In silico Analysis of Pectin Lyase and Pectinase Sequences
P. K. Yadav1,2, V. K. Singh3, S. Yadav2, K. D. S. Yadav2, and D. Yadav1,3*
1Department of Biotechnology, Deen Dayal Upadhyaya Gorakhpur University, Gorakhpur (U. P.), India; E-mail: dinesh_yad@rediffmail.com; pramod0231@rediffmail.com2Department of Chemistry, Deen Dayal Upadhyaya Gorakhpur University, Gorakhpur (U. P.), India
3Department of Molecular Biology and Genetic Engineering, CBSH, G. B. Pant University of Agriculture and Technology, Pantnagar (Uttarakhand), India
* To whom correspondence should be addressed.
Received October 13, 2008; Revision received December 22, 2008
A total of 48 full-length protein sequences of pectin lyases from different source organisms available in NCBI were subjected to multiple sequence alignment, domain analysis, and phylogenetic tree construction. A phylogenetic tree constructed on the basis of the protein sequences revealed two distinct clusters representing pectin lyases from bacterial and fungal sources. Similarly, the multiple accessions of different source organisms representing bacterial and fungal pectin lyases also formed distinct clusters, showing sequence level homology. The sequence level similarities among different groups of pectinase enzymes, viz. pectin lyase, pectate lyase, polygalacturonase, and pectin esterase, were also analyzed by subjecting a single protein sequence from each group with common source organism to tree construction. Four distinct clusters representing different groups of pectinases with common source organisms were observed, indicating the existing sequence level similarity among them. Multiple sequence alignment of pectin lyase protein sequence of different source organisms along with pectinases with common source organisms revealed a conserved region, indicating homology at sequence level. A conserved domain Pec_Lyase_C was frequently observed in the protein sequences of pectin lyases and pectate lyases, while Glyco_hydro_28 domains and Pectate lyase-like β-helix clan domain are frequently observed in polygalacturonases and pectin esterases, respectively. The signature amino acid sequence of 41 amino acids, i.e. TYDNAGVLPITVNSNKSLIGEGSKGVIKGKGLRIVSGAKNI, related with the Pec_Lyase_C is frequently observed in pectin lyase protein sequences and might be related with the structure and enzymatic function.
KEY WORDS: pectin lyase, pectate lyase, polygalacturonase, pectin esterase, domain analysisDOI: 10.1134/S0006297909090144
Abbreviations: MEME, Multiple EM for Motif Elicitation; NCBI, National Center for Biotechnology Information; NJ, Neighbor-Joining method; PCR, polymerase chain reaction; PE, pectin esterase; PG, polygalacturonase; PL, pectate lyase; PNL, pectin lyase; UPGMA method, Unweighted Pair Group method with Arithmetic Mean method.
Pectinases are a group of enzymes involved in degradation of pectin,
that includes various enzymes classified into various classes and
subclasses depending on the substrate specificity and mode of action,
for example, methyl deesterases, hydrolases, and lyases. According to
the cleavage site, pectinases are divided into three groups: (i)
hydrolases consisting of polygalacturonase, PG (EC 3.2.1.15); (ii)
lyase/trans-eliminases comprising pectin lyase, PNL (EC 4.2.2.10), and
pectate lyase, PL (EC 4.2.2.2); (iii) pectin esterase, PE (EC 3.1.1.11)
[1, 2]. Pectin esterase
catalyzes the deesterification of methyl ester linkages of galacturonan
backbone of pectic substances to release acidic pectins and methanol
[3]. The resulting pectin is then acted upon by PG
and PL [4]. Production, biochemical
characterization, and applications of PNL have been reviewed
extensively [1]. Among all pectinases, PNLs are of
particular interest because these degrade pectin polymers directly by a
β-elimination mechanism that results in the formation of
4,5-unsaturated oligogalacturonides, while other pectinases act
sequentially to degrade the pectin molecule completely. Pectin
esterases are found in plants, plant pathogenic bacteria, and fungi [5], while PGs are widely distributed among fungi,
bacteria, and many yeasts [6]. They are also found
in higher plants and some plant parasitic nematodes. Pectate lyases are
mostly produced by bacteria.
The isolation and characterization of pectolytic enzymes is well documented. Numerous studies on fungal pectolytic enzymes have been carried out and several fungal PNL genes have been isolated and characterized from Aspergillus niger [7-9], A. oryzae [10], and Glomerella cingulata [11]. Pectinases have extensive applications in extraction, clarification, and cloud stabilization of fruit juices, in degumming and retting of natural fibers (ramie, hemp, flax, bast), maceration of plant tissues, isolation of protoplasts, and saccharification of biomass [12-17]. A number of PEs have been purified and biochemically characterized [18].
Crystal structures of pectin lyase A (PNLA) from two strains of A. niger, N400 and 4M-147 [19], reveal that PNLA folds into a parallel β-sheet and shares many of the structural features of PL despite not more than 17% sequence identity after pair-wise structure-based alignment. These shared structural features include amino acid stacks and an asparagine ladder. The substrate-binding clefts of these two PNLs are dominated by aromatic residues and are enveloped by negative electrostatic potential. The major difference between these two PNLA structures is in the conformation of the loop formed by residues 182-187. These observed differences are due to the different pH values of crystallization. The three-dimensional structure of pectin lyase B (PNLB) from A. niger has also been determined by crystallographic techniques at resolution of 1.7 Å [20].
In this communication, an attempt has been made to investigate the protein sequences of PNLs from different source organisms along with protein sequences of PNL, PL, PG, and PE common source organisms using various bioinformatics tools to reveal the sequence level similarity. Further in silico domain analysis of these sequences can provide insight into possible functions associated with the existing active site of the enzyme, which might be a target for genetic manipulation for enhanced activity of the enzyme. The multiple sequence alignment of different PNL protein sequences from different organisms can provide us an opportunity to design degenerate primers for PCR amplification of PNL gene family, which can further be used for cloning and overexpression.
MATERIALS AND METHODS
All the sequences of PNL, PL, PG, and PE of different source organisms available in GenBank were downloaded from NCBI (http://www.ncbi.nlm.nih.gov/). A total of 494, 717, 937, and 172 protein sequences of PNL, PL, PG, and PE, respectively, representing major groups of pectinases were downloaded. Only the full-length protein sequences were considered for in silico analysis.
The programs Clustalw [21] and Seaview (http://pbil.univlyon1.fr/software/seaview.html) were used for multiple sequence alignment. Mega 3.1 [22] was used for dendrogram construction by Neighbor-Joining (NJ) [23], Minimum Evolution, and UPGMA methods [24]. For domain search, the Pfam site (http://www.sanger.ac.uk/Software/Pfam/search.shtml) was used. Domain analysis was done using MEME (http://meme.sdsc.edu/meme/meme.html). The conserved protein motifs deduced by MEME were subjected to biological functional analysis using protein BLAST, and domains were studied with Interproscan providing the best possible match based on highest similarity score. The accession numbers of PNL protein sequences along with the source are listed in Table 1, while the accession numbers of PNL, PL, PE, and PG with common source are listed in Table 2.
Table 1. List of pectin lyase protein
sequences reported from different sources

Table 2. List of protein sequences of
pectinases reported from common source organisms
RESULTS AND DISCUSSION
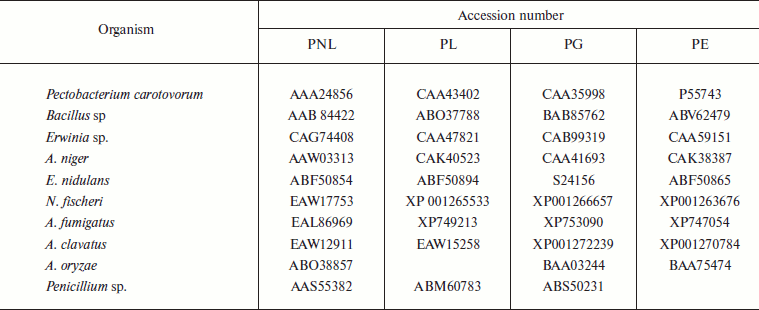
A total of 48 full-length protein sequences of pectin lyase enzyme from different source organisms along with two synthetic sequences were considered for in silico analysis. The phylogenetic tree constructed revealed two distinct clusters for bacterial and fungal PNLs, while multiple accessions of Aspergillus, Pseudomonas, Penicillium, Erwinia, Bacillus, Emericella, Geobacillus, Neosartorya accessions formed distinct clusters showing sequence level similarity (Fig. 1a). Two synthetic sequences (accession numbers CAA01023 and CAA01024) were closely related to A. niger PNL (Fig. 1a). The phylogenetic tree constructed by NJ [23], Minimum Evolution, and UPGMA [24] methods revealed a more or less similar pattern (see Supplement 1 in the PDF version of the article). This clearly indicates the existence of sequence level similarity of PNLs produced from common source organisms. When protein sequence of pectinases produced from common sources (A. niger, A. fumigatus, A. clavatus, A. oryzae, Penicillium sp., Erwinia sp., Bacillus sp., E. nidulans, N. fischeri, and P. carotovorum) were subjected to phylogenetic tree construction using NJ (Fig. 1b), Minimum Evolution, and UPGMA methods (Supplement 1), four distinct clusters of PNL, PL, PG, and PE groups of enzymes were observed.
Fig. 1. a) Phylogenetic trees of PNL sequences of different sources constructed by the NJ method.
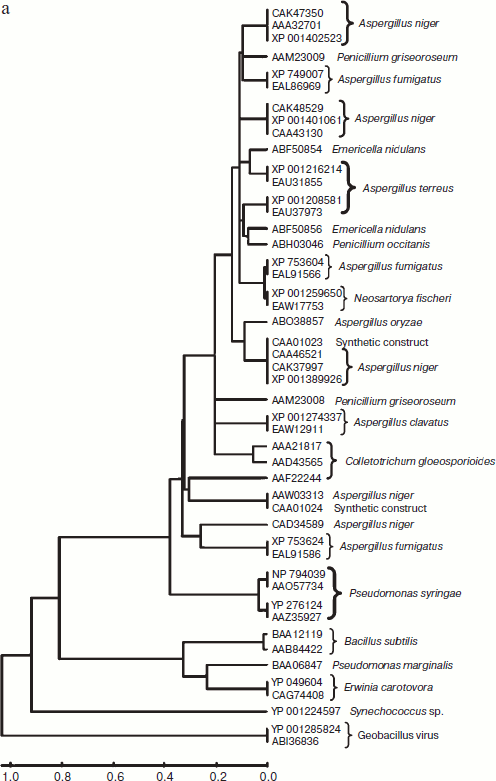
The multiple sequence alignment of 48 protein sequences of PNL proteins revealed a stretch of conserved protein sequences from residues 553 to 679 and from 680 to 806 (see Supplement 2 in the PDF version of the article). Similarly, when protein sequences of pectinases were subjected to multiple sequence alignment, homology was observed at two different locations — from residue 317 to 440 and from 553 to 676 (Supplement 2).Fig. 1. b) Phylogenetic trees of pectinases representing common source organisms constructed by the NJ method.
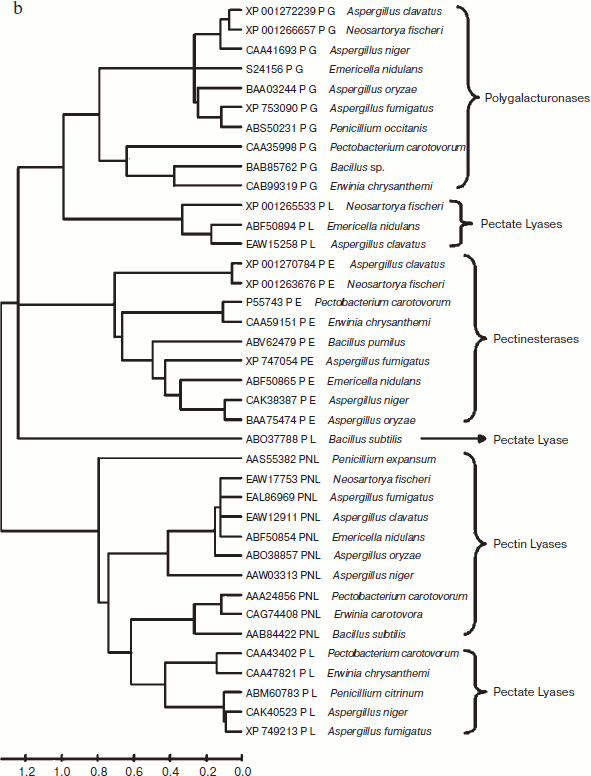
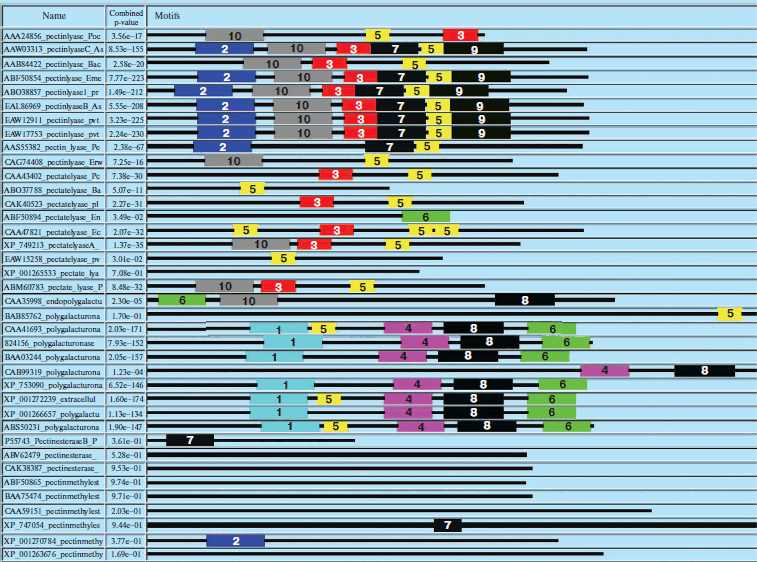
Domain analysis [25, 26] of PNL protein sequences revealed the presence of Pec_Lyase_C domain consistently, irrespective of sources, except YP_001224597 (Synechococcus sp.), ABI36836 (G. virus), and YP_001285824 (G. virus). The ten motifs frequently observed in PNL protein sequences along with the stretch of amino acids and its width is provided (Fig. 2; see color insert). A set of 41 amino acid residues, i.e. TYDNAGVLPITVNSNKSLIGEGSKGVIKGKGLRIVSGAKNI, involved in formation of a right-handed β-helix structure is associated with the Pec_Lyase_C domain (Table 3), which is indicative of its important structural role. The presence and importance of right-handed β-helix in PNLs and PLs has been proved by many workers from time to time by solving crystal structures of PNLA [19], PNLB [20] from A. niger, as well as pectate lyase C [27], pectate lyase E [28] (both from E. chrysanthemi), and also PL crystal structure of B. subtilis [29]. This might explain the stability of the enzyme in hostile extracellular environment during plant virulence [30]. Although the mechanism of pectic cleavage differs for the hydrolases (i.e. PG) and the lyases (i.e. PNL and PL), the substrate binding sites are found within a cleft formed on the exterior of the parallel β-helix fold in both cases [30].
Table 3. Different motifs commonly observed in PNL sequences with best possible match amino acid sequences (see Fig. 2)Fig. 2. Motif analysis of PNL sequences using MEME.
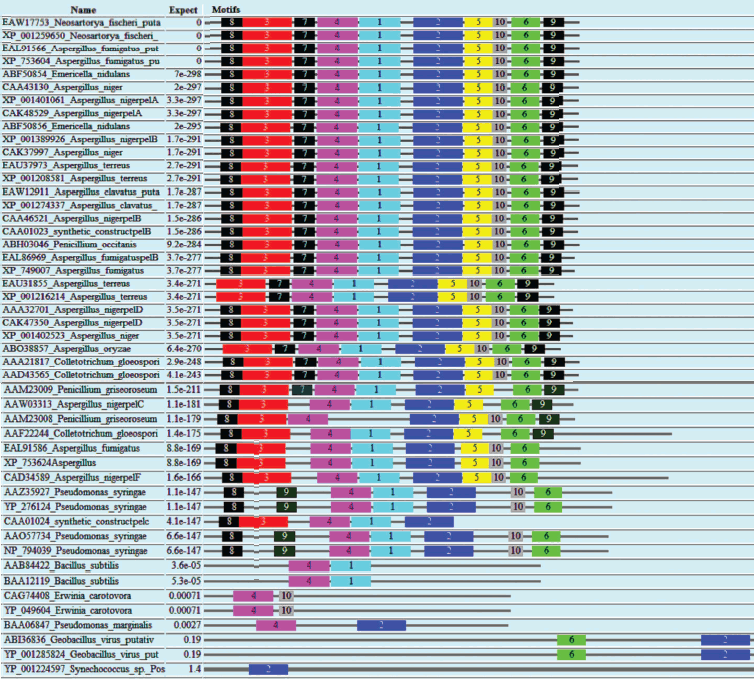

Similarly, domain analysis of PNL, PL, PE, and PG protein sequences from common sources revealed the presence of different conserved sequences. The domain Pec_Lyase_C was frequently observed with PNL and PL belonging to the same group of enzymes, i.e. trans-eliminases. In the case of PGs belonging to glycosyl hydrolase family associated with cell wall metabolism, a domain Glyco_hydro_28 was frequently observed. The ten most frequently observed motifs along with signature protein sequences of maximum matches are shown in Fig. 3 (see color insert) and Table 4. In case of PEs, frequently observed domain represents a member of the Pectate lyase-like β-helix clan. The presence of common and unique domains among different pectinases might confer its structural flexibility, which directly influences its catalytic activity.
Table 4. Different motifs commonly observed in different pectinase protein sequences with best possible match amino acid sequences (see Fig. 3)Fig. 3. Motif analysis of pectinase sequences using MEME.

In silico analysis of PNL protein sequences and its comparison with other pectinases has revealed the sequence-based similarity existing among different pectinases and clustering in distinct groups based on its source organisms and nature of the mechanism of enzymatic activity. In silico domain analysis confirms the existence of the different groups of pectinases based on the presence of unique domains, like PLs and PNLs having a common domain Pec_Lyase_C belong to a common group, i.e. trans-eliminases. While PGs having unique domain Glyco_hydro_28 belong to hydrolases, and PEs with Pectate lyase-like b-helix clan domain belong to the esterase group. The presence and absence of specific domains has direct relation with the structural and functional organization of different groups of pectinases.
The authors wish to acknowledge the support of DBT funded Sub-DIC Bioinformatics Center, Department of Molecular Biology and Genetic Engineering, College of Basic Sciences and Humanities, G. B. Pant University of Agriculture and Technology, Pantnagar for providing software and tools utilized in the present study. P.K.Y. and S.Y. are grateful to the Council of Scientific and Industrial Research (CSIR) and Department of Science and Technology (DST), Government of India, for providing the financial support in the form of Senior Research Fellowship and Women Scientists Fellowship, respectively.
REFERENCES
1.Yadav, S., Yadav, P. K., Yadav, D., and Yadav, K.
D. S. (2009) Process Biochem., 44, 1-10.
2.Visser, J., Bussink, H. J., and Witteveen, C.
(2004) in Gene Expression in Recombinant Microorganisms (Smith,
A., ed.) Marcel Dekker, Inc., New York, pp. 241-306.
3.Cosgrove, D. J. (1997) Ann. Rev. Cell. Dev.
Biol., 13, 171-201.
4.Prade, R. A., Zohan, D., Ayoubi, P., and Mort, A.
J. (1999) Biotechnol. Gene Eng. Rev., 16, 361-391.
5.Jayani, S. R., Saxena, S., and Gupta, R. (2005)
Process Biochem., 40, 2931-2944.
6.Luh, B. S., and Phaff, H. J. (1951) Arch.
Biochem. Biophys., 33, 212-227.
7.Gysler, C., Harmsen, J. A. M., Kester, H. C. M.,
Visser, J., and Heim, J. (1990) Gene, 89,
101-108.
8.Someren, M. A. K., Harmsen, J. A. M., Kester, H. C.
M., and Visser, J. (1991) Curr. Gen., 20, 293-299.
9.Someren, M. A. K., Flipphi, M., de Graaff, L., van
den Broeck, H., Kester, H., Hinnen, A., and Visser, J. (1992) Mol.
Gen. Genom., 234, 113-120.
10.Kitamoto, N., Yasuda, Y. S., Ohmiya, K., and
Tsukagoshi, N. (2001) Biosci. Biotechnol. Biochem., 65,
209-212.
11.Templeton, M. D., Sharrock, K. R., Bowen, J. K.,
Crowhurst, R. N., and Rikkerink, E. H. (1994) Gene, 142,
141-146.
12.Kilara, A. (1982) Process Biochem.,
23, 35-41.
13.Naidu, G. S. N., and Panda, T. (1998)
Bioprocess. Eng., 9, 355-361.
14.Alkorta, I., Garbisu, G., Llama, M. J., and
Serra, J. L. (1998) Process Biochem., 33,
21-28.
15.Blanco, P., Sieiro, C., Reboredo, N. M., and
Villa, T. G. (1997) Arch. Microbiol., 167, 284-288.
16.Takebe, I., Otsuki, Y., and Aoki, S. (1968)
Plant Cell Physiol., 9, 115-124.
17.Beldman, G., Rombouts, F. M., Voragen, A. G. J.,
and Pilnik, W. (1984) Enz. Microb. Technol., 6,
503-507.
18.Whitaker, J. R. (1991) in Microbial Enzymes
and Biotechnology (Fogarty, W. M., and Kelly, C. T., eds.) Elsevier
Applied Science, London-New York, pp. 133-175.
19.Mayans, O., Scott, M., Connerton, I., Gravesen,
T., Benen, J., Visser, J., Pickersgill, R., and Jenkins, J. (1997)
Structure, 5, 677-689.
20.Vitali, J., Schick, B., Kester, H. C. M., Visser,
J., and Jurnak, F. (1998) Plant Physiol., 116,
69-80.
21.Lassmann, T., and Sonnhammer, E. L. (2006)
Nucleic Acids Res., 34 (Web Server Issue),
W596-9.
22.Kumar, S., Tamura, K., and Nei, M. (2004)
Brief. Bioinformatics, 5, 150-163.
23.Saitou, N., and Nei, M. (1987) Mol. Biol.
Evol., 4, 406-425.
24.Shi, G. Y., Jie, T. Y., Bing, T. H., Hua, W. K.,
and Wei, C. K. (2007) Chin. J. Agric. Biotechnol., 4,
33-38.
25.Timothy, L. B., and Gribskov, M. (1997) J.
Comp. Biol., 4, 45-59.
26.Timothy, L. B., and Gribskov, M. (1998)
Bioinformatics, 14, 48-54.
27.Yoder, M. D., Keen, N. T., and Jurnak, F. (1993)
Science, 260, 1503-1507.
28.Lietzke, S. E., Yoder, M. D., Keen, N. T., and
Jurnak, F. (1994) Plant Physiol., 106,
849-862.
29.Pickersgill, R., Jenkins, J., Harris, G., Nasser,
W., and Robert-Baudouy, J. (1994) Nat. Struct. Biol., 1,
717-723.
30.Herron, S. R., Benen, J. A. E., Scavetta, R. D.,
Visser, J., and Jurnak, F. (2000) Proc. Natl. Acad. Sci. USA,
97, 8762-8769.