REVIEW: Pore-Forming Proteins and Adaptation of Living Organisms to Environmental Conditions
Zh. I. Andreeva-Kovalevskaya1, A. S. Solonin1*, E. V. Sineva1, and V. I. Ternovsky2*
1Skryabin Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: solonin@ibpm.pushchino.ru2Institute of Cell Biophysics, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: vternovsky@mail.ru
* To whom correspondence should be addressed.
Received July 16, 2008; Revision received August 4, 2008
Pore-forming proteins are powerful “tools” for adaptation of living organisms to environmental conditions. A wide range of these proteins isolated from various sources, from viruses to mammals, has been used for the analysis of their role in the processes of intra- and inter-species competition, defense, attack, and signaling. Here we review a large number of pore-forming proteins from the perspective of their functions, structures, and mechanisms of membrane penetration. Various mechanisms of cell damage, executed by these proteins in the course of formation of a pore and after its passing to conducting state, have been considered: endo- and exocytosis, lysis, necrosis, apoptosis, etc. The role of pore-forming proteins in evolution is discussed. The relevance of practical application of pore formers has been shown, including application in nanotechnological constructions.
KEY WORDS: pore-forming proteins, adaptation, pore structure, apoptosis, nanotechnologyDOI: 10.1134/S0006297908130087
Abbreviations: GPI, glycosylphosphatidylinositol; lipid II, undecaprenyl-pyrophosphoryl-MurNAc-(pentapeptide)-GlcNAc; RTX, repeats in toxin.
The cell membrane is the primary barrier in contacts of a living
organism with the environment or other species. No wonder that in the
course of evolution most living organisms have acquired the capacity to
secret compounds that alter permeability of membranes of hostile cells
[1, 2]. An intriguing feature
of great importance is secretion of pore-forming proteins that insert
into hostile cell membranes and form pores [2].
The membrane-binding event is the initial step in the action of any pore-forming protein, regardless of its chemical composition, usually aimed to create a hydrophilic channel that helps various compounds (ions, saccharides, and even proteins, provided the pore is large enough) to cross the hydrophobic area of the membrane. Formation of additional pores triggers various mechanisms of cell death. For example, cytolysins make membranes more permeable for ions by shifting osmotic equilibrium of the cell, thereby causing its swelling followed by cytolysis. A disturbed ionic homeostasis can induce massive endocytosis, exocytosis, necrosis, or cell death by apoptosis. Hence, living organisms capable of producing such compounds have the obvious advantage of easier adaptation to environmental conditions, which eventually results in an increase in their number and natural habitat. Pore-forming proteins can be classified in accordance with their function, mechanism of membrane penetration, size of pores or pore-forming subunits, etc. Most informative is classification based on the type of pore-forming structures in the membrane plane, which defines α- and β-pore-forming proteins [2] and reveals common features in pore formation and pore function inherent to evolutionarily remote organisms (Fig. 1 (a and b) and table).
Comparison of pore-forming protein propertiesFig. 1. a) A pore formed by α-pore-forming protein melittin and lipids (side view) is shown using the program RasMol 2.6 [154]. b) The structure of a heptameric pore formed by β-pore-forming protein S. aureus α-hemolysin (side view). The stem-, rim-, and cap-domains [85] are indicated. Schematic illustration of a barrel-stave pore in a lipid bilayer: c) view from above; d) side view. Schematic illustration of a toroidal pore: e) view from above; f) side view. Protein monomers are shown as dark cylinders (c, e) [6] and as dark rectangles (d, f) [9].
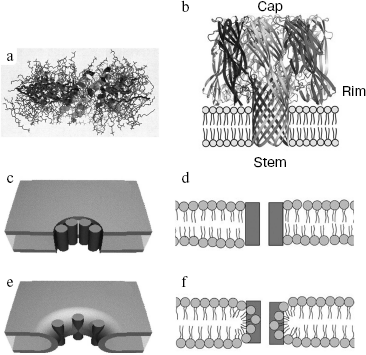
Note: N, number of pore-forming subunits. Symbol “?” means that the pore size and number of pore-forming subunits are unknown.
The current review describes pore-forming toxins and their functional role in adaptation of living organisms from various classification groups to environmental conditions.
PORE-FORMING PROTEINS FROM LIVING ORGANISMS OF VARIOUS
CLASSIFICATION GROUPS
Eukaryotic α-Pore-Forming Proteins
Basically, there are two types of pore-forming cytolysins responsible for adaptation of invertebrates to environmental conditions. The first includes cecropin-like proteins with a helix-bend-helix structure with a low hemolytic activity and pronounced antimicrobial properties, which serves as a component of the insect immune system used against pathogenic microorganisms. Toxins of this type have been detected mainly in hemolymph of various insects, for example, sarcotoxin A in flies, cecropin in butterflies, and spinegirin in termites. The orientation of positively charged N-terminal helices of these toxins allows their electrostatic interaction with negatively charged lipid groups when hydrophobic C-terminal helices of six cecropin dimers insert into the membrane and form a pore [3]. Cecropin A isolated from hemolymph of the moth Hyalophora cecropia consists of 37 amino acids and shows antibacterial activity. Free cecropin A is an amphiphilic monomer with the typical helix-bend-helix structure. At the first step of pore formation, both N- and C-terminal helices are oriented parallel to the membrane, and then C-terminal helices of 12 peptides insert into the membrane and form a pore [4].
The second type of pore-forming cytolysins comprises antimicrobial melittin-like peptides that display a higher hemolytic activity as compared with cecropin-like peptides. They are used by some predators as an immobilizing and killing agent, as well as for defense from other animals or humans. Peptides of this type have been detected in venom of bees, spiders, ants, and scorpions. Structurally, they are amphiphilic α-helical peptides. Melittin (H2N-GIGAVLKVLTTGLPALISTIKRKRQQ-CONH2), a typical member of this group of toxins is the basic component of Apis mellifera bee venom [5]. This polypeptide is capable of killing bacteria and lysing blood cells, as well as various eukaryotic tissues. The cytolytic activity of melittin is underlain by its ability to form pores in membranes using an amphiphilic α-helix formed by two chain regions (residues 1-10 and 13-26). Depending on the type of preferable fatty acids of the membrane bilayer, on melittin phase state, and on the peptide/lipid ratio, melittin binding occurs either predominantly in the membrane-parallel orientation (without pore forming) or in the membrane-orthogonal orientation that subsequently leads to transmembrane pore formation [6]. In the latter case, melittin molecules are brought together by the “helix-helix” interaction, with inwardly directed hydrophilic helix sides and hydrophobic ones exposed to the hydrophobic area of the lipid bilayer. The size of melittin-formed pores depends on toxin concentration and ranges from 1-1.3 to 2.5-3 nm at 0.01 to 0.04 toxin/lipid molar ratios, respectively. To form pores of this diameter, about 6-7 and 10-15 helices, respectively, are required [7, 8]. Pores of this type with the inner surface formed solely by protein monomers are usually termed “barrel-stave”. Later studies demonstrated that most probably melittin yields the so-called toroidal pores formed by invagination of the outer membrane monolayer to involve the hydrophilic bilayer heads, now inwardly directed, in pore formation [9] (Fig. 1, c-f). Due to participation of membrane lipids in toroidal pore formation, pores formed by 4-8 melittin monomers have an inner diameter of 3.5-4.5 nm and an outer diameter of 7-8 nm [10]. Unlike melittin capable of forming both “barrel-stave” and toroidal pores, other antimicrobial α-helical peptides, e.g. alamethicin from the fungus Trichoderma viride used as a biofungicide [11] can form solely “barrel-stave” (6-10 peptide molecules) pores with an inner diameter for the biggest aggregate of 1.8 nm and an outer diameter of 4 nm.
Spiders use their α-helical peptide-based venom not only to immobilize or to kill a prey, but also to digest it. Spider venom contains toxins that disrupt cell membranes, thereby causing tissue necrosis. For example, venom of the wolf spider Lycosa carolinensis contains two amphiphilic α-helical peptides, lycotoxin I (IWLTALKFLGKHAAKHLAKQQLSKL-NH2) and lycotoxin II (KIKWFKTMKSIAKFIAKEQMKKHLGGE-OH), that form membrane pores and cause lysis of prokaryotic and eukaryotic cells (specifically, bacterial, yeast, and red blood cells). As found, these toxins also efflux Ca2+ from rat brain synaptosomes and reduce electrochemical potential across insect muscle membranes. Therefore, these proteins are multifunctional and are involved in a number of events, namely, prey capture and digestion, host protection against infectious microorganisms present in the prey's body, and fight with outward enemies [12].
Five amphiphilic α-helical peptides with antimicrobial, hemolytic, and insecticidal activity have been isolated from venom of the spider Oxyopes kitabensis. These peptides, named oxyopinins, form ion channels in cell membranes. Neurotoxin named oxytoxin 1, isolated from the same spider venom, appeared to be a sodium channel inhibitor [5]. Oxyopinin 1 is composed of 48 amino acid residues and shows sequence homology to the ant insecticidal peptide ponericin L2 and to the frog antimicrobial peptide dermaseptin.
Venom of the spider Latrodectus tredecimguttatus contains α-latrotoxin with a molecular mass of 130 kD. In the absence of divalent cations, it exists in solution predominantly as a dimer incapable of pore formation. Ca2+ or Mg2+ added to the medium at a millimolar concentration makes the dimer oligomerize up to a tetramer. It is in this form that the protein binds to a membrane during pore formation. Its interaction with cells requires the presence of a membrane receptor latrophilin or neurexin [13]. As shown by cryo-electron microscopy, the channel structure resembles a four-vane “propeller” with an inner diameter of 2.5 nm [14] (Fig. 2, a and b). Latrotoxin can interact with cell membranes by two mechanisms: (i) by binding to a receptor without forming its own pores, the toxin induces membrane depolarization through inhibition of voltage-gated potassium channels and activation of L-type Ca2+ channels (most probably this effect is underlain by interaction between the toxin-receptor complex and the G-protein system); (ii) the toxin forms cation-selective transmembrane pores that are permeable for calcium ions. The combined effects of α-latrotoxin on the membrane are manifested by enhanced exocytosis in nerve cell terminals and by massive production of neuromediators, which leads to complete blockage of neuromuscular transmission [15, 16].
Like spiders, scorpions are active predators with venom showing toxicity for both pro- and eukaryotes. Specifically, two toxins isolated from scorpion venom are papabutoporin and opistoporin 1. As shown by whole cell leak current measurement, in cardiac myocyte membranes these toxins form nonselective pores with an effective diameter ranging from 1.38 to 1.78 nm [17]. The pore-forming antibacterial α-helical peptide pandinin 2 (FWGALAKGALKLIPSLFSSFSKKD) with hemolytic activity was found in venom of the African scorpion Pandinus imperator. At the first step of interaction, in its membrane-parallel orientation, the toxin binds to membrane cells; then it forms oligomers, the N-terminal regions of which insert into the membrane and form a pore [18].Fig. 2. a) Cryo-electron microscopy of an α-latrotoxin tetrameric pore formed in a lipid bilayer in the presence of Mg2+; b) a 3D reconstruction of this pore [14]. c) Atomic force microscopy of Clostridium perfringens perfringolysin O pores formed in a cholesterol-containing lipid bilayer [155]. d) Electron microscopy of B. cereus hemolysin II pore in liposomes; e) a model of this pore (view from above) [96].

Sea anemones of the Anthozoa class produce venom containing pore-forming cytotoxins termed actinoporins. The sea anemone Actinia equina secrets equinatoxin II (Eqt-II), whereas sticholysin II (St-II) is a product of Stichodactyla helianthus. Both toxins have a molecular mass of about 20 kD and are folded as a β-sandwich flanked with α-helices. The toxins bind to a lipid bilayer using a cluster of aromatic amino acids located in a loop at the β-sandwich top and in the C-terminal α-helix. The amphiphilic 30-amino-acid N-terminal α-helix, previously parallel to the membrane, inserts into the cell membrane, keeping the β-sandwich undisturbed, and changes its membrane-relative orientation to orthogonal. At the final step, helices of three or four monomers associate to form a toroidal transmembrane pore. Actinoporins bind preferentially to membranes containing sphingomyelin and create cation-selective pores. Actinoporins are highly toxic for fish and shellfish. They play the key role in host protection against enemy attacks and in capture of prey. In vertebrate organisms, these toxins can cause erythrocyte lysis, pulmonary edema, and heart attacks [19, 20].
The marine worm-nemertine Cerebratulus lacteus secrets a protein cytolysin A-III that consists of a polypeptide chain of 95 amino acid residues cross-linked by three disulfide bonds, and possesses an α-helical structure. The C-terminal third of the toxin sequence is postulated to be a helical hairpin structure involved in pore formation that permeabilizes a variety of cells as well as liposomes of various lipid composition. Apparently, the toxin forms large pores as large proteins are released. At sublytic concentrations, the toxin inhibits protein kinase C and voltage-gated sodium and calcium channels occurring in the nervous and cardiovascular systems [21].
Pardaxin is a membrane-lysing peptide isolated from the mucous glands of the fish Pardachirus marmoratus; it is secreted by the fish to repel predatory fish such as sharks. Pardaxin targets the gills of fish, causing irritation at low concentrations and death at high concentrations. Pardaxin also kills bacteria and is capable of lysing red blood cells by perturbing the lipid bilayer of the cell membrane. It is a 33-amino-acid amphiphilic α-helical peptide (GFFALIPKIISSPLFKTLLSAVGSALSSSGGQE) with a “helix-bend-helix” structure. A hinge centered on Pro13 separates the two helices. The composition of the membrane is important for the peptide selectivity. Depending on the membrane composition, amphiphilic C-terminal helices of a number of toxin monomers either have a membrane-parallel orientation and form the so-called “carpet” or insert into the bilayer and form a “barrel-stave” pore. In the former case, the destruction of the membrane results from local defects; in the latter case, the formed pores cause cell lysis. The presence of cholesterol or anionic lipids reduces the ability of this toxin to disrupt bilayers [22].
The α-helical polypeptide magainin with antimicrobial, antifungal, and antitumor activity was detected in the skin of the clawed frog Xenopus laevis. It consists of 23 amino acid residues (NH2-GIGKFLHSAKKFGKAFVGEIMNS-CONH2) and forms solely toroidal (protein-lipid) pores with a 3-5 nm inner diameter and a 7-8 nm outer diameter using only 4-7 magainin monomers [6, 23].
Eukaryotic β-Pore-Forming Proteins
β-Pore-forming proteins are produced by many eukaryotic organisms. For example, snake venom contains β-pore-forming toxins that immobilize and kill the snake prey and help to digest it. Snake food is quite diverse: snakes eat insects, lizards, frogs, birds, and rodents; sea snakes eat fish. Cobra venom causes tissue necrosis, and wound healing in surviving organisms takes a long time. Cardiotoxin CTX A3 from Taiwan cobra venom is a 62-amino-acid β-sheet polypeptide that interacts with cell membranes and forms pores. It is capable of lysing red blood cells and inducing necrosis of cardiomyocytes [24, 25]. Snake venom also contains antimicrobial toxins. Myotoxin II isolated from venom of the Brazilian snake Bothrops jararacussu interacts with bacterial membranes and induces formation of peptidoglycan pores [26]. The presence of antimicrobial toxins in snake venom can be explained by evolutionary closeness of poison and salivary glands. The antibacterial activity of injected venom suppresses growth of bacteria present in the swallowed prey [27].
Pore-forming antimicrobial peptides are a part of the mammalian immune system. Protegrin, an 18-amino-acid β-sheet peptide (NH2-RGGRLCYCRRRFCVCVGR-CONH2) isolated from porcine leukocytes, kills various bacteria and fungi. Apart from prokaryotic cells, protegrin is able to lyse membranes of human erythrocytes. However, it proves ineffective against red blood cells from sheep or goat. Its selectivity is explained by difference in lipid composition of membranes from different species. The amount of positively charged phosphatidylethanolamine ranges from ~33% for human erythrocytes to ~68% for sheep and goat. The presence of negatively charged phospholipids and lipopolysaccharides in prokaryotic membranes underlies their especial sensitivity to the toxin [28]. Protegrin has a one-bend hairpin structure with two β-sheet-stabilizing disulfide bonds formed by four cysteines. Using four or five NCCN parallel dimers, protegrin forms toroidal channels displaying low anion-selectivity with an inner diameter of 2.1 nm and an outer diameter of 4.2 nm [29-31].
The most intriguing pore-forming proteins of the mammalian immune system are perforins. Perforins from vertebrates (including humans) are soluble pore-forming proteins secreted by cytolytic lymphocytes (CTL and NK) that are able to kill both pathogenic microorganisms and host cells, such as cancer cells or cells damaged by viruses [32]. Perforins were first described in 1985 [33, 34]. As shown by recent studies, human perforin is synthesized as a ~67 kD precursor (555 amino acid residues) with a 21-amino-acid N-terminal signaling peptide. The protein acquires its activity after this signaling peptide has been removed, and additionally, after its glycosylated C-terminal peptide has been cleaved by a cysteine protease. The domain structure of perforin is complex and conserved in vertebrates. A monomeric molecule of the mature protein has an L-shaped form and shows high structural homology to cholesterol-dependent bacterial cytolysins [35, 36]. The striking similarity shown by these pore-forming toxins from microorganisms and protective proteins from higher eukaryotes still remains a puzzle. It is unclear whether it results from horizontal gene transfer or from convergence and functional proximity. The C-terminus of mature perforin contains a C2 domain implicated in Ca2+-dependent binding to phospholipid membranes and in oligomerization of perforin monomers. The central α-helical part of the perforin molecule transforms into two antiparallel β-hairpins forming a β-barrel pore [37]. The effective diameter of perforin-induced pores is 5-20 nm. As shown by electron microscopy, a pore formed by 20 subunits has an inner diameter of ~10 nm, an outer diameter of 20 nm, and a height of 16 nm. In the modern view, perforins together with serine proteases called granzymes are exocytosed from cytotoxic lymphocyte granules (modified secretory lysosomes) into the immunological synapse, i.e. the tight space between the lymphocyte- and target cell membranes. Then perforin delivers granzymes into the target cell cytoplasm for apoptosis induction. There are a number of models of the granzyme delivery mechanism. According to one of them, by oligomerizing on the target membrane, perforins form granzyme-permeable pores. Since the presence of Ca2+ in the medium is a prerequisite to pore formation, the formed pores are Ca2+-permeable. A later concept is that at first perforin and granzymes bind to the membrane and then perforin forms pores, thereby increasing Ca2+ concentration in the target cell cytosol and activating endocytosis. After that, perforin oligomers and granzymes pass inside the cell as endosome components. However, it remains unknown how perforin releases granzymes upon passing into the cytoplasm [38].
The mechanism by which lymphocyte cells protect their membrane structures from the perforin effect is of interest. Upon synthesis in the endoplasmic reticulum, perforin probably binds to its inhibitor calreticulin. It is then transported via the trans-Golgi to cytotoxic granules to give, together with granzymes, a complex with the proteoglycan serglycin. Hence, perforin is permanently bound, and the acidic medium inside the granule (pH 5.1-5.4) adds to its inactivation. It is also of importance that inside the granule Ca2+ required for perforin interaction with lipids and for its oligomerization appears to be bound too. Nevertheless, the perforin-activating proteolytic cleavage of the C-terminal peptide occurs most probably inside the granule, because low pH (5.1-5.2) is required for the reaction. When in the immunological synapse, perforin is enabled by neutral pH (7.4-7.5) to separate from serglycin and to acquire its full activity. The mechanism of protection of the lymphocyte outer membrane is explained by a hypothesis that on the membrane surface perforin is cleaved by membrane-bound proteases [39].
Lately, thorough and extensive studies are focused on defensins, eukaryotic peptides playing a key role in innate immune response. In higher multicellular organisms, these low-molecular-weight peptides (~5 kD) act as a primary antimicrobial barrier in mucous membranes of eyes, the respiratory tract, and skin. Also, these peptides have been detected in all kinds of eukaryotes, from unicellular fungi to plants, insects, and mammals [40]. All defensins are believed to be of common evolutionary origin and serve as an ancient means of cell protection. Defensins demonstrate a wide variety of anti-pathogenic properties. As versatile natural antibiotics, defensins prove efficient against gram-positive and gram-negative bacteria, fungi, and some viruses. The mechanisms of their action are diverse, and their study is still a work in progress. However, some of these positively charged peptides are known to kill bacteria by disrupting their membranes through pore formation. Although interactions between defensins and membranes are not receptor-mediated, defensins use their positive charge to bind to membrane surface anion lipids, such as phosphatidylglycerol and cardiolipin, abundant in microorganisms. In contrast, a mammalian cell membrane consists mostly of uncharged phospholipids, such as phosphatidylcholine and sphingomyelin, which determines only slight effect produced by defensins on mammalian cells [41, 42].
In vertebrates, there are three types of defensins: α-, β-, and θ-defensins. Human defensins are synthesized in intestine cells, neutrophils, and mucous membrane cells. Defensins of these three types are mostly β-structural peptides with six cysteines forming structure-stabilizing disulfide bridges. In α-defensins, disulfide bridges are formed between Cys1 and Cys6, Cys2 and Cys4, Cys3 and Cys5; whereas in β-defensins, cysteines are bridged as follows: Cys1 and Cys5, Cys2 and Cys4, Cys3 and Cys6. θ-Defensins have a circular structure and the following order of cysteine bridging: Cys1 and Cys6, Cys2 and Cys5, Cys3 and Cys4. It has been shown that human defensin α-1 forms pores of high conductance in membranes of the parasite Trypanosoma cruzi. Pore diameters range from 3 to 200 nm, with a pronounced 10-20 nm peak. Single pores formed by peptide monomers tend to fusion, which explains their difference in size. Apart from pore formation, human defensin α-1 induces trypanosome DNA fragmentation, thereby showing a dual activity [43]. The best-studied crystal structure is that of β-defensin hBD2 (Fig. 3a; see color insert) [44]. An elementary crystalline unit contains two octameric arrangements with four defensin dimers each. The geometric parameters of an hBD2 octamer are about 25 × 25 × 50 Å. In concentrated solutions, hBD2 exists mainly as a dimer, although a small number of higher aggregates can also be observed. The hBD2-induced pores are permeable for low-molecular-weight (400 daltons) compounds, but transportation of high-molecular-weight compounds (>3000 daltons) through the pores is hindered, though possible. Interestingly, hBD2 is structurally homologous to a peptide from platypus venom [45] and to two sea anemone toxins, although functions of these proteins are significantly different from those of defensins (for a defensin database, see http://defensins.bii.a-star.edu.sg/).
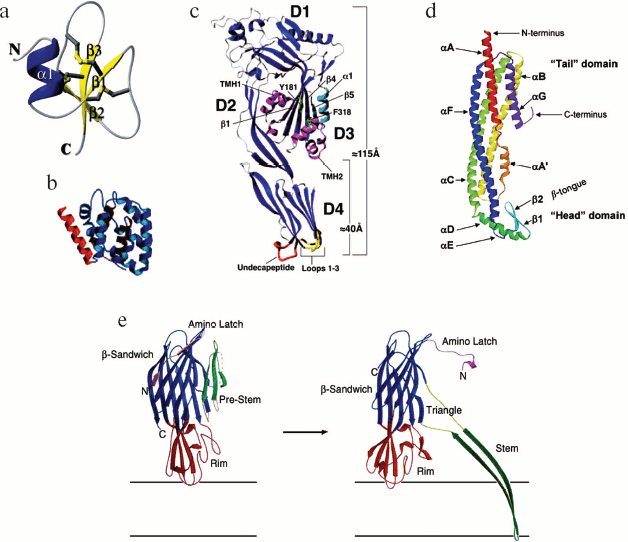
Thus, as mentioned at the beginning of this review, a variety of functions of pore-forming toxins determine an advantage of their host organisms in adaptation to environmental conditions. Some of the toxin-involving events are aimed at host defense against enemies, including infectious microorganisms, others - at prey capture and digestion, which eventually leads to an increased magnitude of population and a larger natural habitat. Besides, pore-forming toxins play an important role in maintaining immunity of eukaryotes, which consists in the ability to kill virally damaged cells or cancer cells of the host organism [32].Fig. 3. a) The 3D structure of human defensin hBD2. The molecule consists of a three-strand β-sheet with its flanking α-helix. The structure is stabilized by three disulfide bonds [44]. b) The structure of a pore-forming domain (domain C) of colicin E1. The pore-forming domain is shown in blue, the hydrophobic hairpin in brown, and α-helix linking domain C with other domains in red [47]. c) Crystal structure of Clostridium perfringens perfringolysin O. Localization of amphiphilic transmembrane β-hairpins TMH1 and TMH2 is shown in pink, three loops of domain four in yellow, undecapeptide in red, amino acid residues Y181 and F318 in green, and β5-α1 in light blue. β-Strands β1 and β4 from domain 3 of the β-sheet core and domains 1 to 4 (D1-D4) [70] are indicated. d) The structure of a water-soluble Escherichia coli HlyE monomer. The tail- and head domains and the N- and C-termini are shown [113]. e) Structures of water-soluble monomeric α-hemolysin and a protomer from the heptameric S. aureus α-hemolysin complex. A conformational change occurring in α-hemolysin during pore formation is presented schematically. The main domains are shown in different colors: the pre-stem and stem domains are green, the rim domain is dark red, and the β-sandwich domain is blue. The amino latch is shown in pink, the triangle in yellow-gray [84]. In all figures, the α-carbon skeleton was drawn using Ribbon graphics.
Prokaryotic α-Pore-Forming Proteins
To date, there are many pore-forming cytolysins produced by both gram-positive and gram-negative microorganisms identified. Typical α-pore-forming toxins are colicins that belong to a family of 60-80 kD antimicrobial proteins secreted by enterobacteria under stress caused either by nutrient deficiency or overpopulation. Colicins kill bacterial strain cells and fall into two groups according to their lethal effect-producing mechanisms. Members of one group (e.g. E1) form ion channels in cytoplasmic membranes, while members of the other group (e.g. E2 and E3) inhibit protein and peptidoglycan synthesis and cause degradation of nucleic acids. Colicins bind to receptors, which are protein components of outer membranes that in norm are responsible for transportation of various nutrients, thereby crossing the outer membrane to enter the periplasm of a foreign microorganism. For example, E1 initially binds to the vitamin receptor BtuB [46]. For translocation across the outer membrane, E1-like colicins use the Tol-system, whereas members of the other colicin group use the Ton-system of the attacked cell. After translocation into the periplasm through the TolC channel formed by three protein molecules, colicin binds to the inner membrane and forms a voltage-gate ion channel.
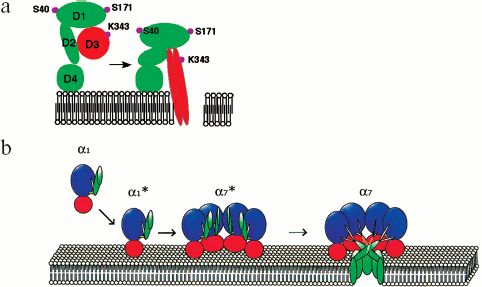
The channel is formed by one colicin E1 molecule. Colicin E1 consists of three functional domains. The middle part of a molecule is responsible for receptor binding, N-terminal domain is in charge of protein translocation towards the inner membrane, while the C-terminal domain of higher hydrophobicity forms the channel (Fig. 3b) [47]. During its translocation as a part of the molecule, the C-terminal domain is in its unfolded state. The C-terminal domain consists of 137 amino acid residues grouped as 5-6 amphiphilic α-helical segments that insert into a bilayer to form a channel [48]. Membrane model-based experiments showed that in 1 M KCl colicin E1 can form pores of two conductance levels: long-lived of ~60 pS and short-lived of ~600 pS. The probability of formation of either channel subtype is determined by thickness of the hydrophobic membrane layer. In “thin” membranes, E1 forms mostly low-conductance channels of 0.8 nm in diameter, while in “thicker” membranes the formed channels are 1.6 nm in diameter. Since channel selectivity is mostly determined by the type of membrane-composing lipids, the authors of [48] believe that colicin forms toroidal pores with not only proteins but also lipids as channel wall material. Low-conductance pore formation may require a lower number of α-helices than that of larger pores [49]. Colicin E1 kills cells by forming ion channels through the inner membrane. Colicin-producing bacterial cells are protected against “self-destruction” by concurrent synthesis of a protein that binds to the colicin C-terminal domain and abolishes its activity.
Diphtheria-causing Corynebacterium diphtheriae also synthesizes a pore-forming toxin used to attack a macroorganism and induce its tissue decay. The diphtheria toxin (58.3 kD) is synthesized as a polypeptide comprising two disulfide-bridged fragments A and B. Fragment A is its N-terminal catalytic domain (C). Fragment B consists of a receptor-binding domain (R) and a membrane-inserting domain (T) [50]. The toxin enters cells by receptor-mediated endocytosis. At acidic pH (~5.5), toxin conformational changes occurring inside the formed endosomes facilitate its insertion into the membrane and pore formation. Subsequent translocation of the catalytic domain into the cell cytosol induces cell death as a result of protein synthesis inactivation. Three of 10 domain T helices, TH5, TH8, and TH9, are involved in pore formation [51]. Depending on toxin concentration, the channel can be formed either by a monomer [52] or, at increased concentrations, by several toxin molecules [53]. Three domain T α-helices from each of four clustered toxin molecules insert into the membrane and form a channel. The channel formed by a diphtheria toxin tetramer provides translocation of domain C into the cell, and after disulfide reductase-induced separation of the two chains this catalytic domain comes out to the cytosol [54].
Cytotoxin VacA is one of major virulent factors secreted by Helicobacter pylori that can reside in the human stomach mucous coat and cause chronic gastritis, stomach ulcer, cancer, and lymphatic tissue tumor. At neutral pH, this 95 kD protein forms a water-soluble flower-shaped complex comprising two hexamers. At pH = 5, VacA interacts with negatively charged lipid bilayers. Initially, at low pH, the water-soluble complex dissociates down to monomers with subsequent VacA oligomerization to give hexameric complexes that insert into a membrane and form there a pore.
Membrane pore formation by VacA oligomers is directly associated with its toxic effect on host cells [55]. A VacA monomer contains two domains linked by a protease-sensitive loop. The N-terminal domain (37 kD) and 150 amino acids from the C-terminal domain are cytotoxic, whereas the C-terminal domain (58 kD) binds on the membrane to a receptor-like protein tyrosine phosphatase β [56]. The amino-terminal α-helical hydrophobic region, essential for pore formation, comprises six glycines and three tandem motifs GxxxG that are required to assemble a homohexameric channel. This toxin forms anion-selective channels and can provoke endosome formation [57]. Channels formed in cell membranes cause osmotic swelling, dissipation of mitochondrial potential, and apoptosis. Channels formed in plasma membranes of stomach cells allow bacteria to access potential metabolic substrates in the host cytosol, e.g. pyruvate and HCO3- [58].
The gram-negative bacterium Escherichia coli often induces extraenteric diseases such as urinary tract infections, pneumonia, and meningitis and leads to sepsis. The immediate cause of these diseases is α-hemolysin, a 117 kD toxin secreted by virulent Escherichia coli strains. It is a member of the RTX (repeats in toxin) family grouped by the mechanism of pore formation that allows for protein homology. This toxin family also includes leucotoxin from Pasteurella hemolytica, hemolysin and leucotoxin from Actinobacillus, hemolysins from Bordetella pertussis, Proteus vulgaris, Morganella morganii, and Moraxella bovis [59], etc. A common structural feature of members of this family is the presence in their C-terminal domains of a number of nanopeptide tandem repeats with a Gly- and Asp-rich consensus sequence X-L-X-G-G-X-X-G-D-D-D. The repeat-composing amino acids are involved in calcium binding. The C-terminal domain of α-hemolysin contains His859 that is crucial in calcium binding. In solution, this protein exists as a monomer tending to aggregate; it can reversibly bind to the membrane in the absence of Ca2+, but Ca2+-complexing is a prerequisite to its conformational changing and membrane-permeabilizing [60]. The effect of α-hemolysin on erythrocytes is achieved at much lower toxin concentrations as compared to liposomes, because erythrocyte membranes contain glycophorin, a specific receptor for α-hemolysin. Hemolysin binds to the receptor using a site near its C-terminal region (aa 914-936) that is conserved for all members of the RTX family [61]. Membrane binding can be performed without the receptor as well, provided the protein concentration is high enough. As shown, the receptor facilitates the binding hundreds-fold. Another common feature of RTX-toxins is the presence in their N-terminal part of nine amphiphilic 21-amino-acid α-helices that permeabilize a cell membrane and form a pore. As found, the pore can be formed by one or several toxin molecules [62]. The diameter of a cation-selective pore is condition- and membrane composition-dependent and is 1-3 nm [63, 64]. There is no signaling peptide in the structure of RTX-hemolysins, and their secretion is assisted by special proteins [65].
Unlike RTX-toxins, pore-forming toxins characteristic of microorganisms of the Serratia family [66] have a typical signaling peptide and do not require either calcium ions or other cofactors to show their activity. Toxins of this family have been detected in pathogenic gram-negative bacteria causing serious human diseases. For example, Serratia marcescens and Proteus mirabilis are responsible for urinary tract infections, and Haemophilus ducreyi - for genital ulcers; Yersinia pestis is known as a bubonic plague and pneumonia pathogen; Yersinia enterocolitica affects the alimentary tract. Moreover, these microorganisms can affect insects (Photorhabdus luminescens) and plants (Xylella fastidiosa). Hemolysin ShlA, a Serratia marcescens-derived 162 kD pore-forming toxin, is believed to be the best-studied representative of this group. Translocation of a newly synthesized toxin from the periplasm to extracellular medium is mediated by a 60 kD protein ShlB pertaining to the outer membrane. Upon ShlB-ShlA interaction, the toxin undergoes a conformational change to adopt an active form [67]. The highest hemolytic activity is displayed by monomers and some dimers. Higher aggregates, e.g. tetramers, are completely devoid of lytic activity. Most probably, toxin monomers initially bind to the membrane and then oligomerize within the bilayer and using their α-helices form a pore with a functional diameter of 2.5-3.0 nm [68]. In model planar bilayers the toxin forms in 1 M KCl nonselective, voltage-independent pores with an average conductance of 1200-1400 pS. To interact with a membrane and to form a pore, the toxin does not require a protein receptor; however, the presence of phosphatidylserine within unilamellar vesicles enhances its activity. The absence of phosphatidylserine from membranes of prokaryotic cells is thought to be the reason why ShlA has no lysing effect on these cells [69].
Prokaryotic β-Pore-Forming Proteins
β-Structural channel-forming cytolysins insert into the membrane using their β-sheet domains. Amphipathic β-hairpins of clustered cytolysins form a membrane-binding β-barrel with hydrophilic inner and hydrophobic outer surfaces. These cytolysins are classified as cholesterol-dependent toxins and include listeriolysin O, the major virulent factor of Listeria monocytogenes that causes listerioses, such as meningitis, encephalitis, and intrauterine infections; streptolysin O from Streptococcus pyogenes causing streptococcal skin infection; pneumolysin O from Streptococcus pneumoniae, leading to meningitis, otitis media, sinusitis, and pneumonia; perfringolysin O from Clostridium perfringens that causes tissue necrosis, gas gangrene [70]; anthrolysin O from Bacillus anthracis, the anthrax pathogen [71], etc. The latter can kill leucocytes, lymphocytes, phagocytes, neutrophils, monocytes, and macrophages, thereby weakening the host immunity [72].
The typical features of cholesterol-dependent cytolysins are: (i) their binding to a membrane cannot occur without cholesterol present in the membrane; (ii) they form unusually large pores with the inner diameter ranging from 30 to 50 nm. These are the largest pores permeable not only for separate ions but also for proteins, the loss of which leads to a rapid colloid-osmotic lysis of the cell. Toxins are secreted into the extracellular medium as soluble monomers. Upon getting in contact with a eukaryotic cell, monomers bind to the cholesterol contained in cell membranes, then laterally diffuse and oligomerize there to give a large ring-shaped membrane-bound pre-pore complex where the β-barrel is not inserted into the bilayer yet; eventually, it induces formation of a membrane-inserted pore complex (Figs. 2c and 4a; see color insert) [73].
Toxin molecules of this type contain one or several cysteines whose SH-groups are crucial in toxin binding to membranes. Their key role is demonstrated by the fact that specific SH-blocking agents inhibit cytolytic activity of the toxin, whereas after treatment with thiol or with other reducing agents, the toxin re-gains its initial activity. Toxins of this type are inactivated by extra membrane cholesterol whose inhibitory effect is underlain by its occupying of the receptor binding site in the toxin molecule, thereby preventing interaction between the toxin and membrane cholesterol. The decreased concentration of cholesterol blocks transition from the pre-pore- to pore state, i.e. prevents insertion of the β-barrel into the membrane [70].Fig. 4. a) Schematic illustration of a conformational change occurring in monomeric Clostridium perfringens perfringolysin O during its transition from the pre-pore to conducting pore state. Main domains D1, D2, D3, D4 and localization of some amino acid residues [73] are indicated. b) Stages (α1-α7) of pore formation by S. aureus α-hemolysin. The water-soluble monomer (α1) binds to the membrane through its rim domain (α1*), which is followed by formation of a pre-pore (α7*) that finally turns into a heptameric pore (α7). The main domains are shown in different colors: the pre-stem and stem domains are green, the rim domain is dark red, the β-sandwich domain is blue, the triangle is gray. For convenience, at the heptameric pre-pore (α7*) and heptameric pore (α7) stages only four protamers are shown [88].
Listeriolysin O, a 58 kD protein, is the only member of this family showing a pH-dependent pore-forming activity. At neutral pH its cytolytic activity is low, while at pH 5.5 it is rather high. Unlike other pH-dependent toxins, listeriolysin O shows pore-forming activity controlled by rapid and irreversible structural denaturing at neutral pH and temperature above 30°C. A rapid denaturing at neutral pH starts with unfolding of domain 3 in the transmembrane β-hairpin that normally forms the β-barrel. A triad of amino acid residues within domain 3 acts as a pH-sensor and initiates listeriolysin denaturing by destabilizing the structure of domain 3.
Listeriolysin O is a pore-forming toxin, and similar to other members of the family of cholesterol-dependent cytolysins, its monomers oligomerize to give a large pore-forming complex. Monomers bind to a membrane and oligomerize as a pre-pore complex that inserts into the membrane and forms the pore β-barrel [74]. Bacteria need listeriolysin O to permeabilize the cytoplasm of eukaryotic cells. Within a phagosome, a bacterial cell enters a eukaryotic cell where it synthesizes toxin active at low pH. The toxin forms pores allow the bacterium invasion into the cell cytoplasm. In the cytoplasm, toxin activity weakens, due to which the cell survives, even though its membrane is damaged, and provides the bacterium nutrition and growth. At suboptimal pH inside the host cell, the toxin retains the ability to form Ca2+-permeable pores in cholesterol-rich membranes. The calcium-dependent cell response is induction of apoptosis. The multiplied bacteria leave the cell, and the process repeats again with other cells involved [75].
Another cholesterol-dependent cytolysin Streptococcus pneumoniae pneumolysin transforms from a soluble 52 kD monomer to a 30-50-subunit pore. Like all other members of this family, pneumolysin has a structure shown in Fig. 3c. Its monomer consists of four β-structural domains with a hydrophilic surface. Domain 4 containing the highly conserved sequence ECTGLAWEWWR is the first to contact the membrane surface. It is this part of the molecule that interacts with cholesterol. Additionally, domain 4 contains loops L1-L3 required for membrane cholesterol binding [76]. This loop-to-membrane binding initiates conformational changes in the toxin molecule and triggers toxin oligomerization [77] that leads to pre-pore formation and then, supported by further conformational changes, to formation of a pore. Structurally, at the pre-pore stage, domain 4, the only one in contact with the membrane, is perpendicular to the membrane surface and linked with domain 2. Domains 2 and 3 interact with each other and are linked with domain 1, a stable structure with which all other mobile domains are linked. In the course of pore formation, domains 2 and 3 move apart, and domain 3 turns about and inserts into the membrane. This domain has two rows of three α-helices each; upon inserting into the bilayer, these helices transform to β-chains to give two β-hairpins. Four chains from each monomer assemble into a large transmembrane β-barrel. The pre-pore-to-pore transition is accompanied by significant conformational changes. Each monomer has two amphiphilic transmembrane β-hairpins contributing to pore formation. In the case of pneumolysin, a pore contains 44 subunits, and its transmembrane cavity is 26 nm in diameter and comprises 176 β-chains that concertedly insert into the membrane and form the channel wall [78, 79].
The group of β-structural channel-forming cytolysins also includes aerolysin-type toxins, such as Staphylococcus aureus α-hemolysin, Aeromonas hydrophila aerolysin, Clostridium septicum α-toxin, Vibrio cholerae Vcc, and Bacillus cereus hemolysin II. These bacteria can cause dermonecrosis, endocarditis, pneumonia (S. aureus), myonecrosis, eczema, gastroenteritis (A. hydrophila), gas gangrene (C. septicum), cholera (V. cholerae), diarrhea and emetic syndromes, eye diseases, and mastitis (B. cereus).
The bacterial exotoxin α-hemolysin secreted by S. aureus is a water-soluble monomer. It is a 33.2-kD 293-amino-acid polypeptide that forms homoheptameric pores in membranes. Its ability to form ion channels in bilayers was first described more than 25 years ago by Krasilnikov and colleagues [80, 81]. In model planar lipid membranes, α-hemolysin induces channels that appear to be slightly anion and voltage-insensitive ones at neutral pH and have an average conductance of 110 pS in 0.1 M KCl [82]. The heptameric complex is mushroom-shaped: it has a cap projecting beyond the membrane surface and a stem piercing the hydrophobic area. The height of a formed pore is 10 nm, the total diameter is 10 nm, and inner diameter of the ion-conducting channel is 1.0-1.4 nm [83, 84]. The stem consists of 14 β-structural chains. The N- and C-termini of the polypeptides form the mushroom cap that is also mainly β-structural.
The modern concept of pore assembly implies three stages. At the first stage, water-soluble monomeric α-hemolysin binds to a membrane due to initial electrostatic interaction. The monomer is sensitive to proteolysis and has two major cleavage sites: one resides in the Gly-rich central domain that later becomes a part of the stem, and the other is close to the N-terminus. The N-terminus prevents untimely oligomerization of the monomers in solution and is essential to pore-forming. Its deletion results in a lower activity of the protein and in a slower forming of the pre-pore and pore [85]. In the membrane-bound monomer its central domain, but not N-terminal, becomes resistant against proteolysis.
At the second stage, seven monomers oligomerize on the membrane to give a nonlytic intermediate pre-pore. Initially, the pre-pore is sensitive to sodium dodecyl sulfate (SDS), but in the presence of non-denaturing detergents (e.g. deoxycholate) these seven subunits remain tightly bound to the membrane. Then, cooperative interactions within the oligomer result in formation of a SDS-insensitive pre-pore. At this stage, central domains of the monomers are still in the process of translocation into the hydrophobic area of the bilayer. The central domain plays the key role in toxin insertion into the membrane. Mutants with a deletion in the central domain can rapidly heptamerize but cannot form pores [86]. The N-terminal region remains proteolysis-sensitive. As shown by atomic force microscopy, the pre-pore is oriented perpendicular to the membrane surface [87].
At the final (third) stage, the heptameric pre-pore turns into a completely assembled pore where the central β-structural Gly-rich domain of each subunit is inserted into the membrane to form an antiparallel β-barrel comprised of 14 chains. Formation of the β-barrel is accompanied by a conformational change enabling each N-terminal region to be fixed in the adjacent subunit, thereby acquiring resistance to protease. The assembled structure is an SDS-resistant heptameric pore (Fig. 4b).
The formed pore has the following structural units: cap and rim domains forming the “mushroom cap” and a stem domain. The cap domain of the heptamer consists of seven β-sheets and amino latches of each protomer. The rim domains participate in protomer-protomer interactions and are very close to the membrane bilayer. The stem domains form a transmembrane channel. In a water-soluble monomer, the amino latch and the pre-stem domain are localized near the β-sandwich domain that later becomes involved in forming of the cap domain. As soon as the pore has been formed, the amino latch makes contact with the adjacent protomer, while the pre-stem domain proceeds to forming a transmembrane β-barrel, now under the name of the stem domain. Between the stem- and β-sandwich domains, there is a triangle region (Fig. 3e) [88]. The area between the tops of the stem and rim domains is implicated in interaction with membrane phospholipid groups.
As shown by high-resolution crystallography, heptameric α-hemolysin assembles on glycerophosphocholine membranes. Phosphatidylcholine binds to each protein subunit in the area between the rim and stem domains. Ammonium groups of the phosphatidylcholine head interact with the tryptophan-179 indole ring, while its phosphate group forms water-mediated H-bonds with arginine-200. Together, the heptameric complex creates a local defect in the bilayer structure with about 14 lipid molecules substituted [89].
The stem domain of α-hemolysin contains seven His144 residues, one from each protomer, protonation of which affects channel conductance. It is believed that the histidine ring may act as a pH-sensor in the αHL-pore structure, and that acidifying the medium reduces lifetime of the open channel [90].
There exist cells, e.g. human granulocytes, resistant against α-hemolysin. It was shown that on granulocyte membranes the toxin forms a SDS-resistant pre-pore that fails to transform to a pore. The mechanism of this resistance is still unclear. The toxin can bind to membranes either at low concentrations through interaction with specific protein receptors or by nonspecific binding that requires its much higher concentrations.
Staphylococcal α-hemolysin is assumed to be the major pathogenic factor in progressing diseases caused by S. aureus [91]. Pore formation in the host cell membranes leads, as a rule, to their lysis by the colloid-osmotic mechanism, and bacteria gain access to nutrients. There is evidence that pore formation by α-hemolysin may initiate apoptosis in host cells [91]. Moreover, S. aureus toxins kill phagocytes and leukocytes, thereby weakening host immunity and providing favorable conditions for the life cycle of the microbe [92]. As shown, α-hemolysin is also required to form biofilms at media interfaces [93], which significantly increases resistance of the microorganisms against antimicrobial agents.
Hemolysin II (HlyII) is one of β-structural channel-forming cytolysins from B. cereus. Its gene, abundant in members of the B. cereus group, is also identified in insect-pathogenic B. thuringiensis [94], therefore used for production of insecticidal agents; besides, it is found in anthrax-causing B. anthracis. Hemolysin II from B. cereus is a pore-forming cytolytic toxin synthesized in bacteria as a precursor with a 31-amino-acid signaling peptide that in the course of processing undergoes splitting off by a signal peptidase to form a 42 kD mature protein [95]. Hemolysin II is secreted by bacteria as a monomer. The binding of HlyII monomer to a membrane is the fastest and temperature-independent stage of pore formation [96]; since other stages occur on the membrane, they are temperature-dependent, and their rates are lower. Upon interaction with the membrane, HlyII oligomerizes and forms anion-selective voltage-gate transmembrane hexa-, hepta-, and octameric pores with a functional diameter of 1.2-1.6 nm and an outer diameter of 8-10 nm (Fig. 2, d and e) [96]. In 0.1 M KCl, average conductance of the pores is 18 ± 6, 31 ± 3, and 46 ± 9 pS, respectively. Since physicochemical properties of B. cereus hemolysin II and S. aureus α-hemolysin are similar, and since these toxins show a 32% sequence homology, it is assumed that the basic stages of pore formation by these two toxins are similar as well (Fig. 5).
As we have shown, the cytolytic and pathogenic effects of HlyII on eukaryotic cells from various tissues and on microorganisms are underlain by formation of ion-conducting channels in cell membranes [95, 97]. A cell is known to be a double Donnan system the osmotic equilibrium of which is determined by both the intracellular protein concentration and ion concentrations inside and outside the cell [98]. When electrolytes and low-molecular-weight compounds pass easily through toxin-formed pores, homeostasis appears to be disturbed, which leads to cell lysis by the colloid-osmotic mechanism. According to this concept, the concentration gradient promotes ion delivery inside the cell using the pores, and the high amount of cellular proteins, remaining unchanged, ceases to be compensated by the outside ion concentration, which provokes an increase in osmotic pressure inside the cell. To neutralize the increase, water starts coming into the cell and proceeds until the membrane disrupts. Having initially permeabilized one organ, bacteria can increase their habitat by capturing other organs through lysis and necrosis of a myriad of cells [97]. Bacteria residing in an animal organism produce hemolysins and use them to release nutrients and other necessary metabolic products. For example, it was shown that B. cereus uses hemolysis-released hemoglobin as a source of ferrum ions [99].Fig. 5. Sequence of events involved in formation of a transmembrane pore by Bacillus cereus hemolysin II.
Aeromonas hydrophila-secreted aerolysin is a 52 kD channel-forming toxin that is synthesized as a pre-protoxin with an N-terminal signaling sequence. Having directed toxin translocation through the inner membrane, this sequence dissociates from the molecule. By type II secretion machinery, pro-aerolysin is translocated from the periplasm to extracellular medium. In solution, this protein may exist as a dimer, but it is exclusively its monomers that bind to the membrane [100]. After binding to glycosylphosphatidylinositol (GPI)-anchored proteins on the host cell surface, pro-aerolysin monomers acquire activity via proteolytic cleavage of the 40-amino-acid C-terminal peptide.
Mature aerolysin is L-shaped, with N-terminus forming its shorter part (1 domain), and usually consists of three domains. Domains 1 and 2 participate in binding to GPI-anchored receptors. Specifically, domain 2 binds to the glycan core of GPI-anchored protein, while domain 1 interacts with a saccharide in the protein part of the receptor molecule [101]. This double binding provides high affinity in aerolysin-receptor interaction. Besides, domain 2 is involved in oligomerization initiation, whereas domains 3 and 4 directly participate in heptamer assembly. The membrane-inserting part of the toxin molecule is a 20-amino-acid loop pertaining to domain 3 that forms an amphiphilic β-hairpin. Proteolytic cleavage is followed by a conformational change required for oligomerization and pore formation, and finally the toxin forms heptameric pores [102].
The pore-forming cytolysin α-toxin secreted by Clostridium septicum shows lytic and necrotic activity. Its primary structure is similar to that of Aeromonas hydrophila aerolysin. It is secreted as inactive protoxin (46.5 kD) that is cleaved at the RGKR motif by host cell proteases to give active monomers (41.3 kD) and 45-amino-acid C-terminal peptides. Like aerolysin, monomeric α-toxin binds to GPI-anchored proteins, oligomerizes into hexameric complexes, and inserts into the cell membrane to form a pore with a functional diameter of 1.3-1.6 nm [103].
The cytolysin Vibrio cholerae VCC is secreted as an 80 kD inactive protoxin as well. Its 15 kD N-terminal prodomain shows homology to the heat shock protein family Hsp90. Functionally, this prodomain is a VCC chaperon required for expression of active toxin. Within the toxin molecule, the N-terminal prodomain interacts with a cytolytic domain and masks the region involved in monomer-monomer interaction during oligomerization. Therefore, toxin activation requires proteolytic cleavage of this prodomain. The cytolytic domain is linked with the pre-stem domain that forms a β-barrel upon insertion into the membrane. The C-terminus of monomeric VCC contains two lectin domains providing interaction between the toxin and receptors. The monomers reversibly bind to the cell surface through carbohydrate receptors located on membrane glycoproteins and glycolipids to form anion-selective heptameric pores by a mechanism similar to that used by staphylococcal α-hemolysin [104].
Like α-hemolysin, β-structural channel-forming cytolysins leucocidin and γ-hemolysin are secreted by Staphylococcus aureus. Dissimilar to α-hemolysin-produced pores, those formed by leucocidin and γ-hemolysin result from pore-forming activity of subunits of two classes, F and S, that form functional heterooligomeric pores. There are six classes of F proteins (LukF-PV, LukF-R, LukD, LukF′-PV, HlgB, and LukF-I) and seven classes of S proteins (LukS-PV, LukS-R, LukE, LukM, HlgA, HlgC, and LukS-I) produced by various strains of S. aureus [105]. Leucocidin pores are formed by HlgB or LukF and HlgC or LukS. Similarly, γ-hemolysin pores are made of HlgB or LukF and HlgA or γHLII. The equimolar LukF-to-LukS ratio is a characteristic feature of leucocidin pores. Four LukF and four LukS subunits form an octameric pore. LukF and LukS, associated with each other, are arranged as a tandem around the central pore axis [106]. The hydrodynamic diameter of a leucocidin pore is 2.1 nm; the inner and outer diameters of the complex, as shown by electron microscopy, are 3 and 9 nm, respectively. Leucocidin displays its specific leucocytolytic activity after LukS has been phosphorylated by protein kinase A to induce membrane binding. Then water-soluble monomers form an inactive oligomeric pre-pore that inserts into the lipid bilayer and creates a conducting pore.
Similarly, staphylococcal γ-hemolysin is a two-component heterooligomeric pore-forming cytolytic toxin. Pores formed by γ-hemolysin are heptameric and consist of components LukF and γHLII in a molar ratio of 3 : 4 or 4 : 3 [107]. A LukF monomer shows structural homology to a monomer of α-hemolysin. It consists of a membrane-binding rim domain, a β-sandwich domain required for γHLII-implicating oligomerization, and a pre-stem domain. The LukF pre-stem domain is initially adjacent to the β-sandwich domain, but in the course of pore formation, it turns about and inserts into the membrane. The LukF N-terminal amino latch resides on the β-sandwich domain of its own protomer, unlike the amino latch of α-hemolysin protomer that interacts with the adjacent protomer.
As found, formation of a pore begins with monomers-to-membrane binding and proceeds with assembly of dimers and small oligomers and creation of a single pore. A prerequisite to pore formation is concerted binding of LukF and γHLII that assemble only into heterodimers and only on the membrane, but not in solution. Also, it was found that the dimer-dimer interaction between complementary sides of LukF and γHLII gives tetramers. Finally, this toxin forms pores with a functional diameter of 2.5 nm. As shown by electron microscopy, the inner and outer diameters of the pore complex are 3 and 7 nm, respectively. At high protein concentrations, three, four, or more single pores assemble into clusters via interaction of amino acids located on their outside surfaces [108]. Like γ-hemolysin-produced pores, homooligomeric pores formed by other toxins of pathogenic bacteria (aerolysin, streptolysin, perfringolysin O, staphylococcal α-hemolysin) can assemble into clusters as well [109, 110].
Escherichia coli secrets a 34 kD pore-forming toxin hemolysin E (HlyE) that is a rod-shaped protein consisting of four long α-helices (Fig. 3d). The N-terminal sequence-comprising tail part of the molecule (tail domain) has an additional shorter helix responsible for HlyE toxic activity. At the opposite end of the chain (head domain) there is a subdomain that includes a short antiparallel double-strand β-sheet flanked with two short helices (β-flap) and the main bundle sandwiched between helices 3 and 4. Hydrophobicity of the β-flap provides interaction between HlyE and membranes. Hemolysin E from E. coli strain JM4660 exists in solution as a dimer, while HlyE from E. coli K-12 was observed to have monomeric, dimeric, and 8-12-meric forms. The dimer is formed by “head-to-tail” arranged subunits. In a membrane, the protein HlyE forms ring-shaped octameric pores with an inner diameter of 4.2-5.2 nm and an outer diameter of 7-10 nm [111-113].
Besides, the group of β-structural channel-forming cytolysins includes two-component AB toxins also called “binary” toxins. Their principle of operation is as follows: component B acts as a pore-forming protein and assists component A in entering the target cell to cause its death. Among proteins of this type, one can mention toxin C2 from Clostridium botulinum (pathogen of botulism, a serious food intoxication accompanied by nervous system affection), VIP toxin from Bacillus cereus, and toxins from Bacillus anthracis causing edema and the lethal outcome. The toxins start their work with specific binding of monomeric component B (C2II C. botulinum, VIP1 B. cereus, PA B. anthracis) to cell receptors followed by its homo-heptamerization on the cell surface. For example, PA-binding receptors are ubiquitin proteins (TEM8 and CMG2), and C2II receptors are glycoproteins. Component B is activated by serine proteases, such as chymotrypsin, trypsin, or furin. Proteolysis cleaves the N-terminal peptide from the toxin molecule, thereby triggering conformational changes followed by homo-heptamerization of component B.
In the active component B, the site responsible for its binding to component A pertains to the N-terminal domain 1, and its receptor binding site is localized in the C-terminal domain 4. Domain 2 is involved in forming a channel in lipid membranes, and domain 3 is responsible for oligomerization. Domain 2 is capable of unfolding to give a β-hairpin that inserts into the membrane, thereby providing transition of the heptameric pre-pore complex to the open pore state. Then the heptameric complex binds to enzymatic component A (C2I C. botulinum, VIP2 B. cereus, EF and LF B. anthracis) that translocates into the cell cytosol.
Components A inhibit functions of normal cells by a variety of mechanisms. C2I and VIP2 perform ADP-ribosylation of G-actin, which results in cytoskeleton destruction and cell death. LF causes MAPKK proteolysis that disturbs the cell signaling function, and EF increases cAMP, which eventually results, at the organism level, in edema or immunodeficiency [114]. Components PA83 of B. anthracis bind to receptors on the cell surface [115]. Then furin-like proteases cleave a 20 kD fragment from the N-terminus, thereby exposing the LF- and EF-binding sites. The next step is PA63 oligomerization into a heptameric pre-pore complex and its binding to three molecules of EF and/or LF [116]. The formed complex permeabilizes the cell by endocytosis [117]. Acidic pH inside the endosome induces insertion of the heptamer into the membrane and channel formation, after which LF and EF dissociate into the cytosol. Toxins from B. anthracis can kill endothelial cells [118], and additionally, LF can cause macrophage necrosis, which reduces immunity and provides conditions for reproduction of bacteria [119].
Bacteriocins are antimicrobial peptides produced by many strains of species of the Eubacteria and Archaea genera that can serve as antibiotic agents against closely related cells. These peptides are secreted into the extracellular medium to be recognized by surface receptors of bacteriocin-sensitive cells. It is postulated that the essential role of bacteriocins consists in regulation of cognate species population dynamics. Bacteriocin toxicity is realized by a variety of mechanisms, mostly by transmembrane pore formation. These peptides are currently in the focus of intensive studies as potential preservative agents useful in food processing industry and for clinical needs. According to the modern classification, bacteriocins are grouped as follows: class I, lantibiotics; class II members fall into pediocin-like bacteriocins (class IIa), two-peptide bacteriocins (class IIb), and one-peptide bacteriocins other than pediocin-like ones (class IIc); class III, thermosensitive bacteriocins [120, 121].
Structurally, bacteriocins are quite diverse: among them, there are α-helical and β-structural peptides, as well as lantibiotics having cyclic structures.
The well-characterized lantibiotic nisin is secreted by Lactococcus lactis. Similar to other lantibiotics, the ribosomally synthesized 34-amino-acid nisin undergoes posttranslational modification to add to its structure amino acid lanthionine consisting of two Ala residues stabilized by thioether-bonded β-carbons [122] (Fig. 6). The 3D structure of nisin comprises two amphiphilic spiral domains consisting of N-terminal A, B, and C rings, and C-terminal D and E rings. The latter are linked by a mobile three-amino-acid sequence.
The antibacterial activity shown by nisin is underlain by its ability to form pores with a diameter ranging from 0.2 to 1-2 nm and a lifetime of a few hundred microseconds in membranes composed mostly of anion lipids. Nisin-induced pore formation requires a negative transmembrane potential. The positive charge of nisin itself is significant. Membranes of bacterial cells contain lipid II (undecaprenyl-pyrophosphoryl-MurNAc-(pentapeptide)-GlcNAc), a receptor displaying high affinity for nisin. In its presence, the pore-forming activity of nisin increases by three orders of magnitude. It is postulated that nisin is a constituent of the formed pore that comprises 5-8 molecules of nisin and as many molecules of lipid II [123].Fig. 6. Structure of Lactococcus lactis nisin [122].
Since in bacterial cells the amount of lipid II ranges widely (from 103 molecules per cell in E. coli to 105 molecules per cell in Micrococcus lysodeikticus), nisin must show a high antibiotic selectivity even for microorganisms [122]. For comprehensive information on these compounds, see the APD database (Antimicrobial Peptide Database http://aps.unmc.edu/AP/main.php).
It is obvious that secretion of pore-forming toxins causes cell death; in doing so, bacteria weaken the host cell immunity and gain access to cell-contained nutrients, which allows their growth and spreading over the host's organism. By pore formation, toxins are able to trigger various mechanisms of cell death, for example necrosis or apoptosis. Besides, bacteria use antimicrobial properties of pore-forming toxins to kill bacterial strains sensitive to these toxins secreted in response to stress, e.g. deficient nutrients or overpopulation. Bacterial pore-forming toxins are also involved in other events. For example, pneumolysin [77] is implicated in a complex process of cannibalism effected by vegetative forms of gram-positive bacteria at the initial stage of spore formation. Cannibalism provides lysis of cells that failed to transit to the spore state [124].
Viral Pore-Forming Proteins
Most probably, the ability to synthesize pore-forming agents appeared at the initial stages of living organism evolution. For example, reoviruses synthesize peptides that form pores in target cell membranes during entry [125]. Viruses of many types, such as hepatitis C virus (HCV), immunodeficiency virus (HIV), and flu virus, produce α-pore-forming proteins viroporins that are close in their basic properties to pro- and eukaryotic pore formers. Viroporin functions are diverse and include releasing of virus particles from the cell.
Viroporins of vertebrate RNA viruses have been extensively characterized, although their role in the life cycle of DNA viruses and bacteriophages is still unclear. Mostly, researchers' efforts are focused on the structure and functions of the following viroporins: p7 from hepatitis virus, Vpu from immunodeficiency virus, and M2 from flu virus.
As a rule, viroporin proteins are small in size (60-120 amino acids). Their transmembrane domains interact with the membrane lipid matrix, oligomerize, and form hydrophilic pores in membranes of the virus-infected cells. The protein p7 from hepatitis C virus consists of 63 amino acids arranged as two 23-amino-acid transmembrane helices separated by a short main loop [126]. As shown by transmission electron microscopy, mass spectroscopy, and experiments on planar lipid membranes, p7 oligomerizes and forms a heptameric pore [127]. Viroporin p7 acts in model membranes as a cation-selective ion channel with an average conductance of 21 pS in 1 M KCl. Viroporins from other viruses are also able to oligomerize in membranes and to form there pores consisting of a variable number of subunits.
Mutations of p7 in the viral genome lead to decreased production of virus particles but have no effect on infectiousness, thereby suggesting that p7 is crucial at the latest stages of virus assembly and uninvolved in its cell entry [128]. In contrast, viroporin M2 from flu virus, as a part of a virion, plays an important role at the initial stages of the viral attack. Expression of some viroporins results in apoptosis.
Thus, it can be concluded that viroporins are strongly involved in interactions between viruses and cells at various stages. The function of several viroporins is in vitro inhibited by the nonspecific antiviral drug amantadine, which suggests a direction in development of novel antiviral medications of extensive action. Most probably, the list of viroporins will soon become longer, and their studies will appear as a hot spot in molecular virology.
PORE-FORMING PROTEINS AS A FACTOR OF EVOLUTION
Appearance of biological membranes is a milestone in the advent of life. It is the presence of a barrier separating self-reproducing living structures from their environment that ushered in the process of life as we understand it now [129]. Many membrane proteins emerged at this stage of membrane formation and then spread widely over all living kingdoms. Ion channels formed with assistance of these proteins provide selective permeability of biological membranes. Apart from membrane-embedded channels, evolution gave rise to a large class of pore-forming proteins that are secreted either inside or outside the cell. They have been found in all organisms, from bacteria to humans, as well as in many viruses. Surprisingly, bacterial cytolytic toxins have principal homology to perforins, innate immunity proteins of higher prokaryotes. The 3D structure of formed pores, as well as the cytolytic function, persisted in evolution, despite low-grade sequence similarity. However, it remains unclear whether these proteins have a common ancestor, and whether their appearance in this or that organism was a result of horizontal transfer of their encoding genes or a product of vertical evolution. Also, it cannot be ruled out that these proteins may show convergent similarity due to their similar functions. This puzzle will probably be solved soon, thanks to compiling information on sequences of the proteins in question from various organisms.
Evolutional features of pore-forming proteins are closely connected with the problem of survival of certain species. With one or two cytolytic proteins available, an organism has an obvious advantage in adapting to varying environmental conditions. As a rule, cytotoxins are synthesized in response to a specific cell-recognized signal, because unleashed synthesis of these components would be both resource-consuming and dangerous for the producer itself. This suggests that synthesis and degradation of pore-forming proteins must be tightly controlled.
Adaptation of a pathogenic organism to its new host is often accompanied not only by acquisition but also by the loss of functions of many genes called antivirulence genes [130]. Genes that are no longer compatible with the novel lifestyle of the pathogen are inactivated by point mutation, insertion, or deletion. Sometimes extreme adaptation conditions lead to the deletion of large regions of the genome generating the so-called “black holes”. Interestingly, the role of antivirulence genes is sometimes played by genes of pore-forming cytotoxins required to display pathogenic activity in other hosts, e.g. insects. For example, hemolysin II gene in the B. anthracis genome is inactivated by point mutation that leads to the reading frame shift, while inhibition of cytolysin K synthesis is performed by inactivation of transcriptional activator PlcR. B. anthracis is virtually devoid of hemolytic activity, which may suggest inactivation of other cytotoxins as well.
In many microorganisms, toxin synthesis is usually controlled by a number of regulators at different levels, from transcription to rapid protein decay. The evolution of bacterial pathogens from nonpathogenic ancestors is accompanied by acquisition of various pathogenicity factors encoded by phages, pathogenicity islands, or plasmids via horizontal gene transfer or through adaptation of the available virulence factor to conditions of its new host organism. From this point of view, host jumping or joining the life cycle of a new host is equivalent to adaptation to a new ecological niche [130].
Many pathogenic and opportunistic microorganisms have more than one cytotoxin in their genomes. Some of them are identified by sequence homology to known pore-forming proteins; expression and cytotoxic functions of many others have been reliably established. B. cereus and S. aureus secret several β-pore-forming toxins that are regulated selectively, although their amino acid sequences are homologous to α-hemolysin from S. aureus. Multiple toxin secretion is characteristic of other bacteria as well. Presumably, these bacteria used duplication of cytotoxic protein genes to adapt to varying environmental conditions. The mode of regulation of expression of each toxin was also subject to evolutional changes. Interestingly, different strains of B. cereus possessing the same cytotoxic protein genes can use different expression regulation mechanisms depending on the isolates. These data might indicate a rapid selection of bacteria that are best adapted to prevailing environmental conditions [131].
Cytotoxin genes have been found in bacteriophage genomes from the Bacillus and Staphylococcus genera. S. aureus bacteriophages phiSLT and tp310-1 are able to transform opportunistic strains to highly pathogenic ones causing system staphylococcal infections that lead to the lethal outcome. The lethal anthrax toxin resides on the plasmid pXO1. Sequences of B. cereus and B. anthracis genomic DNAs are identical, although some differences have been observed (sometimes these are point mutations causing gene knockouts) that show a possibility of anthrax pathogen adaptation to the parasite lifestyle. Interestingly, the machinery required for the biosynthesis of another B. cereus virulence factor cereulide, an emetic valinomycin-like depsipeptide, is also localized on a mega plasmid showing high homology to the anthrax plasmid pXO1 [132]. Genes of insecticidal toxins from B. thuringiensis are plasmid-borne as well. This might mean that there existed a parental plasmid that accumulated pathogenicity factors with time [132].
Mobile elements, e.g. plasmids, are the main means of genetic material transportation used for horizontal gene transfer. A classic example is the colicin-bearing plasmid ColE1 that, due to its borne genes, is capable of being mobilized by various conjugate plasmids [133]. Besides, the colicin gene region on multicopy plasmids coincides with the region of genes pertaining to the DNA restriction-modification systems that have been reported as possibly involved in interplasmid recombination providing vertical gene transfer [134, 135]. ColE1 and its derivatives have been found in virtually all enterobacteria. It can participate in the horizontal transfer of pore-forming toxin genes as well. Thus, plasmids are the main tool for horizontal and vertical transfer of pore-forming protein genes in microorganisms.
PORE-FORMING PROTEINS AND THEIR POSSIBLE APPLICATION IN BIO- AND
NANOTECHNOLOGY
An illustrative example of application of pore-forming proteins in practice is transgenic plants that express a B. thuringiensis Cry toxin gene that confers them resistance against a variety of insects. It should be noted that on-going studies of the pore-forming mechanism have resulted in a much enhanced resistance of previously created plant cultivars. B. thuringiensis spores having an insecticidal effect were also widely used in agriculture. These modes of plant protection are perfectly harmless for warm-blooded animals [136].
For medical purposes, it is important to know the pore-forming mechanisms utilized by various bacterial cytotoxins and by pore-forming peptides responsible for the immune response of a macroorganism. Till recently, treatment and preventive measures against bacterial infections consisted in prevention or suppression of bacterial growth in the organism by antibiotics; in most serious cases, corticosteroid hormones were also used to modulate the immune response. However, in case of intensive infection, when classical approaches prove ineffective due to multiple antibiotic resistance of the microorganisms, it is important to use a therapy aimed either at detoxication or at prevention of the action of already synthesized toxins (many of these are pore formers).
Long-term studies of the pore formation process have been recently crowned by synthesis of compounds that inhibit functions of staphylococcal and anthrax toxins [137]. Most probably, these compounds are also able to inhibit functions of cytotoxic pore formers secreted by other bacteria, because the structure of staphylococcal toxin-formed pore is similar to that formed by other toxins. Currently, novel drugs, whose effectiveness has been demonstrated by experiments on mice, undergo clinical checking. Soon it will be clear whether this approach is promising [138].
Another approach of importance is the use of pore formers as highly selective antibiotics. Some bacterial strains producing, e.g. “Bulgarian bacillus” Lactobacterium bulgaricum or bifidobacteria have been in use for many years. Their healing effect is to a large extent based on bacteriocins that suppress growth of hazardous and dangerous microorganisms without inhibiting the normal intestinal microflora. Purified pore-forming bacteriocins are used in the food processing industry to extend the shelf-life of fresh food, e.g. meat. It is safer than using antibiotics for the same purpose [139, 140].
Currently, special attention is paid to the use of nanometer-scale pores (nanopores) in a great variety of technologies, and specifically, as stochastic sensors for biomolecules and metal ions [141]. The principle of action of these instruments seems simple: using a transmembrane potential, charged molecules pass through a pore and physically block it, which leads to a registered change in pore conductance. Translocation of each molecule can be individually registered. This principle underlies the widely known Coulter counters [142].
The first attempt to use nanopores in detecting single DNA molecules was made using the well-characterized S. aureus α-hemolysin [143]. These pores formed in model membranes continuously remain highly conducting under a wide range of experimental conditions. They have been used to detect single- and double-strand DNA molecules [144]. Adjusting conditions allows distinguishing between DNA and RNA, because their characteristic pore-passing times are different. Also, nanopores can be used for DNA quality control: the difference between short and long DNA fragments is reflected by different channel conductance modulations. Based on these results, several research teams work on creation of nanopores fit for automatic de novo DNA- and RNA sequencing. However, it should be noted that staphylococcal α-hemolysin-formed pores are almost ideal for the purpose of single-strand DNA detection, whereas researchers still run into serious problems when fabricating synthetic nanopores. Using a solitary α-hemolysin channel, a group of authors have created a “mass spectrometer” that quite accurately determines the molecular mass of pore-passing molecules. This technique is based on channel conductance dependence on the molecular mass of a molecule (charged or uncharged) passing through the channel [145].
α-Hemolysin pores were used to build a cell-like vesicular bioreactor aimed at much increased protein production [146]. An E. coli cell-free expression system was encapsulated in a phospholipid vesicle in solution containing ribonucleotides and amino acids. Because expression stopped after 2 h, it was necessary to solve the energy and material limitations and increase the capacity of the reactor. The α-hemolysin pore-forming protein from S. aureus expressed inside the vesicle created a selective permeability for nutrients and made the reactor sustain expression for up to 4 days, thereby having increased the amount of synthesized protein. Detailed studies of the properties of bacterial ion channels allowed creating a portable, durable, single-channel biosensor that could be incorporated with many applicable devices [147, 148]. The core of the biosensor is a “microchip” where a single nanopore is embedded in the membrane sandwiched between two layers of agarose gel, which allows long storage and multiple use of the device. Genetically modified staphylococcal hemolysin was used to make a similar chip for detection of inositol 1,4,5-trisphosphate (second messenger) [147]. Recently, modified staphylococcal hemolysin-based nanopores were used in devices capable of rapid nitrogen mustard detection at concentrations below 50 µM [149]. Many nanotechnology-based devices are underlain by good knowledge of a staphylococcal α-hemolysin-formed pore. However, nanopores produced by other pore-forming proteins are sometimes considered as its alternative. Also, there are reported attempts to use as nanopores porins from E. coli and the ion channel from B. subtilis whose properties are dissimilar to α-hemolysin [150]. For example, the B. subtilis bacterial ion channel has a much larger diameter which allowed translocating a 4.2 kb double-strand plasmid DNA and recording the translocation-caused change in channel conductance. The E. coli outer membrane protein porin OmpF forming a 1.0-1.2 nm trimeric pore was used to detect water-soluble polymers, such as polyethylene glycol [150]. Application of the E. coli mechanosensitive channels MscL and MscS possessing a number of properties likely to increase biosensor sensitivity in future are also of interest [151]. Antimicrobial peptides are already used in medicine as antibiotic agents; thermosensitive pores are also targeted for the use [152]. Thermosensitive pores were obtained by introducing an elastin-like peptide into the β-sandwich domain of a heptamer formed by staphylococcal α-hemolysin. A loop of the elastin-like peptide protrudes inside the channel and in a reversible temperature-dependent manner either keeps folded, thus preserving channel conductance, or unfolds, thereby blocking ion flow through the channel. The temperature of loop transition from one conformational state to the other is about 40°C. At temperature below this threshold value, the polypeptide is unfolded; otherwise, it is folded. Pores of this kind can be embedded either in liposomes for the purpose of temperature-controlled drug release or in cells for controlled compound entry. Intensive studies of the properties of other pore-forming proteins may eventually result in creation of novel devices with radically new properties.
One review can hardly describe all pore-forming proteins known to date. However, even the data presented here are sufficient to show that all living organisms, from viruses to mammals, including humans, secret these compounds and widely use them to survive. Depending on the species type, pore formers may have different functions. Many organisms use them as protective, and specifically, antimicrobial agents. Venom-secreting predators use pore formers to immobilize and to kill a prey. Additionally, the same toxin often protects the host organism against pathogenic microorganisms also present in the prey. It should be emphasized that pore-forming proteins are important components of the immune system. For example, leucocyte-induced pore-forming cytotoxic compounds perforins and defensins are efficient in primary host protecting against intrusion microorganisms. In prokaryotes, the functions of pore-forming toxins are similar. Host cell-lysing bacteria gain access to cell nutrients necessary for their own metabolism, while destruction of tissues and organs allows microorganisms to grow in number and spread over additional areas. Almost all organisms, including microorganisms, synthesize antimicrobial peptides (e.g. bacteriocins) that bring changes to the environmental microflora by inhibiting or killing some types of microorganisms to gain a certain advantage. For example, to combat overpopulation, pore formers are used as antibiotics in intra- and inter-species competition. Although pore-forming proteins are quite diverse, they can be divided into two classes on the basis of their structures, designated α-helical and β-sheet. Members of both classes are synthesized by both pro- and eukaryotes. As a rule, pore formers are amphiphilic and contain a pronounced hydrophobic fragment directly involved in membrane piercing. Initially, pore former molecules bind to the membrane either due to their electrostatic interaction with charged lipid heads or through high affinity interaction with their receptors. Next, most of them undergo oligomerization accompanied by structural rearrangements of the whole complex enhancing its hydrophobicity. The pore-forming part of the oligomer then inserts into the hydrophobic area of the bilayer and forms a pore. All peptide-formed pores fall into barrel-stave, where the inner wall is formed solely by protein monomers, and toroidal, where lipids are also implicated in channel lining. As a rule, the formed pore is an oligomeric complex with widely ranging (1-200 nm) inner and outer diameters depending on the type and number of pore-forming monomers. Moderately sized pores are permeable mainly for ions and low-molecular-weight components. As mentioned above, their toxic action consists in disturbance of cell ionic homeostasis and osmotic equilibrium. In the simplest case, this leads to cell lysis and tissue necrosis. Even if osmotic equilibrium disturbance fails to kill the cell, the increased concentration of some ions (e.g. Ca2+) in its cytoplasm may trigger other dramatic consequences, like exocytosis, endocytosis, or apoptosis. On the other hand, a subject of current intensive studies is the effect of pore-forming cytotoxins on inhibition of apoptosis. It is a crucial moment in protecting a microorganism against a wide-scale attack by viruses and bacteria when the dying cell signals to other cells on the trouble and on the necessity of an adequate response to infection by activating the immune system [153].
Protein-conducting pores of a large diameter are also used both for direct lysis of target cells and for transport to the cell cytoplasm of components displaying enzymatic activity; the latter may lead, for example, to inhibition of protein synthesis in the cell or to initiation of apoptotic reactions. Sometimes, e.g. in the latrotoxin case, the pre-pore is a prerequisite to triggering of cell-destabilizing mechanists. Selectivity of pore-forming toxin action varies widely, from the complete absence to high specificity, depending on the presence of receptors of different nature: lipids (cholesterol, phosphatidylcholine, etc.), membrane proteins (glycolipoproteins), or highly specific protein receptors. As a result, the toxin-damaged cells are either solely prokaryotic or eukaryotic, or cells from certain tissues. This property of pore-forming proteins distinguishes them as an excellent material for nanotechnological constructions. Elucidation of pore former selectivity mechanisms by gene engineering techniques would allow creating toxins that mostly target cells of a particular type, for example, cancer cells.
Interestingly, organisms evolutionarily most remote from one another use pore formers with similar mechanisms of action, like, e.g. the diphtheria toxin and perforins from the mammalian immune system. This is direct evidence for universality of the means of defense and attack, access to nutrients and habitat extension, i.e. everything generally termed as adaptation to environmental conditions.
The authors are most grateful to Zh. I. Budarina for attentive reading of the review and help in its preparation.
This review was supported by the Russian Foundation for Basic Research (grant Nos. 07-04-01706 and 08-04-01424).
REFERENCES
1.Titball, R. W. (1998) Symp. Ser. Soc. Appl.
Microbiol., 27, 127-137.
2.Gonzalez, M. R., Bischofberger, M., Pernot, L., van
der Goot, F. G., and Freche, B. (2008) Cell. Mol. Life Sci.,
65, 493-507.
3.Durell, S. R., Gopalan, R., and Guy, H. R. (1992)
Biophys. J., 63, 1623-1631.
4.Gregory, S. M., Cavenaugh, A., Journigan, V.,
Pokorny, A., and Almeida, P. F. (2008) Biophys. J., 94,
1667-1680.
5.Corzo, G., Villegas, E., Gomez-Lagunas, F.,
Possani, L. D., Belokoneva, O. S., and Nakajima, T. (2002) J. Biol.
Chem., 277, 23627-23637.
6.Yang, L., Harroun, T. A., Weiss, T. M., Ding, L.,
and Huang, H. W. (2001) Biophys. J., 81, 1475-1485.
7.Matsuzaki, K., Yoneyama, S., and Miyajima, K.
(1997) Biophys. J., 73, 831-838.
8.Ladokhin, A. S., Selste, M. E., and White, S. H.
(1997) Biophys. J., 72, 1762-1766.
9.Allende, D., Simon, S. A., and McIntosh, T. J.
(2005) Biophys. J., 88, 1828-1837.
10.Park, S., Kim, J., Shin, S., Jeong, C., Kim, M.,
Shin, S., Cheong, G., Park, Y., and Hahm, K. (2006) Biochem.
Biophys. Res. Commun., 343, 222-228.
11.Zemel, A., Fattal, D. R., and Ben-Shaul, A.
(2003) Biophys. J., 84, 2242-2255.
12.Yan, L., and Adams, M. E. (1998) J. Biol.
Chem., 273, 2059-2066.
13.Van Renterghem, C., Iborra, C., Martin-Moutot,
N., Lelianova, V., Ushkaryov, Y., and Seagar, M. (2000)
Eur. J. Neurosci., 12, 3953-3962.
14.Orlova, E. V., Rahman, M. A., Gowen, B.,
Volynski, K. E., Ashton, A. C., Manser, C., van Heel, M., and
Ushkaryov, Y. A. (2000) Nat. Struct. Biol., 7, 48-52.
15.Ashton, A. C., Volynski, K. E., Lelianova, V. G.,
Orlova, E. V., Renterghem, C. V., Canepari, M., Seagar, M., and
Ushkaryov, Y. A. (2001) J. Biol. Chem., 276,
44695-44703.
16.Lajus, S., Vacher, P., Huber, D., Dubois, M.,
Benassy, M., Ushkaryov, Y., and Lang, J. (2006) J. Biol. Chem.,
281, 5522-5531.
17.Elgar, D., Verdonck, F., Grobler, A., Fourie, C.,
and du Plessis, J. (2006) Peptides, 27, 55-61.
18.Nomura, K., Corzo, G., Nakajima, T., and
Iwashita, T. (2004) Biophys. J., 87, 2497-2507.
19.Malovrh, P., Viero, G., Serra, M., Podlesek, Z.,
Lakey, J. H., Macek, P., Menestrina, G., and Anderluh, G. (2003) J.
Biol. Chem., 278, 22678-22685.
20.Gutierrez-Aguirre, I., Barlic, A., Podlesek, Z.,
Macek, P., Anderluh, G., and Gonzalez-Manas, J. M. (2004) Biochem.
J., 384, 421-428.
21.Kem, W. R. (1994) Toxicology, 87,
189-203.
22.Hallock, K. J., Lee, D., Omnaas, J., Mosberg, H.
I., and Ramamoorthy, A. (2002) Biophys. J., 83,
1004-1013.
23.Matsuzaki, K., Sugishita, K., Ishibe, N., Ueha,
M., Nakata, S., Miyajima, K., and Epand, R. M. (1998)
Biochemistry, 37, 11856-11863.
24.Wang, C. H., and Wu, W. G. (2005) FEBS
Lett., 579, 3169-3174.
25.Wang, C.-H., Liu, J.-H., Lee, S.-C., Hsiao,
C.-D., and Wu, W.-G. (2006) J. Biol. Chem., 281,
656-667.
26.Barbosa, P. S., Martins, A. M., Havt, A., Toyama,
D. O., Evangelista, J. S., Ferreira, D. P., Joazeiro, P. P., Beriam, L.
O., Toyama, M. H., Fonteles, M. C., and Monteiro, H. S. (2005)
Toxicon, 46, 376-386.
27.Nair, D. G., Fry, B. G., Alewood, P., Kumar, P.
P., and Kini, R. M. (2007) Biochem. J., 402, 93-104.
28.Gidalevitz, D., Ishitsuka, Y., Muresan, A. S.,
Konovalov, O., Waring, A. J., Lehrer, R. I., and Lee, K. (2003)
Proc. Natl. Acad. Sci. USA, 100, 6302-6307.
29.Yang, L., Weiss, T. M., Lehrer, R. I., and Huang,
H. W. (2000) Biophys. J., 79, 2002-2009.
30.Mani, R., Cady, S. D., Tang, M., Waring, A. J.,
Lehrer, R. I., and Hong, M. (2006) Proc. Natl. Acad. Sci. USA,
103, 16242-16247.
31.Jang, H., Ma, B., and Nussinov, R. (2007) BMC
Struct. Biol., 7, 21.
32.Voskoboinik, I., and Trapani, J. (2006)
Immunol. Cell Biol., 84, 66-71.
33.Masson, D., and Tschopp, J. (1985) J. Biol.
Chem., 260, 9069-9072.
34.Podack, E. R., Young, J., and Cohn, Z. A. (1985)
Proc. Natl. Acad. Sci. USA, 82, 8629-8633.
35.Hadders, M. A., Beringe, D. X., and Gros, P.
(2007) Science, 317, 1552-1554.
36.Rosado, C. J., Buckle, A. M., Law, R. H.,
Butcher, R. E., Kan, W. T., Bird, C. H., Un, G. K., Browne, K. A.,
Baran, K., Bashtannyk-Puhalovich, T. A., Faux, N. G., Wong, W., Porter,
C. J., Pike, R. N., Ellisdon, A. M., Pearce, M. C., Bottomley, S. P.,
Emsley, J., Smith, A. I., Rossjohn, J., Hartland, E. L., Voskoboinik,
I., Trapani, J. A., Bird, P. I., Dunstone, M. A., and Whisstock, J. C.
(2007) Science, 317, 1548-1551.
37.Lukoyanova, N., and Saibil, H. R. (2008)
Trends Immunol., 29, 51-53.
38.Bolitho, P., Voskoboinik, I., Trapani, J. A., and
Smyth, M. J. (2007) Curr. Opin. Immunol., 19,
339-347.
39.Pipkin, M. E., and Lieberman, J. (2007) Curr.
Opin. Immunol., 19, 301-308.
40.Aerts, A. M., Francois, I. E., Cammue, B. P., and
Thevissen, K. (2008) Cell. Mol. Life Sci., 65,
2069-2079.
41.Verma, C., Seebah, S., Low, S. M., Zhou, L., Liu,
S. P., Li, J., and Beuerman, R. W. (2007) Biotechnol. J.,
2, 1353-1359.
42.Matsuzaki, K. (1999) Biochim. Biophys.
Acta, 1462, 1-10.
43.Madison, M. N., Kleshchenko, Y. Y., Nde, P. N.,
Simmons, K. J., Lima, M. F., and Villalta, F. (2007) Infect.
Immun., 75, 4780-4791.
44.Hoover, D. M., Rajashankar, K. R., Blumenthali,
R., Purii, A., Oppenheim, J. J., Chertov, O., and Lubkowski, J. (2000)
J. Biol. Chem., 275, 32911-32918.
45.Warren, W. C., Hillier, L. W., Marshall Graves, J. A., Birney,
E., Ponting, C. P., Grutzner, F., Belov, K., Miller, W., Clarke, L.,
Chinwalla, A. T., Yang, S. P., Heger, A., Locke, D. P., Miethke, P.,
Waters, P. D., Veyrunes, F., Fulton, L., Fulton, B., Graves, T.,
Wallis, J., Puente, X. S., Lopez-Otin, C., Ordonez, G. R., Eichler, E.
E., Chen, L., Cheng, Z., Deakin, J. E., Alsop, A., Thompson, K., Kirby,
P., Papenfuss, A. T., Wakefield, M. J., Olender, T., Lancet, D.,
Huttley, G. A., Smit, A. F., Pask, A., Temple-Smith, P., Batzer, M. A.,
Walker, J. A., Konkel, M. K., Harris, R. S., Whittington, C. M., Wong,
E. S., Gemmell, N. J., Buschiazzo, E., Vargas, Jentzsch, I. M., Merkel,
A., Schmitz, J., Zemann, A., Churakov, G., Kriegs, J. O., Brosius, J.,
Murchison, E. P., Sachidanandam, R., Smith, C., Hannon, G. J.,
Tsend-Ayush, E., McMillan, D., Attenborough, R., Rens, W.,
Ferguson-Smith, M., Lefevre, C. M., Sharp, J. A., Nicholas, K. R., Ray,
D. A., Kube, M., Reinhardt, R., Pringle, T. H., Taylor, J., Jones, R.
C., Nixon, B., Dacheux, J. L., Niwa, H., Sekita, Y., Huang, X., Stark,
A., Kheradpour, P., Kellis, M., Flicek, P., Chen, Y., Webber, C.,
Hardison, R., Nelson, J., Hallsworth-Pepin, K., Delehaunty, K.,
Markovic, C., Minx, P., Feng, Y., Kremitzki, C., Mitreva, M.,
Glasscock, J., Wylie, T., Wohldmann, P., Thiru, P., Nhan, M. N., Pohl,
C. S., Smith, S. M., Hou, S., Renfree, M. B., Mardis, E. R., and
Wilson, R. K. (2008) Nature, 453, 175-184.
46.Masi, M., Vuong, P., Humbard, M., Malone, K., and
Misra, R. (2007) J. Bact., 189, 2667-2676.
47.Zakharov, S. D., Kotova, E. A., Antonenko, Y. N.,
and Cramer, W. A. (2004) Biochim. Biophys. Acta, 1666,
239-249.
48.Krasilnikov, O. V., Sabirov, P. Z., and
Ternovsky, V. I. (1991) Proteins, Ion Channels, and Regulation of
Ion Transport across Membranes [in Russian], Fan, Tashkent, p.
208.
49.Sobko, A. A., Kotova, E. A., Antonenko, Y. N.,
Zakharov, S. D., and Cramer, W. A. (2006) J. Biol. Chem.,
281, 14408-14416.
50.Kent, M. S., Yim, H., Murton, J. K., Satija, S.,
Majewski, J., and Kuzmenko, I. (2008) Biophys. J., 94,
2115-2127.
51.Senzel, L., Gordon, M., Blaustein, R. O., Oh, K.
J., Collier, R. J., and Finkelstein, A. (2000) J. Gen. Physiol.,
115, 421-434.
52.Gordon, M., and Finkelstein, A. (2001) J. Gen.
Physiol., 118, 471-480.
53.Sharpe, J. C., and London, E. (1999) J. Membr.
Biol., 171, 209-221.
54.D'Silva, P. R., and Lala, A. K. (2000) J.
Biol. Chem., 275, 11771-11777.
55.Czajkowsky, D. M., Iwamoto, H., Cover, T. L., and
Shao, Z. (1999) Biochemistry, 96, 2001-2006.
56.Nakayama, M., Hisatsune, J., Yamasaki, E., Nishi,
Y., Wada, A., Kurazono, H., Sap, J., Yahiro, K., Moss, J., and
Hirayama, T. (2006) Infect. Immun., 74, 6571-6580.
57.Kim, S., Chamberlain, A. K., and Bowie, J. U.
(2004) Proc. Natl. Acad. Sci. USA, 101, 5988-5991.
58.Szabo, I., Brutsche, S., Tombola, F., Moschioni,
M., Satin, B., Telford, J. L., Rappuoli, R., Montecucco, C., Papini,
E., and Zoratti, M. (1999) EMBO J., 18, 5517-5527.
59.Cotte, J. G. (1992) Microbiol. Rev.,
88, 137-162.
60.Cortajarena, A. L., Goni, F. M., and Ostolaza, H.
(2002) J. Biol. Chem., 277, 23223-23229.
61.Cortajarena, A. L., Goni, F. M., and Ostolaza, H.
(2003) J. Biol. Chem., 278, 19159-19163.
62.Menestrina, G., Moser, C., Pellet, S., and Welch,
R. (1994) Toxicology, 87, 249-267.
63.Moayeri, M., and Welch, R. A. (1994) Infect.
Immun., 62, 4124-4134.
64.Bakas, L., Chanturiya, A., Herlax, V., and
Zimmerberg, J. (2006) Biophys. J., 91, 3748-3755.
65.Welch, R. A. (1991) Mol. Microbiol.,
5, 521-528.
66.Hertle, R. (2005) Curr. Protein Pept.
Sci., 6, 313-325.
67.Walker, G., Hertle, R., and Braun, V. (2004)
Infect. Immun., 72, 611-614.
68.Schonherr, R., Hilger, M., Broer, S., Benz,
R., and Braun, V. (1994) Eur. J. Biochem., 223,
655-663.
69.Hertle, R. (2002) J. Membr. Biol.,
189, 1-14.
70.Tweten, R. K. (2005) Infect. Immun.,
73, 6199-6209.
71.Shannon, J. G., Ross, C. L., Koehler, T. M., and
Rest, R. F. (2003) Infect. Immun., 71, 3183-3189.
72.Mosser, E. M., and Rest, R. F. (2006) BMC
Microbiol., 6, 56.
73.Dang, T. X., Hotze, E. M., Rouiller, I., Tweten,
R. K., and Wilson-Kubalek, E. M. (2005) J. Struct. Biol.,
150, 100-108.
74.Schuerch, D. W., Wilson-Kubalek, E. M., and
Tweten, R. K. (2005) Proc. Natl. Acad. Sci. USA, 102,
12537-12542.
75.Bavdek, A., Gekara, N. O., Priselac, D., Aguirre,
I. G., Darji, A.,? Chakraborty, T., Macek, P., Lakey, J. H., Weiss, S.,
and Anderluh, G. (2007) Biochemistry, 46, 4425-4437.
76.Soltani, C. E., Hotze, E. M., Johnson, A. E., and
Tweten, R. K. (2007) Proc. Natl. Acad. Sci. USA, 104,
20226-20231.
77.Heuck, A. P., Savva, C. G., Holzenburg, A., and
Johnson, A. E. (2007) J. Biol. Chem., 282,
22629-22637.
78.Tilley, S. J., Orlova, E. V., Gilbert, R. J. C.,
Andrew, P. W., and Saibil, H. R. (2005) Cell, 121,
247-256.
79.Rossjohn, J., Polekhina, G., Feil, S. C., Morton,
C. J., Tweten, R. K., and Parker, M. W. (2007) J. Mol. Biol.,
367, 1227-1236.
80.Krasilnikov, O. V., Ternovsky, V. I., Musaev, Yu.
M., and Tashmukhamedov, B. A. (1980) Dokl. Akad. Nauk UzSSR,
7, 66-68.
81.Krasilnikov, O. V., Ternovsky, V. I., and
Tashmukhamedov, B. A. (1981) Biofizika, 26, 271-275.
82.Merzlyak, P. G., Yuldasheva, L. N., Rodrigues,
C., Carneiro, C. M., Krasilnikov, O. V., and Bezrukov, S. M. (1999)
Biophys. J., 77, 3023-3033.
83.Gouaux, E., Braha, O., Hobaugh, M., Song, L.,
Cheley, S., Shustak, C., and Bayley, H. (1994) Biochemistry,
91, 12828-12831.
84.Montoya, M., and Gouaux, E. (2003)
Biochim. Biophys. Acta, 1609, 19-27.
85.Jayasinghe, L., Miles, G., and Bayley, H. (2006)
J. Biol. Chem., 281, 2195-2204.
86.Cheley, S., Malghani, M. S., Song, L., Hobaugh,
M., Gouaux, J. E., Yang, J., and Bayley, H. (1997) Protein Eng.,
10, 1433-1443.
87.Gouaux, E. (1998) J. Struct. Biol.,
121, 110-122.
88.Kawate, T., and Gouaux, E. (2003) Protein
Sci., 12, 997-1006.
89.Galdiero, S., and Gouaux, E. (2004) Protein
Sci., 13, 1503-1511.
90.Aksimentiev, A., and Schulten, K. (2005)
Biophys. J., 88, 3745-3761.
91.Bhakdi, S., Walev, I., Husmann, M., and Valeva,
A. (2005) Curr. Top. Genet., 11, 91-110.
92.Menestrina, G., Serra, M. D., Comai, M., Viero,
M. C., Werner, S., Colin, D. A., Monteil, H., and Prevost, G. (2003)
FEBS Lett., 552, 54-60.
93.Caiazza, N. C., and O'Toole, G. A. (2003) J.
Bacteriol., 185, 3214-3217.
94.Budarina, Z. I., Sinev, M. A., Mayorov, S. G.,
Tomashevski, A. Y., Shmelev, I. V., and Kuzmin, N. P. (1994) Arch.
Microbiol., 161, 252-257.
95.Andreeva, Zh. I., Nesterenko, V. F., Yurkov, I.
V., Budarina, Z. I., Sineva, E. V., and Solonin, A. S. (2006)
Protein Expr. Purif., 47, 186-193.
96.Andreeva, Zh. I., Nesterenko, V. F., Fomkina, M.
G., Ternovsky, V. I., Suzina, N. E., Bakulina, A. Y., Solonin, A. S.,
and Sineva, E. V. (2007) Biochim. Biophys. Acta, 1768,
253-263.
97.Kovalevskaya (Andreeva), Zh. I. (2007)
Isolation and Characterization of Bacillus cereus Hemolysin II: Ph.
D. thesis [in Russian], FSUI State Research Institute of Genetics
and Selection of Industrial Microorganisms, Moscow.
98.Klarkson, D. (1978) Ion Transport and Plant Cell Structure
[Russian translation], Mir, Moscow, pp. 85-86.
99.Sato, N., Kurotaki, H., Watanabe, T., Mikami, T.,
and Matsumoto, T. (1998) Biol. Pharm. Bull., 21,
311-314.
100.Fivaz, M., Velluz, M., and van der Goot, F. G.
(1999) J. Biol. Chem., 274, 37705-37708.
101.Fukushima, K., Ikehara, Y., Kanai, M., Kochibe,
N., Kuroki, M., and Yamashita, K. (2003) J. Biol. Chem.,
278, 36296-36303.
102.Diep, D. B., Nelson, K. L., Lawrence, T. S.,
Sellman, B. R., Tweten, R. K., and Buckley, J. T. (1999) Mol.
Microbiol., 31, 785-794.
103.Kennedy, C. L., Krejany, E. O., Young, L. F.,
O'Connor, J. R., Awad, M. M., Boyd, R. L., Emmins, J. J., Lyras, D.,
and Rood, J. I. (2005) Mol. Microbiol., 57,
1357-1366.
104.Olson, R., and Gouaux, E. (2005) J. Mol.
Biol., 350, 997-1016.
105.Jayasinghe, L., and Bayley, H. (2005)
Protein Sci., 14, 2550-2561.
106.Miles, G., Jayasinghe, L., and Bayley, H.
(2006) J. Biol. Chem., 281, 2205-2214.
107.Sugawara-Tomita, N., Tomita, T., and Kamio, Y.
(2002) J. Bacteriol., 184, 4747-4756.
108.Nguyen, V. T., Kamio, Y., and Higuchi, H.
(2003) EMBO J., 22, 4968-4979.
109.Kaneko, J., and Kamio, Y. (2004) Biosci.
Biotechnol. Biochem., 68, 981-1003.
110.Nguyen, V. T., and Kamio, Y. (2004) J.
Biochem., 136, 563-567.
111.Wallace, A. J., Stillman, T. J., Atkins, A.,
Jamieson, S. J., Bullough, P. A., Green, J., and Artymiuk, P. J. (2000)
Cell, 100, 265-276.
112.Wyborn, N. R., Clark, A., Roberts, R. E.,
Jamieson, S. J., Tzokov, S., Bullough, P. A., Stillman, T. J.,
Artymiuk, P. J., Galen, J. E., Zhao, L., Levine, M. M., and Green, J.
(2004) Microbiology, 150, 1495-1505.
113.Tzokov, S. B., Wyborn, N. R., Stillman, T. J.,
Jamieson, S., Czudnochowski, N., Artymiuk, P. J., Green, J., and
Bullough, P. A. (2006) J. Biol. Chem., 281,
23042-23049.
114.Barth, H., Aktories, K., Popoff, M., and
Stiles, B. (2004) Microbiol. Mol. Biol. Rev., 68,
373-402.
115.Rainey, G., Wigelsworth, D. J., Ryan, P. L.,
Scobie, H. M., Collier, R. J., and Young, J. (2005) Proc. Natl.
Acad. Sci. USA, 102, 13278-13283.
116.Mogridge, J., Cunningham, K., and Collier, R.
J. (2002) Biochemistry, 41, 1079-1082.
117.Abrami, L., Liu, S., Cosson, P., Leppla, S. H.,
and van der Goot, F. G. (2003) J. Cell Biol., 160,
321-328.
118.Sherer, K., Li, Y., Cui, X., and Eichacker, P.
Q. (2007) Am. J. Respir. Crit. Care Med., 175,
211-221.
119.Muehlbauer, S. M., Evering, T. H., Bonuccelli,
G., Squires, R. C., Ashton, A. W., Porcelli, S. A., Lisanti, M. P., and
Brojatsch, J. (2007) Cell Cycle, 6, 758-766.
120.Drider, D., Fimland, G., Hechard, Y., McMullen,
L. M., and Herve, P. (2006) Microbiol. Mol. Biol. Rev.,
70, 564-582.
121.Fimland, G., Johnsen, L., Dalhus, B., and
Nissen-Meyer, J. (2005) J. Pept. Sci., 11, 688-696.
122.Chatterjee, C., Paul, M., Xie, L., and van der
Donk, W. A. (2005) Chem. Rev., 105, 633-683.
123.Breukink, E., van Heusden, H. E., Vollmerhaus,
P. J., Swiezewska, E., Brunner, L., Walker, S., Heck, A. J., and de
Kruijff, B. (2003) J. Biol. Chem., 278, 19898-19903.
124.Gilmore, M. S., and Haas, W. (2005) Proc.
Natl. Acad. Sci. USA, 102, 8401-8402.
125.Ivanovic, T., Agosto, M. A., Zhang, L.,
Chandran, K., Harrison, S. C., and Nibert, M. L. (2008) EMBO J.,
27, 1289-1298.
126.Perez-Berna, A. J., Guillen, J., Moreno, M. R.,
Bernabeu, A., Pabst, G., Laggner, P., and Villalain, J. (2008) J.
Biol. Chem., 283, 8089-8101.
127.Clarke, D., Griffin, S., Beales, L., Gelais, C.
S., Burgess, S., Harris, M., and Rowlands, D. (2006) J. Biol.
Chem., 281, 37057-37068.
128.Steinmann, E., Penin, F., Kallis, S., Patel, A.
H., Bartenschlager, R., and Pietschmann, T. (2007) PLoS
Pathogens, 3, 7.
129.Cavalier-Smith, T. (2002) Int. J. Syst.
Evol. Microbiol., 52, 297-354.
130.Maurelli, A. T. (2007) FEMS Microbiol.
Lett., 267, 1-8.
131.Stenfors Arnesen, L. P., Fagerlund, A., and
Granum, P. E. (2008) FEMS Microbiol. Rev., 32,
579-606.
132.Ehling-Schulz, M., Fricker, M.,
Grallert, H., Rieck, P., Wagner, M., and Scherer, S. (2006) BMC
Microbiol., 6, 20.
133.Zakharova, M. V., Beletskaya, I. V.,
Denjmukhametov, M. M., Yurkova, T. V., Semenova, L. M., Shlyapnikov, M.
G., and Solonin, A. S. (2002) Mol. Genet. Genom., 267,
171-178.
134.Summers, D. (1998) Mol. Microbiol.,
29, 1137-1145.
135.Zakharova, M. V., Beletskaya, I. V., Bolovin,
D. V., Yurkova, T. V., Semenova, L. M., and Solonin, A. S. (2003)
Mol. Genet. Genom., 270, 415-419.
136.Chen, M., Zhao, J. Z., Collins, H. L., Earle,
E. D., Cao, J., and Shelton, A. M. (2008) PLoS ONE, 3,
2284-2290.
137.Karginov, V. A., Nestorovich, E. M.,
Schmidtmann, F., Robinson, T. M., Yohannes, A., Fahmi, N. E., Bezrukov,
S. M., and Hecht, S. M. (2007) Bioorg. Med. Chem.,
15, 5424-5431.
138.Moayeri, M., Robinson, T. M., Leppla, S. H.,
and Karginov, V. A. (2008) Antimicrob. Agents Chemother.,
52, 2239-2241.
139.Gillor, O., Kirkup, B. C., and Riley, M. A.
(2004) Adv. Appl. Microbiol., 54, 129-146.
140.Diez-Gonzalez, F. (2007) Curr. Issues
Intest. Microbiol., 8, 15-23.
141.Braha, O., Gu, L., Zhou, L., Lu, X., Cheley,
S., and Bayle, H. (2000) Nature Biotechnol., 18,
1005-1007.
142.DeBlois, R. W., and Bean, C. P. (1970) Rev.
Sci. Instrum., 41, 909-916.
143.Kasianowicz, J. J., Brandin, E., Branton, D.,
and Deamer, D. W. (1996) Proc. Natl. Acad. Sci. USA, 93,
13770-13773.
144.Howorka, S., Cheley, S., and Bayley, H. (2001)
Nature Biotechnol., 19, 636-639.
145.Robertson, J. W., Rodrigues, C. G., Stanford,
V. M., Rubinson, K. A., Krasilnikov, O. V., and Kasianowicz, J. J.
(2007) Proc. Natl. Acad. Sci. USA, 104, 8207-8211.
146.Noireaux, V., and Libchaber, A. (2004) Proc.
Natl. Acad. Sci. USA, 101, 17669-17674.
147.Shim, J. W., and Gu, L. Q. (2007) Anal.
Chem., 79, 2207-2213.
148.Kang, X. F., Cheley, S., Rice-Ficht, A. C., and
Bayley, H. (2007) J. Am. Chem. Soc., 129, 4701-4705.
149.Wu, H. C., and Bayley, H. (2008) J. Am.
Chem. Soc., 130, 6813-6819.
150.Rhee, M., and Burns, M. A. (2007) Trends
Biotechnol., 25, 174-181.
151.Shapovalov, G., and Lester, H. A. (2004) J.
Gen. Physiol., 124, 151-161.
152.Jung, Y., Bayley, H., and Movileanu, L. (2006)
J. Am. Chem. Soc., 128, 15332-15340.
153.Faherty, C. S., and Maurelli, A. T. (2008)
Trends Microbiol., 16, 173-180.
154.Lin, J.-H., and Baumgaertner, A. (2000)
Biophys. J., 78, 1714-1724.
155.Czajkowsky, D. M., Hotze, E. M., Shao, Z., and
Tweten, R. K. (2004) EMBO J., 23, 3206-3215.