Protein Aggregation and Neurodegeneration: Clues from a Yeast Model of Huntington’s Disease
N. Bocharova1, R. Chave-Cox2, S. Sokolov1, D. Knorre3, and F. Severin3*
1Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-4195; E-mail: bona@genebee.msu.ru2University College London, 20 Gordon Street, London WC1H 0AJ United Kingdom; fax: +44 (0) 20-7679-7463; E-mail: r.chave-cox@ucl.ac.uk
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-3181; E-mail: severin@genebee.msu.ru
* To whom correspondence should be addressed.
Received May 29, 2008; Revision received July 30, 2008
A number of neurodegenerative diseases are accompanied by the appearance of intracellular protein aggregates. Huntington’s disease (HD) is caused by a mutation in a gene encoding huntingtin. The mutation causes the expansion of the polyglutamine (polyQ) domain and consequently polyQ-containing aggregates accumulate and neurons in the striatum die. The role of the aggregates is still not clear: they may be the cause of cytotoxicity or a manifestation of the cellular attempt to remove the misfolded proteins. There is accumulating evidence that the main cause of HD is the interaction of the mutated huntingtin with other polyQ-containing proteins and molecular chaperones and most studies based on a yeast model of HD support this point of view. Data obtained using yeasts suggest pathological consequences of polyQ–proteasomal interaction: proteasomal overload by polyQs may interfere with functions of the cell cycle-regulating proteins.
KEY WORDS: Huntington’s disease, aggregation, polyglutamine, yeastDOI: 10.1134/S0006297909020163
Abbreviations: APC, anaphase-promoting complex; HD, Huntington’s disease; polyQ, polyglutamine.
There is a set of human diseases caused by neuronal death or
malfunctioning. These diseases are called neurodegenerative and
typically lead to brain dysfunction. Intracellular protein aggregates
are hallmarks of such neurodegenerative pathologies as
Alzheimer’s, Parkinson’s, and Huntington’s diseases
[1]. The cellular response to the formation of
protein aggregates is rather complex: it includes activation of protein
degradation and re-folding systems and changes in the transcription
level of a significant number of genes. So, which parts of the response
are protective and which ones are self-destructive for the cells? Here
we argue that the data obtained by using a yeast model of
Huntington’s disease (HD) add to our understanding of this
problem.
HD is one of nine diseases caused by the elongation of polyglutamine (polyQ) stretch and accompanied by accumulation of amyloid bodies in neurons. HD is caused by elongation of the glutamine-rich fragment in a protein called huntingtin [2]. The protein is one of the largest known (348 kDa), but it does not contain any conservative domains except for its polyQ domain [3]. Apart from that, huntingtin contains HEAT repeat [4] and several caspase-cleavable sites [5]. The protein is ubiquitous, and the highest levels of expression are detected in brain [6, 7]. Huntingtin is found associated with a number of intracellular structures: the endoplasmic reticulum, mitochondria, and microtubules [8, 9]. It has been shown that huntingtin can interact with more than 50 proteins: kinases, phosphatases, proteases, and transcription factors amongst others [10, 11]. There are data showing that huntingtin regulates transcription by transporting the transcription factors between the cytoplasm and the nucleus [3], plays a role in vesicular traffic [12], and also protects cells against apoptosis [13, 14].
The first exon of huntingtin contains a highly polymorphic region of CAG repeats, which code for glutamine. Normally there are 10-34 repeats and more than 40 almost always leads to the disease [15, 16].
The expanded polyQ fragment is a critical factor driving the formation of intracellular aggregates and neuronal degeneration. Interestingly, unlike the normal one, the mutant huntingtin is often found in the nucleus [8, 9].
PolyQ aggregation and toxicity are currently studied in many cell cultures and model organisms. The yeast Saccharomyces cerevisiae has proven to be a useful experimental model of the disease [17] since the discovery that the mechanisms of aggregate formation in higher organisms and in yeast appeared similar (reviewed in [18]). Moreover, the cytological effects of the expanded polyQ expression in yeast and cultured mammalian cells have a lot in common. The first yeast-based model of HD was described in 2002: it was shown that the expression of the expanded polyQ fragment of human huntingtin slows yeast cell growth, affects the cell cycle, and leads to the formation of cytoplasmic and nuclear aggregates [19].
Why is the expanded polyQ expression toxic for cells? There are two possible answers to this question. First, it may be that the already formed polyQ aggregates are harmful for cells. Indeed, it does not seem surprising that large amounts of misfolded proteins could negatively affect the cell physiology. Alternatively, the aggregate accumulation could be a consequence of the pathogenetic process or reflect a cellular attempt to remove the mutant protein from the metabolism.
On one hand, different models of the disease show that the degree of pathogenesis depends directly on the amount of aggregated protein. Because of that, the reduction of the aggregate level is the main strategy in search for drugs against HD. In particular, with help of the yeast model a number of chemical substances were identified which reduced the amounts of the aggregates and stimulated the survival of the polyQ-expressing yeast cells [20]. On the other hand, the same group has shown that the activation of protein aggregation reduces the toxicities of the expanded polyQ and α-synuclein (Parkinson’s disease protein) [21].
Probably the best way to test whether the formed polyQ aggregates are main contributors to the neurodegeneration is to perform the experiments on neuronal cells. Such a study has been done. Expanded polyQ expression was induced in cultured neuronal cells, and the individual cell fates were monitored under the microscope. Stochastically, each cell in the culture expressed different levels of polyQ and had different degrees of its aggregation. It appeared that the cell survival correlates negatively with the total amount of polyQ and positively with the proportion of polyQ in aggregated form [22]. Therefore, polyQ aggregation seems to reduce the toxicity. How can this contradiction be reconciled? Apparently, only relatively large aggregates were detected by the microscope. At the same time, there is evidence that the small aggregates are the most pathogenic ones [23]. Thus, the next question arises: what is the mechanism of small aggregates-induced toxicity? An obvious possibility is that such aggregates may sequester and inactivate other proteins.
It is known that normally huntingtin localizes to the cytoplasm. It was shown that the polyQ expansion leads to partial cleavage of the mutant huntingtin, accumulation of the polyQ-containing aggregates in the nuclei, and loss of function of transcription factor TBP [24, 25]. The loss of function happens because the polyQ aggregates bind and sequester TBP via interaction with Q-rich domain of the transcription factor. It was shown that the co-aggregation is inhibited by chaperons Hsp40 and Hsp70 [26]. Importantly, similar to TBP, many transcription factors contain Q-rich domains. Therefore, transcription factor inactivation seems to be one of the most probable reasons of the expanded polyQ-induced toxicity.
Our data obtained using the yeast model also point at nuclear protein inactivation as a cause of the toxicity. Nuclear localization of the expanded polyQ (103Q) fragment expressed in yeast depends on yeast metacaspase Yca1. Disruption of YCA1 prevents nuclear accumulation of the aggregates [27] and also improves the growth rate of 103Q-expressing cells (Bocharova et al., manuscript in preparation).
Apparently, transcription factors are not the only proteins whose co-aggregation with huntingtin is expected to contribute to the pathogenesis induced by polyQ expansion. Many of the endocytotic proteins contain polyQ stretches. The yeast model was used to demonstrate the interaction of 103Q with a number of endosome-associated proteins and that the toxicity depends on co-aggregation [19, 20]. Disruptions of endocytosis during HD has also been shown using other experimental models [28, 29].
Obviously, polyQ-induced inactivation of cellular proteins is not the only reason for toxic effects of the expanded polyQs. For instance, polyQ expansion sensitizes cells to proteasomal stress factors [30-32]. This is not surprising because the removal of misfolded proteins is the key proteasomal function.
Here it is important to mention that expression of 103Q in yeast noticeably affects the cell cycle [19, 27]. The main system controlling the cell cycle is anaphase-promoting complex (APC). APC ubiquitinates cyclins leading to their proteasomal degradation [33, 34]. This allows us to suggest that 103Q when expressed in yeast overloads the proteasome, which slows cyclin proteolysis, thus delaying the division step of the cell cycle. This is in accordance with our data showing that 103Q expression in yeast raises the proportion of cells with duplicated DNA and also partially rescues cells from hyper-activation of APC [27]. Moreover, recently we found that the deletion of ASE1 lowers the 103Q toxicity (Bocharova et al., manuscript in preparation). Ase1 is a structural mitotic spindle component and APC substrate. Possibly the deletion of ASE1 in 103Q-expressing cells alleviates APC overload, thus enhancing degradation of cyclins.
The figure illustrates this hypothesis. We speculate that the existing 103Q-containing aggregates are relatively harmless, but the cellular attempt to degrade the aggregates is the main cause of the pathological consequences.
Possible link between proteasomal overload and APC functioning
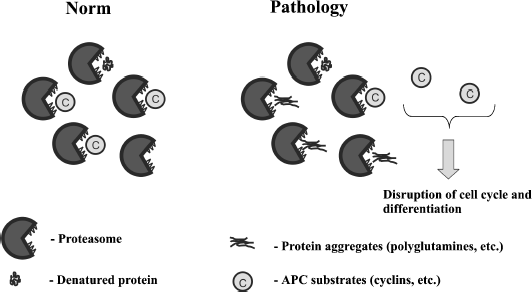
In line with this, it has been shown that inactivation of Hsp104 chaperone reduces the 103Q-induced toxicity in yeast [19, 36]. It appears that in the absence of Hsp104, cells lose [PIN+] thus inhibiting 103Q aggregation [36]. Together with the reasoning presented above, it seems possible that the positive effect of HSP104 deletion could be not only due to [PIN+] conversion. One could speculate that the absence of Hsp104 in 103Q-expressing cells alleviates the proteasomal overload and in this way normalizes the degradation of APC substrates. Indeed, it has been shown that Hsp104 initiates ubiquitination of misfolded cytosolic proteins, which is a necessary step for the proteasomal degradation [37].
Does APC substrate accumulation have any relation to neurodegenerative diseases? It is known that during Alzheimer’s disease neurons frequently attempt abortive mitosis – initiation of DNA duplication followed by cell death. Recently it has been shown that this is caused by cyclin B, accumulation of which is due to APC malfunctioning. It has been suggested that protein aggregates during Alzheimer’s disease act as indirect inhibitors of APC [38].
Does cell cycle disruption contribute to the pathophysiology of HD? On one hand, we are not aware of abortive mitosis happening in HD neurons. On the other hand, cyclins are not the only substrates of APC. It has been shown that the degradation of neuronal differentiation factors Id2 [39] and SnoN [40] is catalyzed by APC/C(CDH1). It can be speculated that in neurons the polyQ expansion might lead to pathological accumulation of Id2 and/or SnoN. If this speculation proves to be correct, then CDH1, SnoN, and Id2 will become potential targets for treatments of expanded polyQ-dependent diseases. In particular, our hypothesis predicts that hyper-expression of CDH1 might reduce the toxicity of the expanded polyQs. Currently we are testing this on 103Q-expressing yeast cells.
This work was supported by the Russian Foundation for Basic Research (grants 06-04-49555 and 07-04-00223).
REFERENCES
1.Taylor, J. P., Hardy, J., and Fischbeck, K. N.
(2002) Science, 296, 1991-1995.
2.The Huntington’s Disease Collaborative
Research Group (1993) Cell, 72, 971-983.
3.Faber, P. W., Barnes, G. T., Srinidhi, J., Chen,
J., Gusella, J. F., and MacDonald, M. E. (1998) Hum Mol Genet.,
7, 1463-1474.
4.Andrade, M. A., and Bork, P. (1995) Nat
Genet., 11, 115-116.
5.Wellington, C. L., Ellerby, L. M., Hackam, A. S.,
Margolis, R. L., Trifiro, M. A., Singaraja, R., McCutcheon, K.,
Salvesen, G. S., Propp, S. S., Bromm, M., Rowland, K. J., Zhang, T.,
Rasper, D., Roy, S., Thornberry, N., Pinsky, L., Kakizuka, A., Ross, C.
A., Nicholson, D. W., Bredesen, D. E., and Hayden, M. R. (1998) J.
Biol. Chem., 273, 9158-9167.
6.Gutekunst, C. A., Levey, A. I., Heilman, C. J.,
Whaley, W. L., Yi, H., Nash, N. R., Rees, H. D., Madden, J. J., and
Hersch, S. M. (1995) Proc. Natl. Acad. Sci. USA, 92,
8710-8714.
7.DiFiglia, M., Sapp, E., Chase, K. O., Davies, S.
W., Bates, G. P., Vonsattel, J. P., and Aronin, N. (1997)
Science, 277, 1990-1993.
8.Trottier, Y., Devys, D., Imbert, G., Saudou, F.,
An, I., Lutz, Y., Weber, C., Agid, Y., Hirsch, E. C., and Mandel, J. L.
(1995) Nat. Genet., 10, 104-110.
9.Velier, J., Kim, M., Schwarz, C., Kim, T. W., Sapp,
E., Chase, K., Aronin, N., and DiFiglia, M. (1998) Exp. Neurol.,
152, 34-40.
10.Harjes, P., and Wanker, E. E. (2003) Trends
Biochem. Sci., 28, 425-433.
11.Li, S. H., and Li, X. J. (2004) Trends
Genet., 20, 146-154.
12.Gauthier, L. R., Charrin, B. C., Borrell-Pages,
M., Dompierre, J. P., Rangone, H., Cordelieres, F. P., de Mey, J.,
MacDonald, M. E., Lessmann, V., Humbert, S., and Saudou, F. (2004)
Cell, 118, 127-138.
13.Rigamonti, D., Bauer, J. H., de Fraja, C., Conti,
L., Sipione, S., Sciorati, C., Clementi, E., Hackam, A., Hayden, M. R.,
Li, Y., Cooper, J. K., Ross, C. A., Govoni, S., Vincenz, C., and
Cattaneo, E. (2000) J. Neurosci., 20, 3705-3713.
14.Hackam, A. S., Yassa, A. S., Singaraja, R.,
Metzler, M., Gutekunst, C. A., Gan, L., Warby, S., Wellington, C. L.,
Vaillancourt, J., Chen, N., Gervais, F. G., Raymond, L., Nicholson, D.
W., and Hayden, M. R. (2000) J. Biol. Chem., 275,
41299-41308.
15.Snell, R. G., MacMillan, J. C., Cheadle, J. P.,
Fenton, I., Lazarou, L. P., Davies, P., MacDonald, M. E., Gusella, J.
F., Harper, P. S., and Shaw, D. J. (1993) Nat. Genet., 4,
393-397.
16.Rubinsztein, D. C., Leggo, J., Coles, R.,
Almqvist, E., Biancalana, V., Cassiman, J. J., Chotai, K., Connarty,
M., Crauford, D., Curtis, A., Curtis, D., Davidson, M. J., Differ, A.
M., Dode, C., Dodge, A., Frontali, M., Ranen, N. G., Stine, O. C.,
Sherr, M., Abbott, M. H., Franz, M. L., Graham, C. A., Harper, P. S.,
Hedreen, J. C., Hayden, M. R., et al. (1996) Am. J. Hum. Genet.,
59, 16-22.
17.Outeiro, T. F., and Giorgini, F. (2006)
Biotechnol. J., 1, 258-269.
18.Vishnevskaia, A. B., Kushnirov, V. V., and
Ter-Avanesian, M. D. (2007) Mol. Biol. (Moscow), 41,
346-354.
19.Meriin, A. B., Zhang, X., He, X., Newnam, G. P.,
Chernoff, Y. O., and Sherman, M. Y. (2002) J. Cell Biol.,
157, 997-1004.
20.Zhang, X., Smith, D. L., Meriin, A. B., Engemann,
S., Russel, D. E., Roark, M., Washington, S. L., Maxwell, M. M., Marsh,
J. L., Thompson, L. M., Wanker, E. E., Young, A. B., Housman, D. E.,
Bates, G. P., Sherman, M. Y., and Kazantsev, A. G. (2005) Proc.
Natl. Acad. Sci. USA, 102, 892-897.
21.Bodner, R. A., Outeiro, T. F., Altmann, S.,
Maxwell, M. M., Cho, S. H., Hyman, B. T., McLean, P. J., Young, A. B.,
Housman, D. E., and Kazantsev, A. G. (2006) Proc. Natl. Acad. Sci.
USA, 103, 4246-4251.
22.Arrasate, M., Mitra, S., Schweitzer, E. S.,
Segal, M. R., and Finkbeiner, S. (2004) Nature, 431,
805-810.
23.Ross, C. A., and Poirier, M. A. (2004) Nat.
Med., 10, S10-S17.
24.Van Roon-Mom, W. M., Reid, S. J., Jones, A. L.,
MacDonald, M. E., Faull, R. L., and Snell, R. G. (2002) Brain Res.
Mol. Brain Res., 109, 1-10.
25.Stevanin, G., Fujigasaki, H., Lebre, A. S.,
Camuzat, A., Jeannequin, C., Dode, C., Takahashi, J., San, C.,
Bellance, R., Brice, A., and Durr, A. (2003) Brain, 126,
1599-1603.
26.Schaffar, G., Breuer, P., Boteva, R., Behrends,
C., Tzvetkov, N., Strippel, N., Sakahira, H., Siegers, K., Hayer-Hartl,
M., and Hartl, F. U. (2004) Mol. Cell., 15, 95-105.
27.Sokolov, S., Pozniakovsky, A., Bocharova, N.,
Knorre, D., and Severin, F. (2006) Biochim. Biophys. Acta,
1757, 660-666.
28.Trushina, E., Singh, R. D., Dyer, R. B., Cao, S.,
Shah, V. H., Parton, R. G., Pagano, R. E., and McMurray, C. T. (2006)
Hum. Mol. Genet., 15, 3578-3591.
29.Hyun, T. S., Li, L., Oravecz-Wilson, K. I.,
Bradley, S. V., Provot, M. M., Munaco, A. J., Mizukami, I. F., Sun, H.,
and Ross, T. S. (2004) Mol. Cell Biol., 24,
4329-4340.
30.Liu, C. W., Giasson, B. I., Lewis, K. A., Lee, V.
M., Demartino, G. N., and Thomas, P. J. (2005) J. Biol. Chem.,
280, 22670-22678.
31.Bossy-Wetzel, E., Schwarzenbacher, R., and
Lipton, S. A. (2004) Nat. Med., 10, S2-S9.
32.Jana, N. R., Zemskov, E. A., Wang, Gh., and
Nukina, N. (2001) Hum. Mol. Genet., 10, 1049-1059.
33.Castro, A., Bernis, C., Vigneron, S., Labbe, J.
C., and Lorca, T. (2005) Oncogene, 24, 314-325.
34.Baker, D. J., Dawlaty, M. M., Galardy, P., and
van Deursen, J. M. (2007) Cell Mol. Life Sci., 64,
589-600.
35.Juang, Y. L., Huang, J., Peters, J. M.,
McLaughlin, M. E., Tai, C. Y., and Pellman, D. (1997) Science,
275, 1311-1314.
36.Kryndushkin, D. S., Alexandrov, I. M.,
Ter-Avanesyan, M. D., and Kushnirov, V. V. (2003) J. Biol.
Chem., 278, 49636-49643.
37.Taxis, C., Hitt, R., Park, S. H., Deak, P. M.,
Kostova, Z., and Wolf, D. H. (2003) J. Biol. Chem., 278,
35903-35913.
38.Aulia, S., and Tang, B. L. (2006) Biochem.
Biophys. Res. Commun., 339, 1-6.
39.Lasorella, A., Stegmuller, J., Guardavaccaro, D.,
Liu, G., Carro, M. S., Rothschild, G., de la Torre-Ubieta, L., Pagano,
M., Bonni, A., and Iavarone, A. (2006) Nature, 442,
471-474.
40.Stegmuller, J., Konishi, Y., Huynh, M. A., Yuan,
Z., Dibacco, S., and Bonni, A. (2006) Neuron, 50,
389-400.