REVIEW: Capping Complex Formation at the Slow-Growing End of the Actin Filament
A. S. Kostyukova1,2
1Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: allakos@gmail.com2Department of Neuroscience and Cell Biology, RWJMS - UMDNJ, 675 Hoes Lane, Piscataway, NJ 08854, USA; E-mail: kostyuas@umdnj.edu
Received April 23, 2008
Actin filaments are polar; their barbed (fast-growing) and pointed (slow-growing) ends differ in structure and dynamic properties. The slow-growing end is regulated by tropomodulins, a family of capping proteins that require tropomyosins for optimal function. There are four tropomodulin isoforms; their distributions vary depending on tissue type and change during development. The C-terminal half of tropomodulin contains one compact domain represented by alternating α-helices and β-structures. The tropomyosin-independent actin-capping site is located at the C-terminus. The N-terminal half has no regular structure; however, it contains a tropomyosin-dependent actin-capping site and two tropomyosin-binding sites. One tropomodulin molecule can bind two tropomyosin molecules. Effectiveness of tropomodulin binding to tropomyosin depends on the tropomyosin isoform. Regulation of tropomodulin binding at the pointed end as well as capping effectiveness in the presence of specific tropomyosins may affect formation of local cytoskeleton and dynamics of actin filaments in cells.
KEY WORDS: actin filament, capping, tropomodulin, tropomyosin, protein-protein interactionsDOI: 10.1134/S0006297908130075
The ability of actin to polymerize and depolymerize is of great importance for many biological functions such as muscle contraction, cell migration, cytoskeletal organization, and organelle transport [1-4]. Lengths of actin filaments differ significantly depending on tissue type and localization in cells. For example, their length in sarcomeres is 1.1 ± 0.025 µm and their length in spectrin network in erythrocyte membrane is 33 ± 5 nm [5, 6]. There are about 160 various actin-binding proteins whose function is capping, stabilizing, cross-linking, and severing filaments [7]. Actin filaments are polar; their ends (slow-growing or pointed and fast-growing or barbed) differ in structure and dynamics of polymerization/depolymerization. Capping proteins for the fast-growing end are gelsolin, CapZ, and adducin [7-9] and for the slow-growing end are tropomodulin [10, 11], acumentin [12], and Arp2/3 complex [13]. Acumentin was found in macrophages; this protein is not well studied and its sequence is still unknown. Arp2/3 plays an important role in polymerization and depolymerization of actin filaments in the leading edge of migrating cells. Its affinity to actin filaments decreases substantially when ATP bound to actin monomers at the pointed end hydrolyzes to ADP [14]. Tropomodulin has been found in different tissues and cells and its role is especially important when actin filaments have to retain constant length [15]. This review concerns regulation of the pointed end by tropomodulin.
Originally, tropomodulin was found in erythrocyte membranes as a tropomyosin-binding protein with a molecular mass of about 40 kD [16]. Tropomodulin binds specifically to the pointed end of the actin filament and inhibits actin polymerization and depolymerization [10, 17]. Tropomodulin affinity to actin filaments is not high in the absence of tropomyosin (Kd ~ 0.3-0.4 µM) but it increases substantially in the presence of tropomyosin (Kd ~ 50 pM) [3]. Despite the fact that in experiments in vitro tropomodulin binds tightly to actomyosin filaments, capping in vivo is a transient and dynamic process. Littlefield et al. [18] showed that molecules of actin and tropomodulin can exchange with free ones at the pointed end. They demonstrated that in spite of the presence of capping proteins, rhodamine-labeled actin injected into myocytes is able to incorporate into myofibrils at the both ends of actin filaments. About 60% of rhodamine-labeled actin incorporates at the pointed end; therefore, it is more dynamic than the barbed end. It was shown that at each moment in time some of the pointed ends are not capped. The authors supposed that there is not enough endogenic tropomodulin to cap all filaments. They suggested the model of dynamic capping when tropomodulin is bound to the pointed end temporarily, which allows accidental depolymerization and polymerization resulting in actin exchange. It was demonstrated that tropomodulin overexpression impeded incorporation of rhodamine-labeled actin and resulted in filament shortening. It was explained that excess of tropomodulin does not prevent depolymerization but impedes polymerization because tropomodulin competes with monomeric actin for binding. Length of actin filaments in myocyte sarcomeres is defined by coordination of different actin-binding proteins, which regulate dynamics of the pointed end. Tropomodulin and tropomyosin are two important proteins in the process of this regulation.
Tropomyosin is an elongated two-chained protein that forms coiled-coil [19]. During formation of the complex with the actin filament, tropomyosin molecules bind along both sides of the filament. The N-terminus of each tropomyosin molecule interacts with the C-terminus of the following one. The N-terminus of tropomyosin is directed to the pointed end of the actin filament. Tropomyosin isoforms are encoded by four genes--α,_β,_γ, and ä. As a result of alternative splicing, both high- (284 residues) and low-molecular-weight (246 residues) tropomyosin isoforms are synthesized at least from two of four genes. Distribution of these isoforms depends on type of tissues and cells and may change during development. For example, in striated muscles high-molecular-weight α- ?and β-tropomyosins interact with actin filaments of contractile apparatus while low-molecular-weight γ-tropomyosin, TM5NM1, is found in cytoskeleton adjoining the Z-line [20].
TROPOMODULIN ISOFORMS
When the tropomodulin sequence was determined, no homology was found with known proteins; therefore, tropomodulins were classified as a new group [21]. At present four tropomodulin isoforms are known [22-24]. Tmod1, previously E(erythrocyte)-Tmod, was detected in many tissues but mainly in erythrocytes and cardiac and skeletal muscles. Tmod2, N(neuron)-Tmod, was found in brain cells. Tmod3, U(ubiquitous)-Tmod, was found in many tissues. Tmod4, Sk(skeletal)-Tmod, was found in skeletal muscles, where it replaces Tmod1 during development. Percent of homology of these isoforms is about 70%.
Based on homology with tropomodulins, proteins with higher molecular mass of about 64 kD, leiomodins, were cloned [24, 25]. There are three leiomodin isoforms: Lmod1 found in many tissues but mainly in smooth muscles, Lmod2 found in cardiac and skeletal muscles, and Lmod3. The role of these proteins is still unknown. They are able to bind tropomyosin [25, 26]. There are evidences that Lmod2 may act as a factor nucleating formation of actin filaments [27].
In muscles, tropomodulin was localized at the slow-growing ends of thin filaments [28]. Tropomodulin overexpression in cardiac myocytes resulted in myofibril degeneration [18, 29]. Decrease of tropomodulin expression resulted in formation of unusually long actin filaments [30]. Tropomodulin function is critical at the late stages of myofibrillogenesis [31]. In mice with knocked out tropomodulin gene the myocardium does not develop, causing lethality at an embryonic stage [32, 33].
Unlike Tmod1 and Tmod4, which bound only with F-actin, Tmod3 is able to bind G-actin as well [34, 35]. Presumably, Tmod2 also is able to bind G-actin. Overexpression of Tmod3 resulted in lower motility of endothelial cells [36]. Tmod2 expression changes under pathological conditions such as epilepsy or cerebral ischemia [37-39].
TROPOMODULIN STRUCTURE
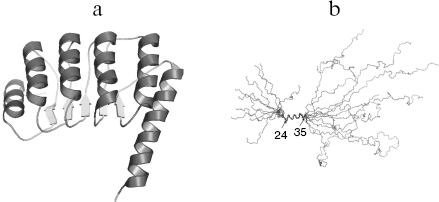
Of all tropomodulin isoforms Tmod1 structure, function, and interaction with other proteins are best studied. In the following text of this review, Tmod1 will be referred as tropomodulin. Tropomodulin is an elongated molecule, its C-terminal half consisting of one compact cooperatively melting domain [40-42]. The crystal structure of this domain is represented by alternating α-helices and β-structures [43] (Fig. 1a). This structure is characteristic for the LRR (leucine rich repeat) motif that usually participates in protein-protein interactions [44-47]. On the contrary, the N-terminal half is unstructured [40-42, 48, 49]. Residues 24-35 form α-helix, and other parts have no regular structure (Fig. 1b).
Fig. 1. a) Ribbon presentation of atomic model of the tropomodulin C-terminal half. b) Alignment of the ten backbone structures of tropomodulin N-terminal fragment (residues 1-92) obtained in solution by nuclear magnetic resonance.
LOCALIZATION OF BINDING SITES WITH TROPOMYOSIN, ACTIN, AND NEBULIN
In spite of the fact that the N-terminal half of tropomodulin has no regular structure, adding the N-terminal fragment (residues 1-91) to tropomyosin drastically changed melting curves as shown by circular dichroism and differential scanning calorimetry [42]. Similar results were obtained using circular dichroism for a longer fragment of tropomodulin (residues 1-130) mixed with the N-terminal tropomyosin fragment [50]. As a result of the complex formation, α-helical content increased substantially as well as the complex melting temperature as compared with the melting temperature of the tropomyosin fragment.
It was supposed that in the tropomodulin molecule there are different sites for binding low- and high-molecular-weight tropomyosins [21, 51]. Residues 6-94 of tropomodulin interact with high-molecular-weight skeletal tropomyosin and residues 90-184 interact with erythrocyte tropomyosin (homodimers or heterodimers of low-molecular-weight α- and γ-tropomyosins [52, 53]). However, some data were in contradiction with this supposition. For example, capping activity of a tropomodulin fragment (residues 95-359) increased 160-fold in the presence of high-molecular-weight tropomyosin [54]. This indicated that there is a second binding site for this tropomyosin. Moreover, tropomodulin fragment 1-92 was able to bind not only high-molecular-weight muscle α-tropomyosin, but also low-molecular-weight non-muscle α-tropomyosin [55]. In recent studies, it was shown that residues 105-127 are important for binding low-molecular-weight γ-tropomyosin [56]. In another study, it was shown that residues Leu134 and Leu135 are key for this binding [57]. None of these studies showed binding of low-molecular-weight tropomyosins to the first binding site or high-molecular-weight tropomyosins to the second binding site. The number of binding sites and their specificity were still unknown.
This issue was resolved by mutagenesis and analysis of tropomyosin-tropomodulin interaction using model peptides. In addition to helix 24-35 determined by NMR, the tropomodulin N-terminal half contains several putative helical regions (residues 65-75 and 126-135) [48]. One side of each helix forms a hydrophobic surface. Replacement of conserved hydrophobic residues in these helices destroys formation of hydrophobic surfaces. This turned out to be crucial for forming binding sites [48, 49, 58]. Changing Leu27 to Glu and Ile131 to Asp resulted in loss of binding of both high- and low-molecular-weight tropomyosins to the first and second binding sites. Change of Leu71 to Asp decreased tropomodulin-capping activity. All together, these three mutations caused 30-fold decrease of tropomodulin capping activity.
Positions of tropomyosin-binding sites in a tropomodulin molecule are shown in Fig. 2 [48, 49, 54, 55, 58]. Two binding sites, residues 1-38 and 109-144, were localized both for high- and low-molecular-weight tropomyosin isoforms. A tropomyosin-dependent actin-capping site was localized to residues 48-92, and a tropomyosin-independent actin-capping site was localized at the C-terminus of tropomodulin. Removal of 15 C-terminal residues resulted in loss of ability to cap actin filaments in the absence of tropomyosin. Regions 1-38, 109-144, and 48-92 are conserved in all tropomodulin isoforms; therefore, most likely other isoforms have the same binding sites. Besides, in Tmod3 two regions (residues 31-40 and 149-169) presumably participate in binding G-actin [35].
In addition to actin and tropomyosin, another protein, nebulin, binds to tropomodulin in striated muscles. Nebulin is a giant protein with molecular mass about 800 kD bound along actin filaments in sarcomeres [59, 60]. The nebulin N-terminus is directed towards the pointed end of the actin filament. Nebulin consists of short repeats [61]. It was suggested that nebulin determines actin filament length in sarcomeres by regulating tropomodulin binding [31, 62]. It acts as a molecular ruler. A nebulin fragment that contains three N-terminal modules M1-M2-M3 binds tropomodulin [63]. The binding site for nebulin is not localized yet, but it is presumably located in the C-terminal half of the tropomodulin molecule (residues 160-344) [43].Fig. 2. Schematic arrangement of tropomyosin-binding and actin-capping sites on the tropomodulin molecule.

ISOFORM SPECIFICITY OF TROPOMODULIN-TROPOMYOSIN BINDING
Specificity of tropomyosin/tropomodulin binding first was shown for tropomyosin isoforms that are expressed in erythrocytes, brain, thrombocytes, and skeletal muscles [64]. Tmod1 formed complexes with all these isoforms but bound preferentially to erythrocyte tropomyosin. It was already known that tropomodulin binds to one of the tropomyosin termini [65]. Therefore, it was supposed that binding correlates with heterogeneity of N- and C-terminal sequences characteristic for tropomyosin isoforms [64]. It was assumed that isoform-dependent interaction may be a mechanism for selective regulation of tropomodulin binding to actin-tropomyosin complex.
Later it was shown that tropomodulin binds to the N-terminal region of tropomyosin [52]. Low-molecular-weight non-muscle γ-tropomyosin is unable to bind tropomodulin without the first 19 residues. Residues 7-14 form a binding site for tropomodulin [66]. The first 14 residues of high-molecular-weight tropomyosins are homologous to residues 6-19 of low-molecular-weight tropomyosins (table) and also form a binding site for tropomodulin [50].
Dissociation constants (Kd) of complexes formed by
tropomyosin and tropomodulin fragments*
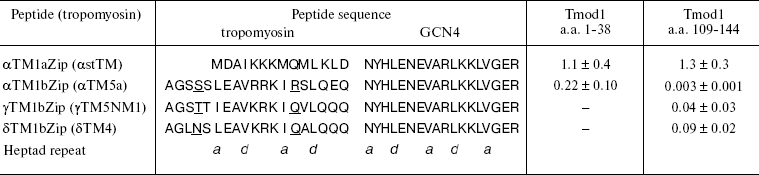
* Kd values (µM) were estimated from the
thermodynamics of denaturation of the complexes compared with
individual fragments [70]. Sequences of
tropomyosin isoforms are aligned according to their homology.
Tropomyosin fragments are chimeric peptides that consist of N-terminal
sequences of low- and high-molecular-weight tropomyosin isoforms
(residues 1-19 and 1-14, correspondingly) and 18 residues of leucine
zipper from yeast transcriptional activator GCN4. Amino acid residues
responsible for isoform specificity of low-molecular-weight
tropomyosins are underlined.
Model chimeric peptides were used for quantitative studies of tropomyosin-tropomodulin binding [48-50, 54, 55, 58, 67]. Originally, these peptides were constructed to determine structure of tropomyosin N-terminal region [68, 69]. In addition to the N-terminal regions of low- and high-molecular-weight tropomyosin isoforms (residues 1-19 and 1-14, correspondingly), chimeric peptides contain 18 residues of so-called leucine zipper from yeast transcriptional activator GCN4 [68] (table). To form stable coiled-coil structure, the amino acid sequence has to contain heptad repeats where hydrophobic residues are in positions a and d. Leucine zipper contains heptad repeats where Leu is in d positions. It forms dimers and stabilizes coiled-coil structure of chimeric peptides. All chimeric peptides were acetylated because N-terminal acetylation stabilizes the structure of high-molecular-weight tropomyosins and is important for interaction with tropomodulin [50].
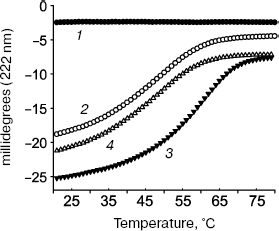
Tropomodulin-tropomyosin interaction was studied in details by circular dichroism (Fig. 3) using two fragments of tropomodulin that contain individual binding sites and four chimeric peptides, αTM1aZip, αTM1bZip, γTM1bZip, and äTM1bZip. These peptides represented high-molecular-weight muscle α-tropomyosin and low-molecular-weight non-muscle α-, γ-, and ä-tropomyosins. The designations 1a and 1b correspond to alternate exons that distinguish high-molecular-weight isoform from low-molecular-weight one. Dissociation constants calculated from melting curves [26, 48, 67] are given in the table. Equations used for the calculation are described in details in [70].
The tropomyosin N-terminal sequence used for design of chimeric peptide αTM1aZip (table) is identical in high-molecular-weight α and β_isoforms. There is only one conserved change (Asp2 to Glu) in a corresponding sequence of high-molecular-weight γ-tropomyosin (bold in the table). High-molecular-weight tropomyosin coded by the ä-gene was not found. Therefore, dissociation constants obtained for complexes of αTM1aZip with tropomodulin fragments should be the same or close to all high-molecular-weight isoforms. As for low-molecular-weight isoforms, their N-terminal regions are different though homologous. In spite of homology, difference in affinity is striking. While αTM1bZip and αTM1aZip bind tightly to both binding sites in tropomodulin (though with different affinity), ?γTM1bZip and äTM1bZip bind well only to the second site. Binding these peptides to the first site is not tight enough to calculate dissociation constants using circular dichroism method. But it can be detected using cross-linking [67]. Amino acid residues responsible for isoform specificity of binding low-molecular-weight tropomyosins to tropomodulin are in positions 4 and 14 of the amino acid sequence shown in the table [67].Fig. 3. Influence of binding of tropomodulin fragment on denaturation of tropomyosin fragment by the example of Tmod1109-144 and γTM1bZip. Temperature dependence of circular dichroism measured at 222 nm obtained for tropomodulin fragment (1), tropomyosin fragment (2), the mixture of the tropomyosin and tropomodulin fragments (3), and the sum of the denaturation curves of the tropomyosin and tropomodulin fragments (4). The increase of helicity and stability in the mixture compared to the sum indicates complex formation.
MODEL OF THE SLOW-GROWING END
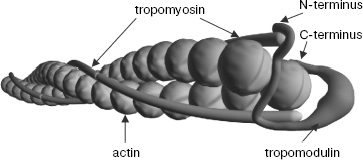
As a consequence of experiments described above, prolonged discussion concerning localization of tropomyosin binding sites in tropomodulin was finished. All tropomyosin isoforms bind both sites. Low-molecular-weight γ- and ä-isoforms bind the first binding site with low affinity. However, if tropomodulin has two binding sites for tropomyosin, how many molecules of tropomyosin can one tropomodulin molecule bind? Tropomodulin was titrated with αTM1bZip, and it was found that one tropomodulin molecule binds simultaneously two peptides [48]. In accordance with the suggested model, one tropomodulin molecule located at the pointed end binds both tropomyosin molecules (Fig. 4).
Complexes of tropomyosin and tropomodulin fragments were studied using nuclear magnetic resonance, and it was shown that structures of the complexes differ significantly [67]. Possible binding modes based on circular dichroism data, mutagenesis, and the effect of complex formation on 1H-15N HSQC spectra of αTM1bZip have been suggested [48, 49, 56, 57, 67]. Tropomodulin residues make a loop around one tropomyosin N-terminus in the first binding site and bind antiparallel to another tropomyosin N-terminus at the second binding site. It is hypothetical model. Structure, arrangement of helices, and significance of amino acid residues of these complexes still have to be determined in future studies.Fig. 4. Model of the slow-growing (pointed) end of the actin filament. Tropomyosin N-termini are directed to the pointed end. Tropomodulin interacts with both tropomyosin molecules.
Mechanisms that regulate capping by tropomodulin are still unknown. It is possible that there are factors directly affecting tropomodulin function by binding or modifying it. Isoform specificity of tropomodulin-tropomyosin interactions might also be one of the regulating mechanisms. Slight differences in amino acid sequences of tropomyosin isoforms have significant influence on tropomodulin binding and therefore may modulate dynamics of the slow growing ends of the actin filaments. High-molecular-weight muscle tropomyosin binds to both sites in the tropomodulin molecule with the same affinity, while binding low-molecular-weight tropomyosins to the second site is tighter than to the first. This difference in affinity might regulate correct positioning of the tropomodulin molecule in the absence of another tropomodulin-binding protein, nebulin, found only in striated skeletal and cardiac muscles. Moreover, affinity of tropomodulin to different tropomyosin isoforms correlates with the capping in the presence of these isoforms. Regulation of tropomodulin binding at the pointed end as well as capping effectiveness in the presence of specific tropomyosins may affect formation of local cytoskeleton and dynamics of actin filaments in cells.
REFERENCES
1.Fowler, V. M. (1996) Curr. Opin. Cell Biol.,
8, 86-96.
2.Fowler, V. M. (1997) Soc. Gen. Physiol.
Ser., 52, 79-89.
3.Weber, A., Pennise, C. R., and Fowler, V. M. (1999)
J. Biol. Chem., 274, 34637-34645.
4.Pollard, T. D., and Borisy, G. G. (2003)
Cell, 112, 453-465.
5.Shen, B. W., Josephs, R., and Steck, T. L. (1986)
J. Cell Biol., 102, 997-1006.
6.Sosa, H., Popp, D., Ouyang, G., and Huxley, H. E.
(1994) Biophys. J., 67, 283-292.
7.Dos Remedios, C. G., Chhabra, D., Kekic, M.,
Dedova, I. V., Tsubakihara, M., Berry, D. A., and Nosworthy, N. J.
(2003) Physiol. Rev., 83, 433-473.
8.Cooper, J. A., and Schafer, D. A. (2000) Curr.
Opin. Cell Biol., 12, 97-103.
9.Kuhlman, P. A., Hughes, C. A., Bennett, V., and
Fowler, V. M. (1996) J. Biol. Chem., 271, 7986-7991.
10.Weber, A., Pennise, C. R., Babcock, G. G., and
Fowler, V. M. (1994) J. Cell Biol., 127, 1627-1635.
11.Schafer, D. A., and Cooper, J. A. (1995) Annu.
Rev. Cell Dev. Biol., 11, 497-518.
12.Southwick, F. S., and Hartwig, J. H. (1982)
Nature, 297, 303-307.
13.Mullins, R. D., Heuser, J. A., and Pollard, T. D.
(1998) Proc. Natl. Acad. Sci. USA, 95, 6181-6186.
14.Blanchoin, L., Pollard, T. D., and Mullins, R. D.
(2000) Curr. Biol., 10, 1273-1282.
15.Weber, A. (1999) Mol. Cell Biochem.,
190, 67-74.
16.Fowler, V. M. (1987) J. Biol. Chem.,
262, 12792-12800.
17.Gregorio, C. C., and Fowler, V. M. (1995) J.
Cell Biol., 129, 683-695.
18.Littlefield, R., Almenar-Queralt, A., and Fowler,
V. M. (2001) Nat. Cell Biol., 3, 544-551.
19.Perry, S. V. (2001) J. Muscle Res. Cell
Motil., 22, 5-49.
20.Gunning, P. W., Schevzov, G., Kee, A. J., and
Hardeman, E. C. (2005) Trends Cell Biol., 15,
333-341.
21.Sung, L. A., Fowler, V. M., Lambert, K., Sussman,
M. A., Karr, D., and Chien, S. (1992) J. Biol. Chem.,
267, 2616-2621.
22.Watakabe, A., Kobayashi, R., and Helfman, D. M.
(1996) J. Cell Sci., 109, 2299-2310.
23.Almenar-Queralt, A., Lee, A., Conley, C. A., de
Pouplana, L. R., and Fowler, V. M. (1999) J. Biol. Chem.,
274, 28466-28475.
24.Conley, C. A., Fritz-Six, K. L., Almenar-Queralt,
A., and Fowler, V. M. (2001) Genomics, 73, 127-139.
25.Conley, C. A. (2001) Am. J. Physiol. Cell
Physiol., 280, C1645-1656.
26.Kostyukova, A. (2007) Arch. Biochem.
Biophys., 465, 227-230.
27.Chereau, D., Boczkowska, M., Skwarek-Maruszewska,
A., Fujiwara, I., Hayes, D. B., Rebowski, G., Lappalainen, P., Pollard,
T. D., and Dominguez, R. (2008) Science, 320,
239-243.
28.Fowler, V. M., Sussmann, M. A., Miller, P. G.,
Flucher, B. E., and Daniels, M. P. (1993) J. Cell Biol.,
120, 411-420.
29.Sussman, M. A., Welch, S., Cambon, N., Klevitsky,
R., Hewett, T. E., Price, R., Witt, S. A., and Kimball, T. R. (1998)
J. Clin. Invest., 101, 51-61.
30.Sussman, M. A., Baque, S., Uhm, C. S., Daniels,
M. P., Price, R. L., Simpson, D., Terracio, L., and Kedes, L. (1998)
Circ. Res., 82, 94-105.
31.McElhinny, A. S., Schwach, C., Valichnac, M.,
Mount-Patrick, S., and Gregorio, C. C. (2005) J. Cell Biol.,
170, 947-957.
32.Chu, X., Chen, J., Reedy, M. C., Vera, C., Sung,
K. L., and Sung, L. A. (2003) Am. J. Physiol. Heart Circ.
Physiol., 284, H1827-1838.
33.Fritz-Six, K. L., Cox, P. R., Fischer, R. S., Xu,
B., Gregorio, C. C., Zoghbi, H. Y., and Fowler, V. M. (2003) J. Cell
Biol., 163, 1033-1044.
34.Fischer, R. S., Sept, D., Weber, K. L., Speicher,
D. W., and Fowler, V. M. (2004) Mol. Biol. Cell, 15,
147a.
35.Fischer, R. S., Yarmola, E. G., Weber, K. L.,
Speicher, K. D., Speicher, D. W., Bubb, M. R., and Fowler, V. M. (2006)
J. Biol. Chem., 281, 36454-36465.
36.Fischer, R. S., Fritz-Six, K. L., and Fowler, V.
M. (2003) J. Cell Biol., 161, 371-380.
37.Yang, J. W., Czech, T., Felizardo, M.,
Baumgartner, C., and Lubec, G. (2006) Amino Acids, 30,
477-493.
38.Iwazaki, T., McGregor, I. S., and Matsumoto, I.
(2006) Brain Res., 1097, 19-25.
39.Chen, A., Liao, W. P., Lu, Q., Wong, W. S., and
Wong, P. T. (2007) Neurochem. Int., 50, 1078-1086.
40.Kostyukova, A., Maeda, K., Yamauchi, E., Krieger,
I., and Maeda, Y. (2000) Eur. J. Biochem., 267,
6470-6475.
41.Fujisawa, T., Kostyukova, A., and Maeda, Y.
(2001) FEBS Lett., 498, 67-71.
42.Kostyukova, A. S., Tiktopulo, E. I., and Maeda,
Y. (2001) Biophys. J., 81, 345-351.
43.Krieger, I., Kostyukova, A., Yamashita, A.,
Nitanai, Y., and Maeda, Y. (2002) Biophys. J., 83,
2716-2725.
44.Kobe, B., and Deisenhofer, J. (1995) Mol. Cell
Neurosci., 6, 97-105.
45.Papageorgiou, A. C., Shapiro, R., and Acharya, K.
R. (1997) Embo J., 16, 5162-5177.
46.Price, S. R., Evans, P. R., and Nagai, K. (1998)
Nature, 394, 645-650.
47.Kobe, B., and Kajava, A. V. (2001) Curr. Opin.
Struct. Biol., 11, 725-732.
48.Kostyukova, A. S., Choy, A., and Rapp, B. A.
(2006) Biochemistry, 45, 12068-12075.
49.Greenfield, N. J., Kostyukova, A. S., and
Hitchcock-Degregori, S. E. (2005) Biophys. J., 88,
372-383.
50.Greenfield, N. J., and Fowler, V. M. (2002)
Biophys. J., 82, 2580-2591.
51.Babcock, G. G., and Fowler, V. M. (1994) J.
Biol. Chem., 269, 27510-27518.
52.Sung, L. A., and Lin, J. J. (1994) Biochem.
Biophys. Res. Commun., 201, 627-634.
53.Sung, L. A., Gao, K. M., Yee, L. J., Temm-Grove,
C. J., Helfman, D. M., Lin, J. J., and Mehrpouryan, M. (2000)
Blood, 95, 1473-1480.
54.Fowler, V. M., Greenfield, N. J., and Moyer, J.
(2003) J. Biol. Chem., 278, 40000-40009.
55.Kostyukova, A. S., and Hitchcock-DeGregori, S. E.
(2004) J. Biol. Chem., 279, 5066-5071.
56.Vera, C., Lao, J., Hamelberg, D., and Sung, L. A.
(2005) Arch. Biochem. Biophys., 444, 130-138.
57.Kong, K. Y., and Kedes, L. (2006) J. Biol.
Chem., 281, 9589-9599.
58.Kostyukova, A., Rapp, B., Choy, A., Greenfield,
N. J., and Hitchcock-DeGregori, S. E. (2005) Biochemistry,
44, 4905-4910.
59.Wang, K. (1996) Adv. Biophys., 33,
123-134.
60.Fock, U., and Hinssen, H. (2002) J. Muscle
Res. Cell Motil., 23, 205-213.
61.Pfuhl, M., Winder, S. J., Castiglione Morelli, M.
A., Labeit, S., and Pastore, A. (1996) J. Mol. Biol.,
257, 367-384.
62.Witt, C. C., Burkart, C., Labeit, D., McNabb, M.,
Wu, Y., Granzier, H., and Labeit, S. (2006) Embo J., 25,
3843-3855.
63.McElhinny, A. S., Kolmerer, B., Fowler, V. M.,
Labeit, S., and Gregorio, C. C. (2001) J. Biol. Chem.,
276, 583-592.
64.Sussman, M. A., and Fowler, V. M. (1992) Eur.
J. Biochem., 205, 355-362.
65.Fowler, V. M. (1990) J. Cell Biol.,
111, 471-481.
66.Vera, C., Sood, A., Gao, K. M., Yee, L. J., Lin,
J. J., and Sung, L. A. (2000) Arch. Biochem. Biophys.,
378, 16-24.
67.Kostyukova, A., Hitchcock-DeGregori, S. E., and
Greenfield, N. J. (2007) J. Mol. Biol., 372, 608-618.
68.Greenfield, N. J., Montelione, G. T., Farid, R.
S., and Hitchcock-DeGregori, S. E. (1998) Biochemistry,
37, 7834-7843.
69.Greenfield, N. J., Huang, Y. J., Palm, T.,
Swapna, G. V., Monleon, D., Montelione, G. T., and Hitchcock-DeGregori,
S. E. (2001) J. Mol. Biol., 312, 833-847.
70.Greenfield, N. J. (2004) Meth. Mol. Biol.,
261, 55-78.