IF bind different membrane components of these organelles. This binding is provided by a large protein family, IFAP [62], but probably direct binding is also possible. Some examples of such interactions will be considered in more detail below.
Mechanical Functions
Interaction with cytoskeleton and plasma membrane. Electron-microscopic investigations show that different cytoskeleton structures are connected to each other and to membrane organelles. “Bridges” discernible on electron microphotographs for a long time were considered as binding structures of unknown nature. Later it became clear that molecular motors, kinesins and dyneins, are responsible for association of many cell organelles with microtubules. The role of kinesin in the interaction of IF with microtubules was shown using antibodies that block activity of this motor protein [55]. The radial distribution of IF along microtubules is due to kinesin-dependent transport.
However, the main role in the interaction of IF with many cell components belongs to IFAP, joining IF with organelles, microfilaments, and microtubules as well as with intercellular contacts [19]. Such proteins as desmoplakin, BPAG1 (bullous pemphigoid antigen 1), and plectin belong to the plakin family. All of them contain a very long central α-helical domain that forms a twisted helix upon dimer complex formation. The central domain is flanked by (i) a non-helical N-terminal domain that may contain sites for actin binding (actin-binding domain, ABD) and/or sites for microtubules (microtubule binding domain, MBD) and (ii) by a C-terminal domain that, in addition to IF binding sites, may contain different amounts of desmoplakin-specific repetitive domains A, B, and C [64].
Plectin is the best-studied protein capable of joining three different cytoskeleton systems and interaction with different proteins (Fig. 5). Mutations in this protein result in serious diseases such as bullous epidermolysis, accompanied by emergence of bullas on the skin, and muscular dystrophy [65]. Electron microscopy revealed that plectin forms approximately 200 nm long and 2-3 nm thick side processes, branched off from IF [63]. The ability of plectin to bind IF by both ends suggests that the ends of its chain have identical properties and its molecules are homotetramers. Quantitative analysis of vimentin-plectin complexes has shown that there are 10 plectin molecules per IF region of 1 µm [63]. In muscles, plectin is co-localized with desmin IF near Z discs and structures forming intracellular myofibrillar skeleton [66]. In plectin-free mice, serious distortions in muscle structure were observed [67], whereas significant alterations of cytoskeleton architecture and dynamics were detected in cultured fibroblasts and astroglial cells obtained from such mice [68].
Desmoplakin, another protein of the plakin family, joins different IF with desmosomes with involvement of plakoglobulin and plakophilins whose family is continuously growing. Expression of plakophilins is thoroughly regulated within cells; thus, the plakophilin-2 gene is expressed simultaneously with genes encoding keratins 8, 18, and 19, which is indicative of expression regulation specific to a certain cell type. Mutations in this protein gene were found in patients with hereditary diseases characterized by skin instability and epidermal dysplasia. Since the plakophilin-2 gene is expressed in all internal organs, mutations in it have very severe consequences [19].Fig. 5. Scheme showing different interactions of cytoskeleton structures with involvement of plectin and kinesin (based on data from [63]).
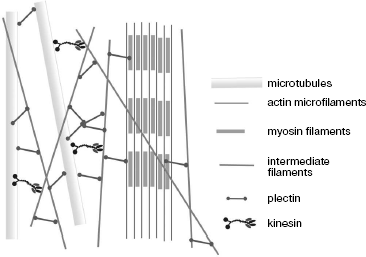
Both plectin and BPAG1 protein are represented by several isoforms that result from alternative splicing of their mRNA [69, 70]. Structural distinctions between these isoforms are probably due their ability to interact with different components in different cell types [70].
Intracellular Distribution of Organelles
Until recently no one supposed that IF could be involved in membrane transport; however it has been recently shown that IF are important both for transport of membrane organelles and their functioning [60].
Mitochondria and IF. Mitochondria are membrane organelles that are most important for cell physiology. They are localized near IF in different cell types, and this probably plays an important role. Thus, it appeared that morphology and intracellular localization of mitochondria are changed in the heart and skeletal muscle of desmin-free mice. Besides, progressive destruction of matrix is observed in such mitochondria, while the latter are organized in groups near sarcolemma [71, 72]. All these alterations result in distortion of mitochondrial functions, i.e. decrease in maximal respiration rate and ADP-stimulated oxygen consumption, disappearance of creatine kinase, and decrease in level of cytochrome c. In addition, anti-apoptotic protein Bcl-2 (B-Cell Lymphoma oncogene) is transferred from the inner mitochondrial membrane to the outer one [72].
Mitochondria and IF interact not only in muscle cells, but in other cell types as well. It was shown in our laboratory that motility of mitochondria in cultured fibroblasts is inhibited by their interaction with vimentin IF [73] under control of protein kinase C (PKC). Another group of researchers has shown that phosphorylated proteins of neurofilaments selectively bind in vitro to mitochondria, and as shown in experiments with potential-dependent dyes, this binding depends on the level of mitochondrial membrane potential [74]. Mutations in the neurofilament protein NF-L, which cause accumulation of mitochondria in the neuronal body, thus lowering the number of mitochondria in axon and axonal transport of neurofilaments, appeared to be a factor that might be responsible for the severe hereditary disease Sharko-Marie-Toose neuropathy [75]. Changes in intracellular distribution of mitochondria were also detected in patients with bullous epidermolysis caused by mutations in keratins K5 or K14 [76].
Thus, it is clear that IF effect the functions of mitochondria, but many details of this interaction are still unclear. It still should be elucidated how the functional relationship is realized and what additional proteins are involved in the interaction of IF with mitochondria [61].
Golgi apparatus and IF. Golgi apparatus is located along IF near the microtubule organization center. The interaction of Golgi apparatus with vimentin IF is well studied, and proteins involved in their association have been found. One of them, formiminotransferase-cyclodeaminase (FTCD), is a histidine cleaving protein. The gene of this protein is expressed in all cell types, but the level of its expression is especially high in liver cells. As shown experimentally, a mutant form of this protein devoid of enzyme activity can bind vimentin [77, 78].
Disintegration of microtubules by nocodazole resulted in altered distribution of Golgi apparatus; another protein of the latter, GM130, is also located along vimentin filaments [77]. A third protein, MICAL (molecule interacting with CasL), directly binds vimentin and GTPase Rab1 that is a main participant in vesicular transport from ER to Golgi apparatus [79].
As shown in experiments on cell culture, the relationship between IF and Golgi apparatus is probably important for cell physiology. However, it is still an enigma that up to now it has not been shown on ex vivo animal models that the absence of IF results in any serious functional disturbance of the Golgi apparatus [61].
Endosomes, lysosomes, and IF. Another example of the involvement of IF in membrane organelle functions is the role of IF in endosome and lysosome transport. It was shown that vimentin, peripherin, and α-internexin bind to adapter protein AP-3 [60, 80]. AP-3 protein is a heterotetrameric adapter complex involved in vesicle transport between endosomal and lysosomal compartments, and it regulates protein delivery to lysosomes and tissue-specific organelles like melanosomes, hematopoietic granules, and synaptic vesicles. Mutations in this protein result in albinism, continuous hemorrhages, and pulmonary fibrosis known as Hermansky-Pudlak syndrome [80]. AP-3 interacts with IF via a specialized domain of β3A and β3B subunits, which is also necessary for the AP-3 function associated with protein sorting, and this is the only AP-3 site interacting with vimentin [80].
The absence of AP-3 or vimentin results in emergence of an identical phenotype, because in both cases distribution of zinc ions is altered. In fibroblasts devoid of AP-3 or vimentin, the zinc ion level is decreased. Intracellular zinc is mainly localized in endocyte vesicles and endosomes, and its storage depends on the flow of chloride through their membranes. The chloride delivering channels regulate both pH in endosomes and zinc ions storage in them. Proteins forming these channels are transferred by membrane vesicles associated with adapter complex AP-3. It is supposed that the absence of AP-3 complex from cells results in alteration of pH level within endosomes, which is defined by the chloride transport. In vimentin-free cells, zinc content in vesicles is 40 times lower [80].
The AP-3 complex is also responsible for transport of another load, the LAMP-2 (lysosome-bound membrane protein) molecule. It is a resident protein of endosomes and lysosomes, required for their fusion with autophagosomal vacuoles that are formed from endoplasmic reticulum, envelope the organelle intended for cleavage, and deliver it to lysosomes or endosomes [81]. It was shown that the alteration in IF assembly results in disturbance of the autophagosomal vacuole formation, but the mechanism of their interaction is still unknown [82]. It is supposed that combined action of IF and AP-3 is connected with LAMP-2 transport. Nevertheless, it is important that localization of endosomes and lysosomes is independent of IF.
Cell nucleus and IF. One of the important functions of IF is localization of cell nuclei. The cell nucleus has a definite position specified by its interaction with different cytoskeleton components. The nuclear envelope is a specialized continuation of endoplasmic reticulum (ER), which has two membranes and a perinuclear space between them separating the cell nucleus from the cytoplasm. The external nuclear membrane is attached to ER, while the lamina is adjacent to the internal membrane. The lamina is a special structure formed by a network of fibrils consisting of type V IF proteins. The external and internal membranes join each other at nuclear pores. The structure of nuclear envelope raises two questions concerning intracellular localization of the nucleus: (i) there should exist proteins able to bind cytoskeleton to the nuclear matrix, i.e. making contact across two membranes and intermembrane space, and (ii) proteins should exist which would allow one to differentiate between external nuclear membrane and ER.
Such proteins have been found in eukaryotic cells. They form two families, KASH and SUN, that join external and internal nuclear membranes and bind cytoskeleton with nuclear laminas [83]. Proteins of the SUN family have a conserved domain at the C-terminus and at least one transmembrane domain. These proteins are supposed to be located on internal nuclear membrane and interact by their C-terminus with KASH proteins, while their N-terminus interacts with laminas and can serve as a linker for chromosomes during meiosis. Proteins of the KASH family have several different functions. They contain at the C-terminus a transmembrane domain that is necessary for localization in nuclear membrane; the N-terminal domain of these proteins is directed outside to the cytoplasm and can interact with actin filaments, centrosomes, and IF [83].
The role of IF in nucleus localization is still not sufficiently studied. Most likely, it is keeping the nucleus in its place and attraction of proteins that would be able to interact with other cytoskeletal components [83]. The effect of cytoplasmic IF on the nucleus localization and shape was studied on the example of vimentin-free cells in culture in which nuclear membrane invagination was observed along with other distortions [84]. It was shown in other works that nuclei in desmin-free myocytes are localized unevenly and form aggregates [85], while in mouse epidermal cells mutations in keratin K10 or its absence result in alterations in the nucleus size and shape [86]. Impressive results have been recently obtained on mice deprived of desmin by knocking its gene out. Although in normal skeletal muscle cells hundreds of nuclei are distributed evenly and separately from each other, nuclei in desmin-free animal muscle cells were gathered in thick clusters. Besides, they were oval and could not line up along the long axis of the cell. Cells with mutant Syne-1 protein of the KASH family had a phenotype [87] that was indicative of involvement of both proteins in nucleus localization in muscle cells.
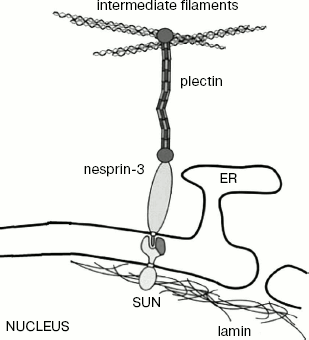
The recently detected protein nesprin-3 of the KASH family probably also joins external nuclear membrane to IF [88]. This protein is known to be localized on the external nuclear membrane, and in the intermembrane space it interacts with SUN protein localized in the internal nuclear membrane (Fig. 6). Besides, it interacts with plectin that, as mentioned above, can be bound to IF and actin. Since nesprin-3 joins the actin-binding plectin domain, this protein provides for its interaction with IF.
Other Functions of IFFig. 6. Scheme showing the supposed participation of nesprin-3 in the interaction of the nuclear membrane with the IF network (based on a scheme from [83]). ER, endoplasmic reticulum; SUN, integral protein of nuclear membrane.
Other known functions of IF include their participation in maintenance of the membrane lipid composition. Thus, the absence or alteration of vimentin IF structure lowered triglyceride stability in pre-adipocytes and resulted in changes in cholesterol metabolism in fibroblasts and adrenal cells [89]. Quite a number of data show that alterations in the membrane lipid composition are caused by damages emerging in the late stages of the endosomal pathway. It appeared that one of the key enzymes of cholesterol metabolism, hydroxysterol-binding protein, interacts with vimentin IF [90]. Enhanced cholesterol synthesis and lowered generation of its ester are observed in IF-free cells. It is supposed that this is also due to alterations in cholesterol transfer to endosomal and lysosomal membranes. Maturation of glycosphingolipids in IF-free cells is also retarded because their transport from Golgi apparatus to the endosome system is hampered. Alterations in lipid transport suggest the extremely important role of the transport vesicle interactions with IF.
In conclusion, the diversity of intracellular functions of IF should be noted. Along with other cytoskeleton components, they provide for the mechanical strength of cells, take part in correct arrangement of intracellular organelles and the cell nucleus, and in protein transport. However, some details and fine mechanisms of these interactions as well as the role of many participants are still not clear.
PHOSPHORYLATION OF INTERMEDIATE FILAMENTS REGULATES THEIR
FUNCTION
All the above-said shows that the basis of diverse intracellular functions of IF is their interaction with many structures and individual proteins. Numerous data show that these interactions are under strict control of external factors and are quickly rearranged in response to different signals [91-93]. One of the best studied methods of regulation of IF function is phosphorylation of IF proteins by different protein kinases. Thus, phosphorylation plays the main role in regulation of IF structure and assembly [94, 95]. In addition, phosphorylation may be involved in control of such functions as IF association with IFAP and tissue-specific interactions [96, 97], as well as changes in IF structure under stress conditions [98-100]. The direct involvement of IF proper in signal transduction has been shown in a number of works, because activity of many regulatory proteins appear to be dependent on interaction with IF. The equilibrium between polymeric and depolymerized vimentin under phosphorylation control regulates activity and localization of Rho kinase [101, 102]. To understand different mechanisms of regulation of IF function by phosphorylation we shall analyze some examples in more detail.
In vitro IF Phosphorylation
Early works on IF structure and functions have shown that in vitro phosphorylation causes immediate depolymerization of IF [103]. Simultaneous analysis of phosphopeptides obtained from phosphorylated vimentin has revealed that protein kinase A (PKA) and protein kinase C (PKC) phosphorylate this protein at different sites [103]. Further investigations have shown that site-specific phosphorylation in vitro by a number of protein kinases also resulted in disintegration of different IF. As is seen in the table, different authors have shown that practically all IF proteins are phosphorylated by the main protein kinases, which in most cases results in their in vitro depolymerization. The table also shows that phosphorylation sites are located mainly on the N-terminal protein domains, which according to the above-said are important for IF assembly. In this connection, it was proposed already in early works that intracellular phosphorylation of IF proteins is a mechanism of regulation of their structure [103].
In vivo IF Phosphorylation
Data obtained on cells entering mitosis showed for the first time that phosphorylation causes significant alterations in the IF network [104-106]. Protein kinase cdc2 (cell division cycle) undergoes activation at early stages of mitosis, and vimentin phosphorylation by this kinase results in fragmentation and collapse of the IF network in many cell types. Besides, it became clear that, along with cdc2 kinase, PKC can also be involved in vimentin phosphorylation in mitotic cells [107]. It should be noted that not all numerous vimentin phosphorylation sites, found in in vitro experiments for quite a number of protein kinases, were detected in experiments in vivo. Thus, PKC activation by phorbol esters in interphase cells did not result in vimentin phosphorylation at the sites specific for this enzyme [107]. At the same time, vimentin phosphorylation was observed after injection of a constitutively-activated PKC mutant. Based on these data, the authors concluded that the involvement of PKC in vimentin IF phosphorylation depends on localization of the kinase proper, and that in interphase cells the IF network is inaccessible for this enzyme.
Other protein kinases injected into cells also caused IF network alteration or collapse. Thus, IF collapse and fragmentation were observed after microinjection of constitutively activated PKA mutant into fibroblasts [125] or after foreign CaMKII (Ca-calmodulin-sensitive protein kinase) expression in astrocytes [91]. Similar alterations were also observed in cells treated by phosphatase inhibitors, which increased the level of IF protein phosphorylation [126]. However, the role in cell physiology of IF protein phosphorylation by various protein kinases is still unclear because the above-mentioned alterations in IF are not usually observed in interphase cells. These reactions may be local and involved in slight rearrangements of the IF network.
The recent pronounced progress in investigation of the protein kinase regulatory role in IF rearrangement is due to the use of specific antibodies able to recognize different sites of vimentin and some other IF proteins phosphorylated by known protein kinases. Inagaki and Izawa [127] used such antibodies to show that after vimentin Ser55 phosphorylation by cdc2 kinase, protein kinase Polo 1 binds to this site and undergoes activation, and then, in turn, phosphorylates Ser82. Detection in the same laboratory of two sites of vimentin phosphorylation during cytokinesis is another example of successful usage of phospho-specific antibodies [127]. It was shown that successful separation of two daughter cells required the involvement of Ser38 and Ser71 phosphorylation in vimentin by Rho kinase [128] and Ser72 phosphorylation by Aurora B kinase [129].
Another very fruitful approach to investigation of intracellular phosphorylation of IF proteins was the application of mass spectroscopy. Analysis of phosphopeptides, formed upon vimentin cleavage enables very precise determination of the level of phosphorylation of this protein at the sites specific for known protein kinases. Goldman et al. [93] used mass spectroscopy to study the role of vimentin phosphorylation by PKA at Ser38 and Ser72 residues for maintenance of equilibrium between polymeric and soluble vimentin.
It is important that one and the same amino acid residue in vimentin can be phosphorylated by different protein kinases. This follows from data of in vitro experiments shown in the table. The participation of protein kinase PAK in regulation of smooth-muscle cell contraction is an example of one and the same amino acid residue being involved in different signal chains during in vivo vimentin phosphorylation [92]. This kinase phosphorylates Ser56 in vimentin of these cells (corresponds to Ser55 of human vimentin), which results in partial disassembly of IF and their intracellular re-orientation. It is supposed that partial disassembly of vimentin IF causes redistribution of associated proteins, like Rho kinase and adaptor protein p130, involved in regulation of contraction [92]. So, phosphorylation of this amino acid residue is the key to IF rearrangements in mitosis, but it can also take part in other not less important events.
In vitro phosphorylation of IF proteins by some protein kinases

Thus, now many examples of IF protein phosphorylation, connected with assembly-disassembly of these cytoskeleton structures, are known. However, the involvement of IF protein phosphorylation in regulation of their interactions with different cell components is far less studied. Progress in this field should be expected in the nearest future.
The need for more thorough investigation of principles and mechanisms of IF functioning in different cell types is defined first of all by the fact that changes associated with these cytoskeleton structures are responsible for many pathological states. Mutations in IF protein genes are now known to be responsible for some severe hereditary diseases. Among them there are hereditary bullous epidermolysis caused by mutations in keratins 5 and 14; myopathy and cardiomyopathy caused by desmin alterations; severe neurological diseases like amyotrophic lateral sclerosis and Alexander's disease due to peripherin and GFAP mutations, respectively; Parkinson's disease, and some other severe cases of neuropathy caused by neurofilament alterations (detailed information is available on the site of the Society of IF Researchers www.interfil.org). The aim of active investigations in this field is to help or make easier the life of such patients. In this connection the recently proposed methods of gene therapy that would be able to compensate for the damaged gene seem to be quite promising.
Another application of fundamental knowledge concerning IF was successful usage of their proteins as markers of various malignant tumors. The IF immunocytochemical analysis, proposed for the first time by K. Weber in 1981 [130], is still used in cancer diagnostics. It is based on the fact that normal differentiated cells contain IF types characteristic of a certain tissue, while expression of other IF protein genes begins in tumor cells after malignization. For many kinds of tumors the type of “incorrect” IF protein can serve as a diagnostic marker. Recently, due to increased interest in the problem of the use of stem cells, many researchers have paid attention to IF as a convenient marker of cell differentiation. In fact, detection of expressed IF protein makes possible easy and rapid determination of cell type.
Thus, IF represent an important cytoskeleton component exhibiting many unique properties and playing an important role in cell physiology. It is paradoxical that animals do not survive in the absence of most IF proteins, while knock-out of such proteins like vimentin [131] and GFAP [132] is not lethal. Mice without these genes not only attain sexual maturity but produce progeny, and cells of these animals usually containing vimentin or acidic glial filaments are completely free of IF. Quite a number of various pathologies were found in vimentin-free animals [133-135], while no distinctions from the wild-type animals were detected in mice containing no GFAP [132]. This suggests that in the absence of these proteins some of their functions are carried out by different cell components. Nevertheless, even the possibility to obtain a full-value animal free of a single cytoskeleton component in a single vital tissue seems surprising and makes the investigation of IF even more intriguing.
This work was supported by the Russian Academy of Sciences Presidium Program “Molecular and Cell Biology” and by the Russian Foundation for Basic Research (grant No. 06-04-48452-a).
REFERENCES
1.Fuchs, E., and Weber, K. (1994) Annu. Rev.
Biochem., 63, 345-382.
2.Kreplak, L., and Fudge, D. (2007) Bioessays,
29, 26-35.
3.Toivola, D. M., Tao, G. Z., Habtezion, A., Liao,
J., and Omary, M. B. (2005) Trends Cell Biol., 15,
608-617.
4.Herrmann, H., and Aebi, U. (2000) Curr. Opin.
Cell Biol., 12, 79-90.
5.Coulombe, P. A., and Wong, P. (2004) Nat. Cell
Biol., 6, 699-706.
6.Hesse, M., Magin, T. M., and Weber, K. (2001) J.
Cell Sci., 114, 2569-2575.
7.Moll, R., Franke, W. W., Schiller, D. L., Geiger,
B., and Krepler, R. (1982) Cell, 31, 11-24.
8.Steinert, P. M., Idler, W. W., and Zimmerman, S. B.
(1976) J. Mol. Biol., 108, 547-567.
9.Lazarides, E. (1982) Annu. Rev. Biochem.,
51, 219-250.
10.Osborn, M. (1983) J. Invest. Dermatol.,
81, 104s-109s.
11.Portier, M. M., de Nechaud, B., and Gros, F.
(1983) Dev. Neurosci., 6, 335-344.
12.Monteiro, M. J., and Cleveland, D. W. (1989)
J. Cell Biol., 108, 579-593.
13.Steinert, P. M., Idler, W. W., Cabral, F.,
Gottesman, M. M., and Goldman, R. D. (1981) Proc. Natl. Acad. Sci.
USA, 78, 3692-3696.
14.Lewis, S. A., and Cowan, N. J. (1985) J. Cell
Biol., 100, 843-850.
15.Pachter, J. S., and Liem, R. K. (1985) J. Cell
Biol., 101, 1316-1322.
16.Levy, E., Liem, R. K., D'Eustachio, P., and
Cowan, N. J. (1987) Eur. J. Biochem., 166, 71-77.
17.Gerace, L., and Burke, B. (1988) Annu. Rev.
Cell Biol., 4, 335-374.
18.Perng, M. D., Zhang, Q., and Quinlan, R. A.
(2007) Exp. Cell Res., 313, 2180-2188.
19.Herrmann, H., and Aebi, U. (2000) Curr. Opin.
Cell Biol., 12, 79-90.
20.Herrmann, H., and Aebi, U. (2004) Annu. Rev.
Biochem., 73, 749-789.
21.Styers, M. L., Salazar, G., Love, R., Peden, A.
A., Kowalczyk, A. P., and Faundez, V. (2004) Mol. Biol. Cell,
15, 5369-5382.
22.Tang, H. L., Lung, H. L., Wu, K. C., Le, A. H.,
Tang, H. M., and Fung, M. C. (2007) Biochem. J., 410,
141-146.
23.Gao, Y., and Sztul, E. (2001) J. Cell
Biol., 152, 877-894.
24.Styers, M. L., Kowalczyk, A. P., and Faundez, V.
(2005) Traffic, 6, 359-365.
25.Herrmann, H., Bar, H., Kreplak, L., Strelkov, S.
V., and Aebi, U. (2007) Nat. Rev. Mol. Cell Biol., 8,
562-573.
26.Steinert, P. M., Marekov, L. N., and Parry, D. A.
(1993) J. Biol. Chem., 268, 24916-24925.
27.Herrmann, H., Strelkov, S. V., Feja, B., Rogers,
K. R., Brettel, M., Lustig, A., Haner, M., Parry, D. A., Steinert, P.
M., Burkhard, P., and Aebi, U. (2000) J. Mol. Biol., 298,
817-832.
28.Wu, K. C., Bryan, J. T., Morasso, M. I., Jang, S.
I., Lee, J. H., Yang, J. M., Marekov, L. N., Parry, D. A., and
Steinert, P. M. (2000) Mol. Biol. Cell, 11,
3539-3558.
29.Brown, J. H., Cohen, C., and Parry, D. A. (1996)
Proteins, 26, 134-145.
30.Michalczyk, K., and Ziman, M. (2005) Histol.
Histopathol., 20, 665-671.
31.Parry, D. A., Steven, A. C., and Steinert, P. M.
(1985) Biochem. Biophys. Res. Commun., 127,
1012-1018.
32.Geisler, N., and Weber, K. (1982) EMBO J.,
1, 1649-1656.
33.Phillips, G. N., Jr. (1992) Proteins,
14, 425-429.
34.Meng, J. J., Khan, S., and Ip, W. (1994) J.
Biol. Chem., 269, 18679-18685.
35.Strelkov, S. V., Herrmann, H., Geisler, N.,
Wedig, T., Zimbelmann, R., Aebi, U., and Burkhard, P. (2002) EMBO
J., 21, 1255-1266.
36.Herrmann, H., and Aebi, U. (1999) Cell Mol.
Life Sci., 55, 1416-1431.
37.Letai, A., Coulombe, P. A., and Fuchs, E. (1992)
J. Cell Biol., 116, 1181-1195.
38.Budamagunta, M. S., Hess, J. F., Fitzgerald, P.
G., and Voss, J. C. (2007) Cell Biochem. Biophys., 48,
45-53.
39.Traub, P., Scherbarth, A., Wiegers, W., and
Shoeman, R. L. (1992) J. Cell Sci., 101, 363-381.
40.Herrmann, H., Hofmann, I., and Franke, W. W.
(1992) J. Mol. Biol., 223, 637-650.
41.Shoeman, R. L., Hartig, R., Berthel, M., and
Traub, P. (2002) Exp. Cell Res., 279, 344-353.
42.Geisler, N., Hatzfeld, M., and Weber, K. (1989)
Eur. J. Biochem., 183, 441-447.
43.Herrmann, H., Haner, M., Brettel, M., Muller, S.
A., Goldie, K. N., Fedtke, B., Lustig, A., Franke, W. W., and Aebi, U.
(1996) J. Mol. Biol., 264, 933-953.
44.Heitlinger, E., Peter, M., Haner, M., Lustig, A.,
Aebi, U., and Nigg, E. A. (1991) J. Cell Biol., 113,
485-495.
45.Geisler, N., Schunemann, J., and Weber, K. (1992)
Eur. J. Biochem., 206, 841-852.
46.Geisler, N. (1993) FEBS Lett., 323,
63-67.
47.Svergun, D. I., and Koch, M. H. (2002) Curr.
Opin. Struct. Biol., 12, 654-660.
48.Koch, M. H., Vachette, P., and Svergun, D. I.
(2003) Q. Rev. Biophys., 36, 147-227.
49.Sokolova, A. V., Kreplak, L., Wedig, T., Mucke,
N., Svergun, D. I., Herrmann, H., Aebi, U., and Strelkov, S. V. (2006)
Proc. Natl. Acad. Sci. USA, 103, 16206-16211.
50.Soellner, P., Quinlan, R. A., and Franke, W. W.
(1985) Proc. Natl. Acad. Sci. USA, 82, 7929-7933.
51.Herrmann, H., Haner, M., Brettel, M., Ku, N. O.,
and Aebi, U. (1999) J. Mol. Biol., 286, 1403-1420.
52.Helfand, B. T., Chang, L., and Goldman, R. D.
(2004) J. Cell Sci., 117, 133-141.
53.Prahlad, V., Yoon, M., Moir, R. D., Vale, R. D.,
and Goldman, R. D. (1998) J. Cell Biol., 143,
159-170.
54.Yoon, M., Moir, R. D., Prahlad, V., and Goldman,
R. D. (1998) J. Cell Biol., 143, 147-157.
55.Gyoeva, F. K., and Gelfand, V. I. (1991)
Nature, 353, 445-448.
56.Helfand, B. T., Mikami, A., Vallee, R. B., and
Goldman, R. D. (2002) J. Cell Biol., 157, 795-806.
57.Yoon, K. H., Yoon, M., Moir, R. D., Khuon, S.,
Flitney, F. W., and Goldman, R. D. (2001) J. Cell Biol.,
153, 503-516.
58.Brown, A. (2000) Nat. Rev. Mol. Cell
Biol., 1, 153-156.
59.Roy, S., Coffee, P., Smith, G., Liem, R. K.,
Brady, S. T., and Black, M. M. (2000) J. Neurosci., 20,
6849-6861.
60.Styers, M. L., Kowalczyk, A. P., and Faundez, V.
(2005) Traffic, 6, 359-365.
61.Toivola, D. M., Tao, G. Z., Habtezion, A., Liao,
J., and Omary, M. B. (2005) Trends Cell Biol., 15,
608-617.
62.Goldman, R. D., Chou, Y. H., Prahlad, V., and
Yoon, M. (1999) FASEB J., 13, Suppl. 2, S261-265.
63.Svitkina, T. M., Verkhovsky, A. B., and Borisy,
G. G. (1996) J. Cell Biol., 135, 991-1007.
64.Ruhrberg, C., and Watt, F. M. (1997) Curr.
Opin. Genet. Dev., 7, 392-397.
65.Wiche, G. (1998) J. Cell Sci., 111,
2477-2486.
66.Hijikata, T., Murakami, T., Imamura, M.,
Fujimaki, N., and Ishikawa, H. (1999) J. Cell Sci., 112,
867-876.
67.Andra, K., Lassmann, H., Bittner, R., Shorny, S.,
Fassler, R., Propst, F., and Wiche, G. (1997) Genes Dev.,
11, 3143-3156.
68.Andra, K., Nikolic, B., Stocher, M.,
Drenckhahn, D., and Wiche, G. (1998) Genes Dev., 12,
3442-3451.
69.Yang, Y., Bauer, C., Strasser, G., Wollman, R.,
Julien, J. P., and Fuchs, E. (1999) Cell, 98,
229-238.
70.Elliott, C. E., Becker, B., Oehler, S., Castanon,
M. J., Hauptmann, R., and Wiche, G. (1997) Genomics, 42,
115-125.
71.Paulin, D., and Li, Z. (2004) Exp. Cell
Res., 301, 1-7.
72.Milner, D. J., Mavroidis, M., Weisleder, N., and
Capetanaki, Y. (2000) J. Cell Biol., 150, 1283-1298.
73.Nekrasova, O., Kulik, A., and Minin, A. (2007)
Biochemistry (Membrane and Cell Biology), 1, 108-113.
74.Wagner, O. I., Lifshitz, J., Janmey, P. A.,
Linden, M., McIntosh, T. K., and Leterrier, J. F. (2003) J.
Neurosci., 23, 9046-9058.
75.Brownlees, J., Ackerley, S., Grierson, A. J.,
Jacobsen, N. J., Shea, K., Anderton, B. H., Leigh, P. N., Shaw, C. E.,
and Miller, C. C. (2002) Hum. Mol. Genet., 11,
2837-2844.
76.Bonifas, J. M., Rothman, A. L., and Epstein, E.
H., Jr. (1991) Science, 254, 1202-1205.
77.Gao, Y., and Sztul, E. (2001) J. Cell
Biol., 152, 877-893.
78.Gao, Y. S., Vrielink, A., MacKenzie, R., and
Sztul, E. (2002) Eur. J. Cell Biol., 81, 391-401.
79.Suzuki, T., Nakamoto, T., Ogawa, S., Seo, S.,
Matsumura, T., Tachibana, K., Morimoto, C., and Hirai, H. (2002) J.
Biol. Chem., 277, 14933-14941.
80.Styers, M. L., Salazar, G., Love, R., Peden, A.
A., Kowalczyk, A. P., and Faundez, V. (2004) Mol. Biol. Cell,
15, 5369-5382.
81.Shintani, T., and Klionsky, D. J. (2004)
Science, 306, 990-995.
82.Blankson, H., Holten, T., Oksendal, A. N., and
Jynge, P. (1995) Acta Radiol. Suppl., 399, 135-141.
83.Starr, D. A. (2007) Mol. Biosyst.,
3, 583-589.
84.Sarria, A. J., Lieber, J. G., Nordeen, S. K., and
Evans, R. M. (1994) J. Cell Sci., 107, 1593-1607.
85.Shah, S. B., Davis, J., Weisleder, N.,
Kostavassili, I., McCulloch, A. D., Ralston, E., Capetanaki, Y., and
Lieber, R. L. (2004) Biophys. J., 86, 2993-3008.
86.Fuchs, E., Esteves, R. A., and Coulombe, P. A.
(1992) Proc. Natl. Acad. Sci. USA, 89, 6906-6910.
87.Ralston, E., Lu, Z., Biscocho, N., Soumaka, E.,
Mavroidis, M., Prats, C., Lomo, T., Capetanaki, Y., and Ploug, T.
(2006) J. Cell Physiol., 209, 874-882.
88.Wilhelmsen, K., Litjens, S. H., Kuikman, I.,
Tshimbalanga, N., Janssen, H., van den Bout, I., Raymond, K., and
Sonnenberg, A. (2005) J. Cell Biol., 171, 799-810.
89.Schweitzer, S. C., and Evans, R. M. (1998)
Subcell. Biochem., 31, 437-462.
90.Wang, C., JeBailey, L., and Ridgway, N. D. (2002)
Biochem. J., 361, 461-472.
91.Ogawara, M., Inagaki, N., Tsujimura, K., Takai,
Y., Sekimata, M., Ha, M. H., Imajoh-Ohmi, S., Hirai, S., Ohno, S.,
Sugiura, H., et al. (1995) J. Cell Biol., 131,
1055-1066.
92.Li, Q. F., Spinelli, A. M., Wang, R.,
Anfinogenova, Y., Singer, H. A., and Tang, D. D. (2006) J. Biol.
Chem., 281, 34716-34724.
93.Eriksson, J. E., He, T., Trejo-Skalli, A. V.,
Harmala-Brasken, A. S., Hellman, J., Chou, Y. H., and Goldman, R. D.
(2004) J. Cell Sci., 117, 919-932.
94.Eriksson, J. E., Opal, P., and Goldman, R. D.
(1992) Curr. Opin. Cell Biol., 4, 99-104.
95.Ku, N. O., Liao, J., Chou, C. F., and Omary, M.
B. (1996) Cancer Metastasis Rev., 15, 429-444.
96.Foisner, R., Traub, P., and Wiche, G. (1991)
Proc. Natl. Acad. Sci. USA, 88, 3812-3816.
97.Liao, J., and Omary, M. B. (1996) J. Cell
Biol., 133, 345-357.
98.Caulin, C., Ware, C. F., Magin, T. M., and
Oshima, R. G. (2000) J. Cell Biol., 149, 17-22.
99.Feng, L., Zhou, X., Liao, J., and Omary, M. B.
(1999) J. Cell Sci., 112, 2081-2090.
100.Giasson, B. I., and Mushynski, W. E. (1996)
J. Biol. Chem., 271, 30404-30409.
101.Goto, H., Kosako, H., Tanabe, K., Yanagida, M.,
Sakurai, M., Amano, M., Kaibuchi, K., and Inagaki, M. (1998) J.
Biol. Chem., 273, 11728-11736.
102.Sin, W. C., Chen, X. Q., Leung, T., and Lim, L.
(1998) Mol. Cell Biol., 18, 6325-6339.
103.Inagaki, M., Nishi, Y., Nishizawa, K.,
Matsuyama, M., and Sato, C. (1987) Nature, 328,
649-652.
104.Chou, Y. H., Bischoff, J. R., Beach, D., and
Goldman, R. D. (1990) Cell, 62, 1063-1071.
105.Chou, Y. H., Rosevear, E., and Goldman, R. D.
(1989) Proc. Natl. Acad. Sci. USA, 86, 1885-1889.
106.Tsujimura, K., Tanaka, J., Ando, S., Matsuoka,
Y., Kusubata, M., Sugiura, H., Yamauchi, T., and Inagaki, M. (1994)
J. Biochem., 116, 426-434.
107.Takai, Y., Ogawara, M., Tomono, Y., Moritoh,
C., Imajoh-Ohmi, S., Tsutsumi, O., Taketani, Y., and Inagaki, M. (1996)
J. Cell Biol., 133, 141-149.
108.Ando, S., Tanabe, K., Gonda, Y., Sato, C., and
Inagaki, M. (1989) Biochemistry, 28, 2974-2979.
109.Ando, S., Tokui, T., Yamauchi, T., Sugiura, H.,
Tanabe, K., and Inagaki, M. (1991) Biochem. Biophys. Res.
Commun., 175, 955-962.
110.Kusubata, M., Tokui, T., Matsuoka, Y., Okumura,
E., Tachibana, K., Hisanaga, S., Kishimoto, T., Yasuda, H., Kamijo, M.,
Ohba, Y., et al. (1992) J. Biol. Chem., 267,
20937-20942.
111.Chou, Y. H., Ngai, K. L., and Goldman, R.
(1991) J. Biol. Chem., 266, 7325-7328.
112.Goto, H., Tanabe, K., Manser, E., Lim, L.,
Yasui, Y., and Inagaki, M. (2002) Genes Cells, 7,
91-97.
113.Nakamura, Y., Takeda, M., Aimoto, S., Hojo, H.,
Takao, T., Shimonishi, Y., Hariguchi, S., and Nishimura, T. (1992)
J. Biol. Chem., 267, 23269-23274.
114.Inagaki, M., Gonda, Y., Nishizawa, K.,
Kitamura, S., Sato, C., Ando, S., Tanabe, K., Kikuchi, K., Tsuiki, S.,
and Nishi, Y. (1990) J. Biol. Chem., 265, 4722-4729.
115.Sekimata, M., Tsujimura, K., Tanaka, J.,
Takeuchi, Y., Inagaki, N., and Inagaki, M. (1996) J. Cell Biol.,
132, 635-641.
116.Geisler, N., and Weber, K. (1988) EMBO
J., 7, 15-20.
117.Kitamura, S., Ando, S., Shibata, M., Tanabe,
K., Sato, C., and Inagaki, M. (1989) J. Biol. Chem., 264,
5674-5678.
118.Kusubata, M., Matsuoka, Y., Tsujimura, K., Ito,
H., Ando, S., Kamijo, M., Yasuda, H., Ohba, Y., Okumura, E., Kishimoto,
T., et al. (1993) Biochem. Biophys. Res. Commun.,
190, 927-934.
119.Sihag, R. K., and Nixon, R. A. (1991) J.
Biol. Chem., 266, 18861-18867.
120.Gonda, Y., Nishizawa, K., Ando, S., Kitamura,
S., Minoura, Y., Nishi, Y., and Inagaki, M. (1990) Biochem. Biophys.
Res. Commun., 167, 1316-1325.
121.Vallano, M. L., Buckholz, T. M., and DeLorenzo,
R. J. (1985) Biochem. Biophys. Res. Commun., 130,
957-963.
122.Link, W. T., Dosemeci, A., Floyd, C. C., and
Pant, H. C. (1993) Neurosci. Lett., 151, 89-93.
123.Guan, R. J., Khatra, B. S., and Cohlberg, J. A.
(1991) J. Biol. Chem., 266, 8262-8267.
124.Hocevar, B. A., Burns, D. J., and Fields, A. P.
(1993) J. Biol. Chem., 268, 7545-7552.
125.Lamb, N. J., Fernandez, A., Feramisco, J. R.,
and Welch, W. J. (1989) J. Cell Biol., 108,
2409-2422.
126.Eriksson, J. E., Brautigan, D. L., Vallee, R.,
Olmsted, J., Fujiki, H., and Goldman, R. D. (1992) Proc. Natl.
Acad. Sci. USA, 89, 11093-11097.
127.Izawa, I., and Inagaki, M. (2006) Cancer
Sci., 97, 167-174.
128.Kosako, H., Goto, H., Yanagida, M., Matsuzawa,
K., Fujita, M., Tomono, Y., Okigaki, T., Odai, H., Kaibuchi, K., and
Inagaki, M. (1999) Oncogene, 18, 2783-2788.
129.Goto, H., Yasui, Y., Kawajiri, A., Nigg, E. A.,
Terada, Y., Tatsuka, M., Nagata, K., and Inagaki, M. (2003) J. Biol.
Chem., 278, 8526-8530.
130.Altmannsberger, M., Osborn, M., Holscher, A.,
Schauer, A., and Weber, K. (1981) Virchows Arch. B Cell Pathol.
Incl. Mol. Pathol., 37, 277-284.
131.Holwell, T. A., Schweitzer, S. C., and Evans,
R. M. (1997) J. Cell Sci., 110, 1947-1956.
132.Pekny, M., Leveen, P., Pekna, M., Eliasson, C.,
Berthold, C. H., Westermark, B., and Betsholtz, C. (1995) EMBO
J., 14, 1590-1598.
133.Colucci-Guyon, E., Portier, M. M., Dunia, I.,
Paulin, D., Pournin, S., and Babinet, C. (1994) Cell, 79,
679-694.
134.Schiffers, P. M., Henrion, D., Boulanger, C.
M., Colucci-Guyon, E., Langa-Vuves, F., van Essen, H., Fazzi, G. E.,
Levy, B. I., and De Mey, J. G. (2000) Arterioscler. Thromb. Vasc.
Biol., 20, 611-616.
135.Terzi, F., Henrion, D., Colucci-Guyon, E.,
Federici, P., Babinet, C., Levy, B. I., Briand, P., and Friedlander, G.
(1997) J. Clin. Invest., 100, 1520-1528.
136.Alberts, B., Brey, D., Lewis, J., Raff, M.,
Roberts, K., and Watson, J. D. (1993) Molecular Biology of Cell,
Vol. 2 [Russian translation], 2nd Edn., Mir, Moscow.