REVIEW: Free Radicals and Cell Chemiluminescence
Yu. A. Vladimirov* and E. V. Proskurnina
Faculty of Basic Medicine, Lomonosov Moscow State University, Lomonosovsky pr. 31/5, 117192 Moscow, Russia; E-mail: yuvlad@mail.ru; proskurnina@fbm.msu.ru; proskurnina@gmail.com* To whom correspondence should be addressed.
Received January 11, 2009
Application of chemiluminescence (CL) for study of free-radical reactions in human and animal cells and tissues is considered in this review. Historically, the study of intrinsic (ultraweak) luminescence gave place to the measurement of CL in the presence of chemical activators (CL probes) and physical activators (sensitizers) of luminescence, which made the method much more sensitive and specific. The methods of CL and EPR are direct methods of radical investigation, though the advantage of the CL method consists in the fact that CL intensity is directly proportional to a steady-state concentration of the radicals responsible for luminescence (first of all, lipid and oxygen radicals) irrespective the activity of these radicals. The mechanisms of CL reactions in the absence of activators and in the presence of luminol and lucigenin are considered. Examples of various applications of the CL method in medical, biological, and clinical investigations are given including those for estimation of the phagocytic activity of cells, antioxidant activity, determination of toxicity, and other purposes.
KEY WORDS: chemiluminescence, reactive oxygen species, luminol, lucigenin, cell luminescence, lipid peroxidationDOI: 10.1134/S0006297909130082
Abbreviations: CL, chemiluminescence; CL probe, chemiluminescent probe (a chemical compound that enters into a CL reaction with active molecules: free radicals, peroxides, hypochlorite, etc.); CL reaction, chemiluminescence reaction (a chemical reaction accompanied by light radiation – in visible, UV, or near IR spectral region); Coum-CL, isoquinolizine coumarin C-525-enhanced chemiluminescence; EPR, electron paramagnetic resonance spectroscopy; ICL, intrinsic chemiluminescence (ultraweak luminescence); LCL, chemiluminescence in the presence of luminol; LPO, free-radical chain (peroxide) lipid oxidation; Luc-CL, chemiluminescence in the presence of lucigenin; ROS, reactive oxygen species; SAR, superoxide anion-radical (·OO–); TAR, total antioxidant reactivity; TRAP, total reactive antioxidant potentials.
INTRODUCTION
Mitogenetic Rays and Ultraweak Luminescence
The idea that living human and animal cells could radiate weak light in the UV region of the spectrum was first formulated by a Russian scientist Alexander Gavrilovich Gurvich who called it mitogenetic radiation [1, 2]. Gurvich’s experiments were based on the recording of radiation by its action on biological objects, which were called biological detectors including budding yeast and dividing cells. The use of a sensitive physical detector, a photomultiplier cooled with carbon dioxide snow [3-5] or liquid nitrogen [6] revealed luminescence of plantlets [3-5], animal cells and tissues [6-9], ground tissue fragments, and isolated mitochondria [10-13]. The luminescence of all those objects was called ultraweak chemiluminescence [14]. The works by Robert Allen, who discovered the ultraweak chemiluminescence of bacteria-induced human blood leukocytes in 1973 [15, 16] and proposed luminol as activator of macrophage chemiluminescence [17], became an important stage in this series of experiments.
Free Radicals
In all cited instances, luminescence was associated with production and transformation of free radicals [11, 18, 19], which are worthy of mention.
Primary and secondary radicals. The main source of all radicals in normally functioning human and animal cells is the reaction of one-electron reduction of molecular oxygen, which brings about production of superoxide anion-radical ·OO– (SAR). This radical is generated by either the NADPH-oxidase complex of cytoplasmic membrane or endoplasmic reticulum membranes [20, 21], or the respiratory chain of the inner membrane of mitochondria [22]. The other radical, not less significant for the life of cells, is nitrogen monoxide NO, generated by NO-synthase. Both radicals are generated by enzymatic systems and are called primary radicals [23].
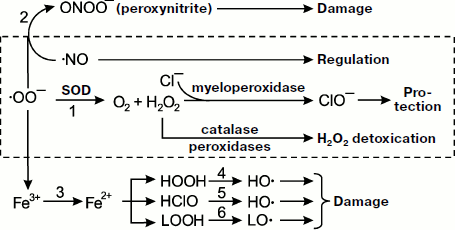
All radicals are highly reactive, so primary radicals quickly transform into molecular products (see the scheme in Fig. 1). The special enzyme superoxide dismutase (SOD) transforms SAR to hydrogen peroxide HOOH (reaction 1). In the presence of SAR, NO reacts with it to form the toxic ion peroxynitrite ONOO– (reaction 2). Superoxide is able to reduce trivalent iron stored in ferritin or contained in iron-sulfur complexes of electron transfer chains to bivalent iron (reaction 3), and it occurs under conditions unfavorable for the cell [24, 25]. Bivalent iron reacts well with HOOH or hypochlorite producing the extremely reactive hydroxyl radical, ·OH (reactions 4 and 5) [26], and is able to branch lipid oxidation chains reacting with lipohydroperoxides (reaction 6). Hydroxyl radical can trigger the process of lipid peroxidation and generation of lipid radicals. The result of all these reactions is formation of a set of quite aggressive compounds in cells (·OO–, HOOH, ·OH, etc.) that are called reactive oxygen species (ROS) [27] to which hypochlorite ClO– is sometimes referred and reactive nitrogen species associated with NO transformation, mainly higher nitrogen oxides and peroxynitrite. Hydroxyl and lipid radicals are called secondary radicals [23].
Reactive oxygen species and oxidative stress. In a number of papers referring to the terms “reactive oxygen species” (ROS) and “oxidative stress” in the international database PubMed (http://www.ncbi.nlm.nih.gov/sites/entrez), these terms are used in research medicine literature as often as the terms “radicals” and “chemiluminescence” (for December 3, 2008, the number of papers was 40112, 60470, 37014, and 44951, respectively) though they do not possess the specificity.Fig. 1. Metabolism of primary radicals (see explanations in the text).
Various researchers include in the concept of ROS a different number of compounds somehow related to the primary formation of superoxide radical. In a well-known monograph by Halliwell and Gutteridge, the following compounds are quoted as ROS: superoxide anion-radical ·OO–, hydrogen peroxide HOOH, hydroxyl radical ·OH, peroxyl radicals ROO·, alkoxyl radicals RO·, hydroperoxyl radical HOO·, hypochlorite ClO–, O3, singlet oxygen 1O2, peroxynitrite ONOO– [28]. Such a list does not seem ideal. Superoxide, hydroxyl radical, hydrogen peroxide, singlet oxygen, and ozone can undoubtedly be considered as ROS. Hypochlorite can also be called a reactive chlorine species. Peroxynitrite, as well as the initial radical – nitrogen monoxide, and numerous radical and nonradical species of higher nitrogen oxides should rather be called reactive nitrogen species and not ROS. Finally, the participants and products of chain lipid oxidation, alkyl radicals L·, alkoxyl LO·, and peroxyl LOO· differ significantly from the group of purely oxygen ROS by localization, formation, and action mechanisms and rather deserve the name “reactive lipid species”, though the term lipid radicals seems to be preferable. In any case, the general notion of ROS is rather convenient though quite indistinct, and shades the fact that different representatives of this group by no means possess identical properties, carry out different functions in the cell, and their proportion can be quite different in different situations.
The term oxidative stress was introduced by Helmut Sies in 1991 and officially entered the Mesh PubMed dictionary in 1995. According to the determination given in PubMed, oxidative stress is an imbalance between pro- and antioxidants in favor of prooxidants, which can bring about damage. Oxidative stress manifests itself in the accumulation of damaged DNA bases, protein oxidation and lipid peroxidation products, as well as in a decreased level of antioxidants and the associated increased susceptibility of membrane lipids and lipoproteins to the action of prooxidants including Fe2+ and H2O2. The ambiguity of the term “oxidative stress” is connected with the fact that either both notions – prooxidants and antioxidants – are quite vague, or it is not clear when the balance transforms to an imbalance.
Nevertheless, in specific situations the notions of ROS and oxidative stress can describe the situation more clearly. For instance, on initiation of phagocytes, by ROS are generally implied all the released reactive oxygen, nitrogen and chlorine species, lipid radicals, and hydroperoxides, and by oxidative stress is meant damage of other cells by these products.
Chemiluminescence as a Method of Radical Detection
An immediate chemical analysis of radicals is impossible since, unlike ordinary molecules, they can neither be isolated nor purified due to their very high reactivity. Stable molecular products of the reactions with the participation of radicals are commonly determined. In this review, neither the methods of determination of these markers, nor those based on the inhibition of the processes by radical quenchers, such as phenol antioxidants (for hydroxyl and lipid radicals and other organic molecules) or antioxidant enzymes (superoxide dismutase for SAR and catalase for H2O2) [29], are considered. These methods are often quite effective but do not enable immediate determination of the nature or concentration of radicals.
A direct method of analyzing radicals is the method of electron paramagnetic resonance (EPR). In principle, it enables not only detection but also identification of many radicals by analyzing the hyperfine structure of the EPR signals. Nevertheless, in biological systems, it often turns out to be insufficiently sensitive due to an extremely low steady-state concentration of radicals in cells and tissues. For instance, it became possible to detect radicals generated during the interaction between Fe2+ and lipid hydroperoxides immediately with the EPR method only in a flow system with a great flow rate of reagents [30] or using spin traps [31]. The traps can, however, effect the biochemical reactions proceeding in the system or decompose in the course of some of them. The method of chemiluminescence (CL) offers such an advantage that, first, it is not generally associated with changes in the course of the processes in solutions, cells, or even whole tissues in which luminescence is recorded, and second, it is very sensitive in detecting particularly highly reactive radicals.
The fact is that no radical concentration is immediately determined with the chemiluminescence method but the rate of the reaction in which they are produced. In the very general case, the reaction in which radical generation brings about CL can be presented by the following scheme:
![]()
ICL = K·ke[P*], (1)
where K is a coefficient characterizing the sensitivity of the device to radiation with particular quantum yield and spectrum. Due to a high rate of radical (R·) transformation reactions, a stable state sets in instantly in the system in which the rates of all successive reactions are equal. It is now clear that the intensity of CL is proportional to the rate of radical formation v1:
ICL = K·ke[P*] = K·v1. (2)
It is seen that, under other equal conditions, there exists a direct relation between the radical formation rate and radical steady-state concentration, since
ICL = K·v1 = K·k2[R·]. (3)
Thus, the CL method reflects radical concentration in the system along with the methods of spectrophotometry and EPR though, unlike EPR, the indications of a chemiluminometer do not depend on radical reactivity.
In this scheme, this value is determined by the radical R· disappearance reaction rate constant k2. The steady-state radical concentration is determined by the following equation:
K·v1 = K·k2[R·], whence [R·] = v1/k2. (4)
It is the steady-state concentration of a substance (in this case that of radicals [R·]) that is determined with the EPR method as well as with other spectroscopic or fluorometric methods. With increasing radical reactivity, i.e. with the growth of k2, [R·] value decreases, and the recorded signal decreases as well. Even if many active radicals are generated, they will not be observed due to high k2 values. On the contrary, CL intensity does not depend on radical reactivity since, as the radical reactivity increases, the steady-state radical concentration decreases simultaneously and to the same extent, their product remains constant, and, moreover, no changes in the CL intensity take place (Eq. (3)). In other words, the CL method records even the most active radicals, whose concentration in the system under study can be vanishingly small, and this is in what its uniqueness and advantage over other methods of radical detection in chemical and biological systems consists. The more active is a radical, the more difficult is to detect it directly with the EPR method, and no such restriction exists for CL.
Chemiluminescence Measurement Instrumentation
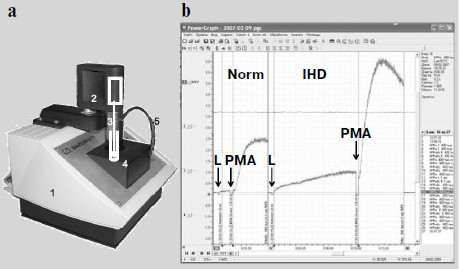
The time when a photomultiplier had to be cooled with carbon dioxide ice [3-5] or liquid nitrogen [6-12] when measuring weak luminescence seems to have become a thing of the past. Modern photomultipliers and current methods of electronic signal processing enable recording of even cell and tissue intrinsic luminescence, not to mention enhanced CL. However, an insufficient number of specialized devices meant for direct recording of the kinetics of weak CL accompanying free-radical formation are produced. A portable chemiluminometer has recently been developed in our laboratory in common with a small-scale enterprise (InterOptika, Russia); it possesses sufficient sensitivity and is provided with the modern data processing program PowerGraph (Fig. 2).
Intrinsic and Activated ChemiluminescenceFig. 2. Lum-5773 portable chemiluminometer (http://www.powergraph.ru/hard/lum.asp). a) Appearance of the device: 1) case; 2) electric motor for a stirrer; 3) stirrer; 4) cuvette inside a light-isolated thermostat; 5) light-isolated tube for introduction of solutions in the course of measurement. The receiving device is under the cuvette. b) Computer screen image showing the recorded chemiluminescence curve of phagocytes prepared from the monocytes of a healthy donor (left) and a patient with ischemic heart disease (IHD) (right). The moments of luminol (L) and phorbol myristate acetate (PMA) addition are shown by the arrows. The curves are recorded and processed with the PowerGraph program (http://www.powergraph.ru/hard/lum.asp). Measurements are conducted together with Bilenko and Pavlova [225].
The discovery of an ultraweak, or better to say intrinsic, cell and tissue chemiluminescence (ICL) [6-9] initiated a series of investigations both in the USSR (see monographs [11, 32, 33]) and abroad [14, 34-46], since its application gave a new stimulus to the study of free radicals in the life of cells and development of pathological states [33]. However, the method has two shortcomings [47, 48]. First, due to a low CL intensity, the measurement of ICL requires not only the application of very sensitive and therefore expensive equipment but a great amount of material for analysis, which is very undesirable and often even impossible in biology and medicine. Second, the method is not very specific since the reactions of many active particles (radicals and peroxides) can be responsible for the ultraweak luminescence in biological objects [11, 33, 49].
The way out consists in the use of certain substances called CL enhancers. By the mechanism of action, the enhancers are divided into two distinctly differing groups that can be called chemical and physical enhancers [50].
Chemical CL enhancers, also called CL probes, represent compounds that enter into reactions with ROS or organic free radicals to form molecules of products in their excited electronic state. The luminescence observed in the course of these reactions can be explained by transition of the molecules to the ground state, which results in photon fluorescence [51]:
Enhancer + radicals → product* → product + photon.
Well-known representatives of such enhancers are luminol (5-amino-1,2,3,4-tetrahydro-1,4-phthalazinedione, 3-aminophthalic acid hydrazide) and lucigenin (10,10′-dimethyl-9,9′-biacridinium dinitrate). The schemes of their transformations resulting in luminescence will be considered below.
Physical enhancers do not enter into chemical reactions and have no effect on the course of luminescence-accompanied reactions but, nevertheless, they greatly enhance CL intensity. A physical process of energy transfer (energy migration) from a CL reaction product molecule to an enhancer underlies their action:
Radicals → product* → product + photon 1 (inactivated CL);
Product* + enhancer → product + enhancer* → photon 2 (enhanced CL).
The enhancement of luminescence by an enhancer occurs if the quantum yield of radiation transition (luminescence) is greater in the second case than in the first. The most efficient physical CL enhancer during lipid peroxidation is a coumarin C-525 derivative; its addition to a system where a lipid-peroxidation reaction proceeds enhances luminescence by more than 1500 times [52].
INTRINSIC CHEMILUMINESCENCE OF CELLS
A very weak (“ultraweak”) fluorescence [6, 47] is observed during the interaction between ROS (see literature in [11]), in the reaction of hypochlorite with HOOH, in the interaction of peroxynitrite with proteins [53-55], and in the chain (peroxide) lipid oxidation [33].
Mechanism of Formation of Molecules in Electron-Excited State
The luminescence accompanying chemical reactions is called CL. The presence of such a luminescence means that the energy released at one of the stages of a chemical process proceeding in the system is sufficient to form one of the reaction products in its electron-excited state:
A + B → P* + other products (chemiluminescence reaction);
P* → P + hν (luminescence).
The spectrum of CL and the quantum yield Qlum of the second reaction are the spectrum and quantum yield of the ordinary photoluminescence of the product. In most biochemical reactions, the products have a very low (near zero) quantum yield of photoluminescence. Formation of excited molecules in CL reactions Qex, however, also has a low yield as most of the molecules in the chemical reactions proceeding in aqueous medium at normal temperatures are not generated in the excited state but in the ground electronic state: A + B → P. The total quantum yield of CL, QCL = Qex·Dlum, in such reactions as chain oxidation of organic molecules (including, evidently, lipid peroxidation) has an order of magnitude of QCL = 10–4·10–4 = 10–8, i.e. it is very small (ultraweak luminescence) [11].
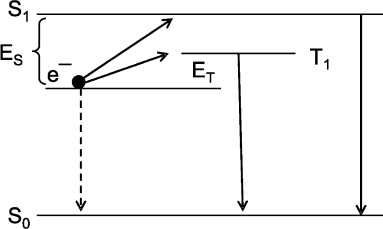
The cause of a low yield of excited product molecules in CL reactions becomes clear from consideration of the scheme of radiative and nonradiative electron transitions in an aromatic amino acid molecule in a simplest CL reaction of recombination of cation-radical AH+ and solvated electron e– generated during the photo-ionization of aromatic amino acids under the action of UV-radiation. The scheme in Fig. 3 shows the electron transitions and is built on the base of investigations of the thermo- and photoinduced luminescence of UV-irradiated samples [11]. The scheme helps to explain the main properties typical of CL reactions: a low quantum yield of CL, summation of CL reaction heat energy and luminescence enhancement energy (the Audubert rule), and the fact that the spectrum of CL in most CL reactions coincides with the spectrum of phosphorescence and not fluorescence of the reaction product [56] (for details see [11, 49]).
Intrinsic Chemiluminescence in Lipid PeroxidationFig. 3. Scheme of electron levels and electron transitions in aromatic amino acids exposed to UV-radiation at 77°K. S0, S1, and T1 are amino acid in the ground state, in the excited singlet state, and in the triplet state; e– is solvated electron produced during amino acid photo-ionization on UV-radiation. Electron transitions e– → S1 → S0 and e– → T1 → S0 are accompanied by photon luminescence. A direct transition e– → S0 is not accompanied by luminescence. The ratio of radiative transition probability to nonradiative transition probability is equal to the quantum yield of (chemi)luminescence. The difference between e– and T1 (or S1) levels is equal to the luminescence activation energy on recombination of solvated electron and cation-radical. It is seen that the activation energy for photon radiation from the triplet state (T1) is considerably lower than the activation energy for radiation during transition through the singlet excited state. For this reason, the probability of radiative transition, i.e. the quantum yield of chemiluminescence, through the triplet state is approximately 100,000 times higher than that on the transition through the singlet state. This scheme also shows that the energy of a photon is equal to the total energy of activation and heat of the recombination reaction of solvated electron and cation-radical (i.e. the reaction of e– → S0 transition). For details see [11], pp. 54-62.
The excited state generated during free radical interaction can be generally described by the following equation of the reaction:
![]()
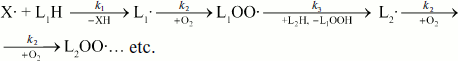
![]()
In these reactions, excited molecules are generated in the interaction of two peroxide radicals. It was shown in special experiments with oxygen-free solutions that formation of only L· and LO· radicals is not sufficient for CL appearance; LOO· radicals generated in the presence of oxygen are necessary [63]. It is most probable that CL in lipoperoxidation is associated with LOO· radical disproportionation:
![]()
This mechanism is proved in the reactions of hydrocarbon chain oxidation that are also accompanied by CL (see bibliography in [11]). Tetroxide is evidently generated first:
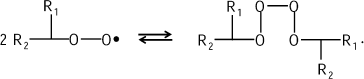
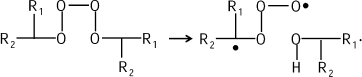
![]()
Recording of CL during chain oxidation enables estimation of the kinetics of changes in the steady-state concentration of free radicals in the system in time and, moreover, measurement of LPO reaction rate at each certain instant in time. The relation between radical concentration LOO· and luminescence intensity ICL can be found as follows from the reaction equation (see above):
ICL = QCLk6[LOO·]2. (5)
Here, ICL stands for the total CL luminous flux in all directions and at all wavelengths, Einstein/sec; QCL denotes CL quantum yield.
The rate of the lipid peroxidation process is the rate of formation of hydroperoxide oxidation products in the following reaction:
![]()
![]()
Kinetics of Iron-Induced Chemiluminescence
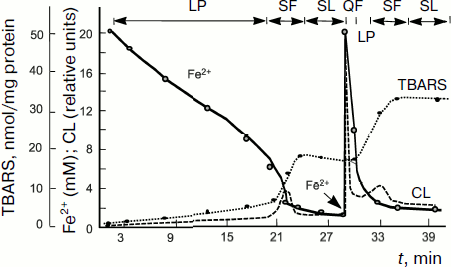
The above-described parallelism between the intensity of intrinsic CL accompanying lipid chain oxidation and chain oxidation rate is clearly revealed in the process of studying of a rather complex CL kinetics in lipid-containing systems including liposomes and mitochondria to the suspension of which some amount of Fe2+ salts is added. A typical CL kinetics in the chain lipid oxidation reaction initiated by divalent iron salts added to the system is shown in Fig. 4 and includes a series of successive stages [77, 78]. After the addition of Fe2+ ions, a quick flash of luminescence is immediately observed, which is related to the decomposition of lipid hydroperoxides if they were contained in the objects, and then it gives place to the luminescence inhibition phase. This stage is due to Fe2+ antioxidant properties at rather high Fe2+ concentrations (of the order of 50 µM in mitochondrial and liposome suspensions). When Fe2+ concentration is reduced to lower than this critical value, Fe2+ ions begin acting as prooxidants due to the chain branching reaction; as a result, the so-called slow flash of luminescence develops. It subsides after full oxidation of Fe2+ to Fe3+. However, the process is not over since it is followed by the so-called stationary luminescence which develops gradually in liposome and mitochondrial suspension and in blood plasma (see Fig. 4) [57].
Investigation of the kinetics of such a CL in combination with the determination of Fe2+ oxidation, oxygen consumption, and mathematical modeling of reactions [79] made it possible to decipher the system of equations of branched chain lipid oxidation, to determine the rate constants of basic reactions, and to study the mechanism of action and activity of different antioxidants, both natural [74, 80-82] and synthetic [71]. These results are considered in detail in reviews [33, 47, 48, 58, 67, 68, 76], and we shall not dwell on them herein.Fig. 4. Kinetics of oxidation of Fe2+ ions (solid line), production of lipid peroxidation products (thiobarbituric acid reactive substances, TBARS) (dotted line), and chemiluminescence (dashed line) in a suspension of mitochondria to which 0.2 mM Fe2+ is added (the moment of introduction is shown by the arrow) [57]. Incubation mixture composition: 1 ml of a mitochondrial suspension in 10 ml of 105 mM NaCl and 20 mM Tris-buffer, pH 7.5; t = 20°C. LP, latent period; QF, quick flash; SF, slow flash; SL, stationary luminescence.
Intrinsic Chemiluminescence (Ultraweak Luminescence) of Tissues and Cells
As is has already been mentioned, the ultraweak luminescence of tissues and cells was first described in 1959-61 [7, 8, 11].
A series of investigations on the ICL of mitochondria, homogenates, and a rat liver suspension was carried out in 1964-66, in which a distinct correlation between the level of intrinsic luminescence and accumulation of lipid peroxidation products reacting with 2-thiobarbituric acid [10, 12, 83-85] was revealed. It was also shown that the presence of Fe2+ admixtures greatly initiated luminescence and lipid peroxidation, while chelator EDTA eliminated them completely. At the same time, a relationship between metabolism and ICL was revealed: ICL was inhibited by amytal and cyanide. All these data summarized in the monograph of 1972 [33] suggested that luminescence was related to lipid peroxidation in cells and isolated mitochondria, and that the key role in activation of this process belonged to Fe2+ ions (see also [57]).
However, chain lipid oxidation is not the only possible source of the intrinsic CL of animal cells and tissues. It was shown in a large series of studies that the reactions with the participation of oxygen radicals were also accompanied with CL, probably as the result of singlet oxygen production (see bibliography in [11]). The intensity of that luminescence, however, was generally much lower than that in lipoperoxidation reactions. One more constituent of cell and tissue “ultraweak luminescence” was described in the experiments by the group headed by Turrens in perfused lung and model systems [53, 86], and it was associated with NO biosynthesis. On the other hand, it was shown in paper [87] that CL was observed when protein interacted with peroxynitrite. Under the action of peroxynitrite on individual amino acids, the most intensive CL was obtained with tryptophan and a little lower one with phenylalanine [86]. It could be thought that generation of excited molecules took place under tryptophan oxidation. In the experiments in perfused lung, NO-dependent ICL was observed to be normal, while during inflammatory processes in lung the main constituent was CL associated with lipid peroxidation [88].
The intrinsic luminescence of initiated phagocytes was discovered in 1972 [15, 16]. The dynamics of CL development in a macrophage suspension is shown in Fig. 2b. When a suspension of phagocytizing cells is placed into a cuvette, a relatively weak background CL is observed, but after the addition of a stimulus initiating the production of superoxide radical and other ROS, CL flash development is observed (CL-response).
It should be admitted that intrinsic CL is at present seldom used for studying radical production by cells because of weakness of the luminescence. After the studies by Allen and Loose of 1976 [17] in which luminol was used as an activator of phagocyte CL, the LCL (chemiluminescence in the presence of luminol) method practically displaced the ICL analysis method. Normally, the addition of luminol enhances the CL intensity of cells by more than 1000 times, and it means that the amount of biological material taken for analysis can be 1000 times less.
Nevertheless, is should be kept in mind that the information given by intrinsic CL and LCL is not identical as the intensity of LCL is determined mainly by the concentration of hypochlorite + H2O2 or that of hydroxyl radicals + superoxide, while ICL normally depends on the rate of lipid peroxidation in the system. The papers by Klebanov et al. can be mentioned as a study where intrinsic CL was successfully used. In those papers, activation of lipid peroxidation of blood plasma low-density lipoproteins by initiated neutrophils was shown with the methods of TBARS analysis (the analysis of thiobarbituric acid reactive products) of lipid peroxidation [89] and intrinsic CL [90]. The activation was performed with water-soluble ROS penetrating through a dialysis membrane, but only in the presence of Fe3+ ions in the water-soluble form (in complex with ADP). The sequence of events in this case can be presented as follows:
1) Initiation of cells by zymosan → ·OO– release → HOOH generation;
2) Fe3+ + ·OO– → Fe2+ + O2;
3) HOOH + Fe2+ → Fe3+ + HO– + ·OH → chain lipid oxidation.
The argument in favor of such a scheme is that LPO activation was abrogated by both superoxide dismutase (inhibition of stage 2) and catalase (inhibition of reaction 3).
There exist many data that the described events are of great importance for production of oxidized forms of blood plasma lipoproteins, and this is, in its turn, very important for development of atherosclerosis (see reviews [91-94]).
LUMINOL-DEPENDENT CHEMILUMINESCENCE OF CELLS
Measurement of luminol-dependent CL of blood cells is widely used for study of the functional state of the immunity phagocytic link.
The appearance and interaction of a foreign material with the phagocytizing cell triggers a complex cascade of physiological and metabolic processes which starts with the interaction of cell surface receptors with ligands of different kind including immune complexes, and ends with the formation of superoxide radicals during the transfer of an electron from NADPH to O2, metabolism of these radicals, breakdown of a bacterial cell, and initiation of the biosynthesis of numerous physiologically active compounds in phagocytes (see reviews [20, 21, 95, 96]). LCL arises as the result of oxidation of luminol by reactive oxygen and chlorine species, and interaction of oxidized forms of luminol with superoxide radical or hydrogen peroxide. To comprehend information obtained from LCL investigations, it would be useful to consider the mechanism of these reactions in more detail.
Mechanism of Reactions Responsible for Luminol Luminescence
Literature data about the mechanism of the reactions responsible for luminol luminescence are generalized in the article by Roshchupkin et al. who also proposed the schemes of appropriate reactions ([97], Fig. 5). In a simplified form, the process takes in three stages. The first stage is oxidation of luminol by a strong oxidant, for instance, a radical or a variable-valence metal in oxidized form (Fig. 5, reactions 1 and 2):
![]()
·LH + InH → LH2 + In·.
The second and a very important stage in the development of a CL reaction is the appearance of a key hydroperoxide product (4-hydroperoxy-1-oxy-5-aminophthalazin-4-olate) which is generally produced in biochemical systems as the result of interaction of a luminol radical with superoxide (Fig. 5, reaction 3):
![]()
LHOOH– → perhydro acid → endoperoxide-N=NH → endoperoxide-H + N2↑.
At the final stage, the bond between oxygen atoms in the endoperoxide group breaks, which generates a molecule of monoprotonated aminophthalic acid in the electron-excited state with the following light quantum emission (reactions 7 and 8 in Fig. 5).Fig. 5. One-electron luminol oxidation (see explanations in the text).
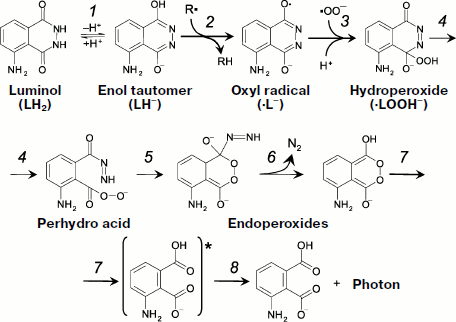
The path of transformation of the hydroperoxide key product to the electron-excited aminophthalic acid (reactions 4-8 in Fig. 5) is one and the same under the action of many oxidants [97, 98], but hydroperoxide production itself can follow different pathways which will be considered below in more detail.
Application of Luminol-Dependent CL for Determination of Total Antioxidant Reactivity
Antioxidants affect the intensity of luminol-dependent CL. It is the basis for a well-known method of analysis of the total content of antioxidants in in vitro experiments with the use of such radical sources as 2,2′-azo-bis(amidinopropane)dihydrochloride (ABAP) [99-102] for water-soluble antioxidants, or 2,2′-azo-bis(2,4-dimethyl-valeronitrile) (AMVN) for hydrophobic antioxidants [103]. In these investigations, the action of antioxidants is manifested either in a decreased CL intensity at a certain stage of the CL response, or in detained CL development the measure of which can be a latent period, or induction period. In the presence of oxygen, a most probable chain of CL-producing reactions looks like this:
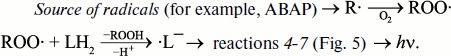
The scheme of CL-producing reactions in such systems is not completely known. Since both superoxide anion-radical and luminol hydroperoxide anion participating in the chain of luminol transformations (Fig. 5) are stable only in alkaline medium, it is not surprising that the quantum yield of luminol-dependent CL increases greatly with increasing pH [99]. Apparently, the fraction of CL-unaccompanied side reactions increases in neutral and acidic medium. In model experiments, media with pH 8.3 to 9.0 and higher are usually applied, which is far from the physiological standard. This fact greatly restricts application of the LCL method in biology.
Luminol as a Method for Determination of Peroxidase Activity
A rather bright CL is observed during the interaction of luminol with peroxidases in the presence of hydrogen peroxide, and its mechanism was investigated with many methods including the stop-flow method [106].
It is known that Complex I is produced on mixing HOOH with horseradish peroxidase (EC I.II.I.7) and transforms to Complex II on standing (see literature in [107]). Literature data on the stoichiometry of those transformations and our own measurements of CL rapid kinetics and light absorption in the system of peroxidase (E), hydrogen peroxide, and luminol (L) at close-to-stoichiometric concentrations enabled Cormier to draw up a scheme of reactions, which is presented below with some changes in designations [106]:
![]()
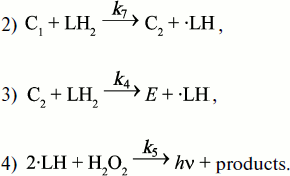
The second-order rate constants of reactions (2) and (3) were determined and were found to be 2.3·106 and 7.2·104 mol–1·sec–1, respectively.
In our investigations, LCL was used to determine the peroxidase activity of cytochrome c–cardiolipin complexes. A relationship was shown between the activity of the complexes and cardiolipin content [59, 61, 62, 108], the presence of antioxidants [109, 110], and nitrogen monoxide, and a reduced activity of the complexes inhibited by NO under the action of laser radiation [111].
Chemiluminescence of Luminol in the Presence of Hypochlorite
Hypochlorite is a natural compound that forms in the reaction catalyzed by the enzyme myeloperoxidase (MPO):
![]()
![]()
Hypochlorite oxidizes luminol (LH2), probably via the stage of its chlorine derivative (LHCl) [97], to aminobenzodiazaquinone (ABDQ) (Fig. 6):
LH2 + HOCl → H2O + LHCl → HCl + L (ABDQ).
In the presence of H2O2, luminol hydroperoxide can be produced, which is a key link of CL reaction [97]:
L + H2O2 → LHOOH.
In biological systems, hypochlorite is formed in a reaction with the participation of H2O2, and thus its appearance brings about without fail the CL whose intensity will, however, be reduced both in the presence of substances reacting with hypochlorite (for instance, amino group-containing compounds) and under the action of catalase (removing H2O2).Fig. 6. Hypochlorite oxidation of luminol (see explanations in the text).
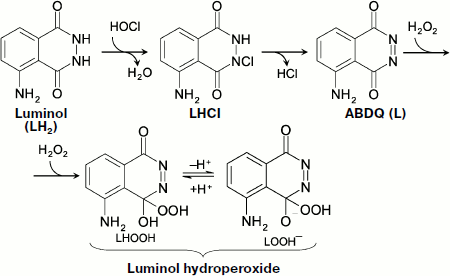
Therefore, an important difference exists between the “hypochlorite” and “radical” ways of initiating luminol-dependent CL: the production of a key compound, luminol hydroperoxide, requires H2O2 in the first case (uninhibited SOD, or catalase-sensitive LCL) and superoxide radical in the second case (inhibited SOD, or SOD-sensitive, catalase-insensitive LCL). It is not easy to apply these criteria in practice because, for example, the removal of H2O2 by catalase can be followed by cessation of the radicals initiating the reaction with luminol, and this way of CL will thus also turn out to be sensitive to the action of catalase.
The known independence of two ways of luminol oxidation is corroborated by the data of the action of different biochemical substances on CL observed in a model luminol-and hypochlorite-containing system [112]. It was shown that the greatest inhibiting activity was demonstrated by thiol compounds: thiourea > cysteine > human serum albumin > reduced glutathione > N-acetyl-L-cysteine (half inhibition in concentrations of (1.2-8.5)·10–7 M). Much less active were amino acids and oxidized glutathione, i.e. amino group-containing compounds (2.5·10–6-3.2·10–5 M). Methionine, ascorbate, taurine, and desferal had an intermediate activity (1.1·10–6-5.3·10–5 M) [112]. This order of activities corresponds to the ability of these compounds to bind hypochlorite, whose concentration in the experiments was 2.5·10–6 M. On the other hand, it was shown in the cited and other papers that quenchers of singlet oxygen and hydroxyl and organic radicals, such as mannitol, glucose, and dimethylurea, had practically no effect on hypochlorite-produced LCL. This confirms the fact that the “hypochlorite way” of luminol CL reaction requires no radical participation. On the other hand, a very effective LCL inhibitor in this system was catalase [112]. In another paper by the same group of authors, it was shown that CL intensity in the system containing hypochlorite (10–6 M) and luminol (5·10–5 M) was directly proportional to the amount of H2O2 added in concentrations of 3·10–9 to 3·10–6 M [113].
Relative Contribution of Two Ways of Luminol Oxidation to Cell Chemiluminescence
The question about a relative role of the two LCL mechanisms is not simple since the contribution of each mechanism depends on the concentrations of all participants of the process: superoxide, H2O2, hydroxyl radicals and other compounds, transient-valence metals, compounds reacting with hypochlorite and antioxidants, as well as on the peroxidase activity in the biological system under study.
One of the most often investigated objects is polymorphonuclear leukocytes (PMNL). The physicochemical basis of the luminol-enhanced CL of initiated (activated) PMNL was studied in the work by Roshchupkin et al. [97]. In particular, during the investigation of the nature of the primary oxidizer of luminol, it was shown that 20 µM luminol-activated CL of phorbol myristate acetate (PMA)-initiated leukocytes showed no changes in the presence of dimethyl sulfoxide (DMSO) in concentrations of 0.02 to 2.6 mM at which it must reveal a specific ability to quench hydroxyl radicals. This indicates that no generation of hydroxyl radicals with the participation of superoxide-anion and myeloperoxidase-synthesized hypochlorite takes place during stimulation of PMNL. At high DMSO concentrations (260 mM), a significant CL reduction was observed due to a direct reaction of DMSO with hypochlorite. On the other hand, exogenous amino acids and taurine in significant concentrations (3-15 mM) weakened CL. It was concluded that CL in the investigated system was due mainly to the initial reaction of hypochlorite with luminol; and the intensity of luminescence was enhanced as the result of oxidation of luminol-transformation products by H2O2.
It is curious that not only hypochlorite but also chloramine, for instance, such as N-chlorophenylalanine, gives rise to luminol CL in the presence of H2O2 [114]. This indicates that the initial stage of luminol oxidation by the two-electron oxidation mechanism (see the scheme in Fig. 6) can be performed by both hypochlorite and chloramines that are produced in living systems on the appearance of hypochlorite. When added to PMNL, chloramines gave rise to a luminescence flash of unstimulated cells (ibid).
Intra- and Extracellular ROS Pools
ROS generation during cell activation (i.e. as the result of a stimulus) is supposed to take place both inside and outside the cell. It should be admitted that it is not easy to prove this statement with the CL methods if a ROS penetrates through cell membranes and thus redistributes among intra- and extracellular pools in the course of CL measurement. Generally speaking, it is possible to measure steady-state ROS concentrations and not the site of their appearance. With this reservation, it should be dwelt on work [115] in which the action of several SH-compounds was investigated, which, as it is known, are active quenchers of ROS and hypochlorite on the luminol-enhanced PMNL chemiluminescence [112]. It is shown that cells themselves are the source of approximately half luminescence, and this luminescence is quenched by penetrating sulfhydryl compounds such as dithiothreitol and N-acetyl cysteine. The other half of luminol luminescence is completely inhibited by nonpenetrating reduced glutathione. This approach seems to be very promising in such investigations.
LCL as a Method for Determination of Myeloperoxidase Activity
With respect to the applicability of the LCL method, a special role among peroxidases belongs to myeloperoxidase (MPO) under whose action luminol undergoes chemical attack not so much by the enzyme as by its substrate, hypochlorite. MPO is a heme-containing enzyme released by stimulated blood granulocytes in sites of inflammation. Hypochlorite generated by this enzyme is missioned to attack bacterial walls, though it can simultaneously damage its own tissues. The properties and mechanisms of action of this enzyme and the CL methods applied for estimation of its activity are considered in review [116].
The method of a specific MPO analysis by luminol-dependent CL in the presence of bromide was introduced in 1999 [117]. In the presence of 0.1 mM luminol, 10 mM H2O2, and 10 mM DTPA (diethylenetriaminepentaacetate), CL was observed in a medium containing neutrophil extract and 5 mM KBr, and was absent in a KCl-containing medium. The elaborated method made it possible to detect MPO at a neutrophil content of above 100 cells in a sample, and was specific since the presence of other peroxidases and hemoproteins made no effect on the results of the analysis.
LUCIGENIN-DEPENDENT CELL CL
Lucigenin-Dependent CL as a Method for Quantitative Determination of Superoxide Radicals
Detection of superoxide radical in living cells is extremely important due to the leading role of this radical in redox signaling inside the cell and in the development of numerous pathological states. Due to a high sensitivity of lucigenin as a probe for superoxide radical [118], lucigenin-activated CL (Luc-CL) was applied for detection of ·OO– in oxidation of xanthine or hypoxanthine by xanthine oxidase [119], by microsomal NADPH cytochrome reductase [119], by NADPH oxidase of phagocyte cells [120, 121], and by diphenyleneiodonium-sensitive NADPH oxidases of endothelial cells, fibroblasts [122], and smooth muscle cells of blood vessel walls [123, 124]. It was also proposed to use Luc-CL in studies on ·OO– generation by mitochondria in intact cells [125, 126]. Such a popularity of the method is clear since the amount of biological material necessary for the ·OO–-induced signal to exceed background 10 times as much was 3750 times less while using Luc-CL than the method of Fridovich based on the reduction of acetylated cytochrome c (Fe3+) by superoxide [119].
However, Faulkner and Fridovich in 1993 [127] and Liochev and Fridovich in 1997 [128] cast doubt on the possibility to use lucigenin as an appropriate method of ·OO– detection on the grounds that, under certain conditions, lucigenin itself can be a source of superoxide radicals in the reaction of its one-electron oxidation by air oxygen. To comprehend the essence of the opened discussion, attention should be drawn to the scheme of the reactions responsible for lucigenin chemiluminescence [129]. The first of these reactions is production of cation-radical (·Luc+) from lucigenin, which is a bivalent cation (Luc2+) in a neutral aqueous solution (see also Fig. 7).
1) Luc2+ + e– → ·Luc+.Fig. 7. Lucigenin chemiluminescent reaction (taken from [129] with minor alterations).
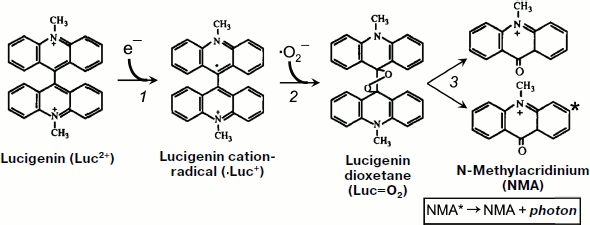
During the interaction of lucigenin cation-radical with superoxide, an unstable compound is produced – lucigenin dioxetane (Luc=O):
2) ·Luc+ + ·OO– → Luc=O.
Two N-methylacridinium molecules are produced in a spontaneous reaction of Luc=O decomposition, one of which turns out to be in the electron-excited state:
3) Luc=O → methylacridinium* + methylacridinium;
4) methylacridinium* → methylacridinium + photon.
It follows from this scheme that the rate of chemiluminescent reaction (3) depends both on ·OO– concentration and on the rate of lucigenin one-electron reduction reaction (1).
Under stationary conditions, the rates of all stages are equal:
k1[Luc2+][e–] = k2[·Luc+][·OO–] = k3[Luc=O] = v. (7)
And
ICL = ηCL·v. (8)
Therefore, the intensity of Luc-CL is proportional both to lucigenin [Luc2+] and superoxide-radical [·OO–] concentration and the rate of lucigenin reduction k1[e–], where [e–] designates the concentration of one-electron reductants in the system and k1 stands for the reaction rate constant.
Luc-CL as a method for determination of ·OO– concentration is subjected to criticism based on the fact that the autooxidation of lucigenin cation-radical by air oxygen can become a source of superoxide radicals [128, 130].
The main reason in favor of such an assumption lies in the fact that cytochrome c reduction in the xanthine/xanthine oxidase system was accelerated by lucigenin, and this acceleration was abrogated by SOD. The development of the events can be presented as the following scheme [118, 128]:
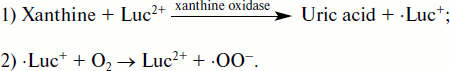
![]()
![]()
If there were no such competition with cytochrome c, “a greater increase in lucigenin-provided superoxide production would have to be observed”, the authors say [130].
However, the conclusion that reaction (2) can make a significant contribution to the total rate of superoxide radical production and, consequently, to Luc-CL became, in its turn, a subject of severe criticism [129, 131]. First of all, it was shown that at low lucigenin concentrations (below 20 µM), at which the contribution of reaction (2) was insignificant, superoxide-radical production was observed in different biochemical systems. A parallelism between three indices of superoxide formation was then noted: (i) Luc-CL intensity, (ii) oxygen consumption rate estimated by polarography, and (iii) production of a specific spin adduct in the reaction of ·OO– with a trap 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide [129]. Lucigenin concentrations varied in the range of 1 to 5 µM, and xanthine + xanthine oxidase, lipoamide dehydrogenase + NADH, isolated mitochondria, mitochondria in intact cells, and phagocyte NADPH-oxidase were the objects. A directly proportional relationship between CL intensity and SOD-dependent ferricytochrome reduction was observed in the xanthine + xanthine oxidase system and phagocyte NADPH-oxidase system. Luc-CL practically took no place in two enzymatic oxidizing systems producing H2O2 and no superoxide (glucose oxidase + glucose and xanthine oxidase + NADH).
The authors of the cited paper came to the conclusion that the so-called lucigenin recycling (i.e. reaction (2)) made no effect on CL in those cases. However, they make a reservation that this occurs at small (below 20 µM) lucigenin concentrations in the system, implying that the situation could be different at higher concentrations [129].
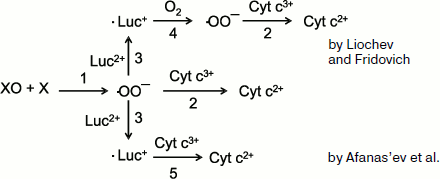
Afanas’ev et al. [131] raised their stronger objections against the role of lucigenin “recycling” in the development of Luc-CL. First of all, their calculations showed that the equilibrium in the reversible reaction
Luc+ + O2 ⇔ Luc2+ + ·OO–
is strongly shifted to the left (the equilibrium constant is 106), and thus superoxide in the “recycling” reaction will be scarcely produced in substantial amounts. On the other hand, data were obtained which showed that the interpretation by Liochev and Fridovich of their own results was erroneous. The scheme of the reactions under consideration is presented in Fig. 8. According to this scheme, superoxide radical produced in the xanthine oxidase (XO) + xanthine (X) system (reaction 1) reduces cytochrome c to bivalent state (2), and this underlies the classic method of ·OO– detection. In the interaction of ·OO– with lucigenin (Luc2+), cation-radical (·Luc+) is produced whose steady-state concentration is proportional to the intensity of Luc-CL (see Eq. (8)). According to Liochev and Fridovich [128, 130], ·Luc+ is able to reduce oxygen, thus forming cation-radical (reaction (4)), which results in the additional SOD-dependent reduction of cytochrome3+ (reaction 2). Afanas’ev and coworkers explained this effect by a direct reduction of cytochrome by lucigenin cation-radical (reaction 5) [131]. Their conclusion obtained on the base of calculations and experimental data is unambiguous: lucigenin (in the form of cation-radical) participates in the reduction of cytochrome c and does not in the production of the additional amount of superoxide radicals. All these data confirm the adequacy of the CL method for superoxide detection in chemical and biological systems.
Luc-CL in the Study of Isolated MitochondriaFig. 8. Reactions in the xanthine–xanthine oxidase system in the presence of lucigenin and cytochrome c (see explanations in the text).
The problem of the application of Luc-CL for the study of superoxide-radical formation in mitochondria was specially considered in paper [129]. The addition of lucigenin did not increase significantly oxygen consumption by mitochondria whose cytochrome oxidase was completely blocked by potassium cyanide. No additional oxygen consumption was observed after the addition of lucigenin at concentrations of 20 µM and below to a suspension of such mitochondria. This fact makes impossible an additional superoxide formation in the system as the result of lucigenin “recycling” at least at its small concentrations.
The site of superoxide-radical formation in the respiratory chain is the object of special consideration (see, in particular, papers [22, 132-135]), and we shall briefly dwell on only some works in which this problem was solved with different methods including the Luc-CL method.
When succinate was used as a source of electrons for the electron transfer chain in mitochondria, cyanide enhanced Luc-CL greatly by increasing the concentration of electron carriers in the reduced form, and thus a possibility of superoxide formation due to their interaction with dissolved oxygen. Rotenone and mexidol, which blocked electron transfer from respiratory Complex I to coenzyme Q and from coenzyme Q to Complex III, correspondingly, completely eliminated Luc-CL development [129].
In another paper by the same group of authors [136], it was shown with the Luc-CL method that the inhibitor of NADH-oxidoreductase diphenyleneiodonium (DPI) had no effect on the production of superoxide by isolated mitochondria if succinate served as the substrate (the reduction substrate for respiratory Complex II) and inhibited Luc-CL if pyruvate or NADH (able to reduce Complex I) were substrates. These data corroborate the existence of at least two superoxide production sections in mitochondria: at the level of respiratory Complex I (DPI-sensitive and rotenone- and mexidol-insensitive), and the section of ubiquinone–respiratory Complex III (DPI-insensitive and rotenone- and mexidol-sensitive). According to the data of paper [134], it is ubisemiquinone that reduces oxygen in this case. A possibility of superoxide production in other links of the respiratory chain is probable.
Taking into consideration an extensive application of Luc-CL for the analysis of superoxide radicals in mitochondria and intact cells, possible shortcomings of this method should be taken into proper account. Being concentrated on the problem of lucigenin recycling as a possible, though unlikely, cause of errors in the quantitative determination of superoxide by CL, researchers quite often overlook two more sources of possible errors: (i) the effect of lucigenin one-electron reduction rate at the level of cation-radical that determines the intensity of Luc-CL, and (ii) the effect of membrane potential on lucigenin and its cation-radical intramitochondrial concentration.
The first source of the faults is to be considered first. The way of lucigenin metabolism in the system under study begins from its transformation to cation-radical under the action of an intracellular reductant, for example, the respiratory Complex I reduced form:
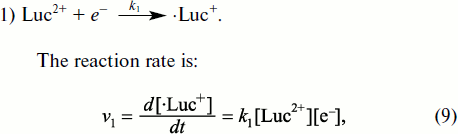
Lucigenin cation-radical can enter into several reactions, including the reaction of recycling:
![]()
![]()
![]()
v2 = –k2[O2][·Luc+],
v3 = –k3[Ox][·Luc+],
v4 = –k4[·OO–][·Luc+].
The total rate of changes of lucigenin cation-radical concentration in the system is equal to the sum of all these rates:
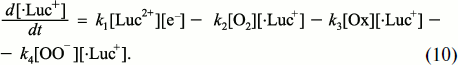
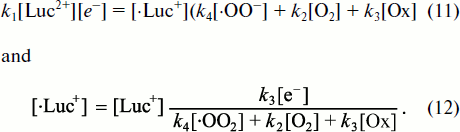
![]()
– CL intensity increases linearly with lucigenin concentration [Luc2+];
– at low superoxide concentrations, when the first item in the denominator is slight, Luc-CL intensity is directly proportional to superoxide concentration [·OO–];
– at high concentrations and at a small contribution of the dark lucigenin radical reactions in comparison with CL reaction (the greater part of the first item in the denominator), the increase of CL with the growth of superoxide concentration becomes less expressed;
– under stationary conditions, CL intensity is directly proportional to lucigenin one-electron reduction rate, i.e. to k1[Luc2+][e–].
The latter conclusion shows that CL intensity in the systems with a low lucigenin one-electron reduction rate will be smaller than that at a high rate of this reaction at the same superoxide concentrations. And it is a matter of fact. For example, it is noted in paper [137] that the difference between the data on superoxide concentration determined by calibration (Luc-CL intensity as a function of superoxide concentration) made during superoxide production by Complex I (which is able to reduce lucigenin to cation-radical), and by calibration with the use of the xanthine oxidase–hypoxanthine system (which is not able to) can reach one order of magnitude.
The other circumstance is a quite probable effect of the electric field on Luc-CL in mitochondria. Lucigenin is a bivalent cation penetrating through the lipid membrane of mitochondria. A simple calculation with the Nernst equation shows that Luc2+ concentration in the mitochondrial matrix at a membrane potential of –175 mV is approximately one million times higher than in the extramitochondrial space, and thus, the release of a relatively small superoxide amount into matrix will cause a much greater chemiluminescence burst than even a significant superoxide amount released outwards. Changes in the membrane potential can change Luc-CL intensity irrespectively of superoxide radical production by the respiratory chain. This circumstance should be taken into consideration when interpreting the data of chemiluminescent studies.
Study of Lucigenin-Dependent CL of Cells and Tissues
In most cells, Luc-CL is determined by superoxide production by the respiratory chain of mitochondria. It is partially related to the fact that lucigenin is accumulated in the mitochondrial matrix [126] where ·OO– produced by respiratory Complex I enters, and also to the fact that in nonphagocytizing cells mitochondria produce more superoxide than cell membrane NADPH-oxidase. It is shown in paper [126] that the inhibitors of respiratory Complex I (rotenone) and III (antimycin) inhibit Luc-CL, while the inhibitor of Complex IV (cyanide at a concentration below 100 µM) slightly enhances CL in a rat alveolar macrophage suspension. It corresponds to the behavior of isolated mitochondria [129] in which superoxide is produced by respiratory Complex I and in a Complex III-preceding point (probably at the level of ubisemiquinone) [134]. The authors conclude that it is mitochondria that are the source of superoxide radicals in macrophages [126].
Neutrophils are poor in mitochondria, unlike tissue macrophages and their precursors, blood monocytes. In the latter, it is mitochondria that also serve as the main source of superoxide. The arguments in favor of this fact were obtained in work [129] in which Luc-CL indices (such as CL dependence on cyanide, rotenone and myxothiazol) were completely identical in intact cells and in mitochondria isolated from these cells. Judging by Luc-CL measurements and oxygen consumption by cells, respiratory Complex I (NADH-oxidoreductase) was the main source of ·OO– since CL and oxygen consumption both in isolated mitochondria and in intact cells (monocytes) were inhibited by micromolar concentrations of DPI [136], a well-known inhibitor of flavoprotein enzymes including Complex I.
In our investigations, lucigenin-activated and oxygen-dependent chemiluminescence was also observed in whole liver tissue fragments. Superoxide-binding nitrogen monoxide inhibited Luc-CL, and the total light of NO-quenched CL was proportional to the amount of added monoxide [138].
OTHER CL PROBES FOR REACTIVE OXYGEN SPECIES
A number of papers describe the luminol-like compound 8-amino-5-chloro-7-phenylpyrido[3,4-d]pyridazin-1,4-(2H, 3H)dione (L-012) whose structure is shown in Fig. 9 as a CL probe for ROS. Under physiological conditions, the CL intensity of opsonized zymosan-stimulated neutrophils was approximately 100 and 10 times greater in the presence of L-012 than in the presence of luminol and luciferin analog MCLA (2-methyl-6-[p-methoxyphenyl]-3,7-dihydroimidazo[1,2-a]pyrazin-3-one), respectively [139].
Superoxide radical can be detected by CL with coelenterazine (2-(4-hydroxybenzyl)-6-(4-hydrophenol)-8-benzyl-3,7-dihydroimidazo[1,2-alpha]pyrazin-3-one) [140]. In the cited paper, the contribution of the extracellular component to superoxide-radical generation caused by epileptic attack was studied, and the role of NADPH-oxidase as a source of this radical was determined. It was shown that epileptic seizure activated membranous NADPH-oxidase and enhanced superoxide-radical production.Fig. 9. Superoxide-sensitive CL probes.
MCLA is also used for superoxide detection. Paper [141] describes in detail the mechanism of action of this CL probe; it is shown that it helps to detect and record superoxide production in whole perfused organs of laboratory animals. A drawback of MCLA is that it also reacts with dissolved oxygen, thus giving a background CL. Moreover, this compound acts as an antioxidant due to capture of free radicals [142]. Comparison of different CL methods of analysis of ROS released by blood vessels can be found in review [143].
The luminescence produced in the reaction with ozone is used for NO detection. A bright CL is observed while mixing nitrogen oxide and ozone:
NO + O3 = NO2 + O2 + photon.
The use of CL for the analysis of NO produced by vasodilating drugs is described in paper [144]. NO produced by endothelial cells of vessels is detected in studies of the mechanisms of vascular endothelium pathology. CL methods for measurement of NO production by cell cultures, which provide reliable and sensitive nitrogen monoxide detection, are described along with electrochemical methods and EPR [145].
COUMARIN-ACTIVATED CHEMILUMINESCENCE
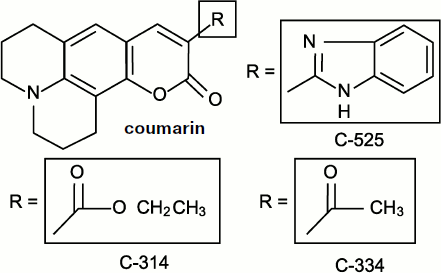
Chemiluminescent (or chemiluminogenic) [46, 146, 147] probes are chemically active molecules; moreover, their molecules bind free radicals and consequently influence the processes proceeding in living cells, i.e. those which we study. Discussion concerning the role of lucigenin interference in superoxide-radical production has already been considered above. Meanwhile, in the case of organic radicals including those of polyunsaturated fatty acids, a fundamentally different method of luminescence enhancement exists that is based on the electron excitation energy transfer from a free-radical reaction product molecule to a CL activator molecule. At present, isoquinolizine coumarin C-525 can be considered the most efficient physical luminescence activator, which enhances CL accompanying chain lipid oxidation in liposomes by more than 1500 times but has no effect on the CL in the interaction of oxygen radicals (hydroxyl and superoxide) [52]. Two other related coumarins, C-314 and C-334, turned out to be a little less active [148]. The formulas of these compounds are given in Fig. 10.
Coumarin C-525-activated CL was initially discovered in the experiments with a suspension of phospholipids (liposomes) in which peroxidation was initiated by the addition of Fe2+ salts. It was shown later that C-525 in micromolar concentrations also enhanced CL in rat liver microsomes where lipid peroxidation was initiated by the addition of tert-butyl hydroperoxide (t-BHP). As it is shown, C-525 is a more efficient CL activator than the earlier known chlorophyll a [149]. Moreover, activation took place only in peroxidation systems where carbonyl products were generated in the electron-excited state, and no activation was observed when horseradish peroxidase acted.Fig. 10. Quinolizine coumarins – physical CL activators [148].
In paper [150], C-525-activated CL was estimated on the addition of Fe2+ ions to low-density lipoproteins (LDLP) oxidized with different methods and to different extent. A good correlation (0.97-0.98) was revealed between the amplitude of the quick flash and the amount of hydroperoxides detected with the method of recording of conjugated double bonds (diene conjugation). The CL amplitude itself was more than two orders higher in the presence of C-525 than in the absence of the activator.
Activation of CL during lipid peroxidation of blood lipoproteins (LP) was also investigated in detail in work [151]. C-525 in a concentration of 4 µM enhanced the CL flash after the addition of Fe2+ ions to oxidized LDLP by 2000 times. Fluorescence measurements showed that the dye was in the hydrophobic phase of LP, and thus, the real concentration of the activator was rather high for an effective energy transfer from the excited molecules of lipoperoxidation carbonyl products.
It was shown in paper [148] that dyeing of cells in a culture with coumarin dyes C-525 and C-334 enabled detection of CL in those cells caused by a free-radical oxidation initiator AMVN (2,2′-azo-bis(2,4-dimethylvaleronitrile)). Neither cell viability nor the content of reduced glutathione in cytoplasm changed, and the added dye underwent no chemical degradation. C-525 in the used concentrations also had no effect on the lipid peroxidation level. A luminescence was also observed during the peroxidation caused by tert-butyl and cumene (isopropylbenzene) hydroperoxides.
However, the attempt to enhance CL by introducing C-525 on blood plasma peroxidation failed. This is explained by binding of hydrophobic C-525 by plasma proteins, first of all by serum albumin.
CL in the presence of C-525 has recently been used by our group for study of lipid radical production during oxidation of natural cardiolipin (which contains polyunsaturated fatty acids) catalyzed by the cardiolipin–cytochrome c complex [109, 110]. It was found in the process of the research that the formation of a complex of cytochrome c with cardiolipin and other anionic lipids as itself confers peroxidase properties to this hemoprotein [59-62]. In the presence of natural cardiolipin, lipid radicals are produced, which is manifested in Coum-CL. This process could proceed in the absence of H2O2 if the system contained other hydroperoxides including cardiolipin peroxides, but additional H2O2 introduction accelerated the peroxidation reaction. Antioxidants inhibited production of both luminol radicals in the peroxidase cytochrome c–cardiolipin system and lipid radicals. Since cardiolipin peroxidation can be considered as a key stage in apoptosis triggering [152], these data can be used in elaboration of the methods for regulation of apoptosis with medicines.
APPLICATION OF CELL CL IN MEDICINE
An important role in the pathogenesis of many diseases and pathological processes belongs to oxidative stress [153]. The CL method is efficient in the study of such pathologies as it enables measuring the level of free radicals (ROS, NO) and estimating antioxidant protection parameters and antioxidant action. Chemiluminescence is successfully applied in the study of immune derangements, metabolic disorders, endothelium dysfunction, myocardial and cerebral ischemia, and oncological and inflammation diseases, as well as many other diseases whose pathogenesis is related to oxidative stress.
Ischemic Damage to Myocardium and Brain
Damage to cells on reperfusion of ischemic tissue is mediated by many factors, and free-radical mechanisms are the key factor since the reestablished blood flow enhances greatly oxidant processes during a reduced activity of the antioxidant system. Oxidative stress is a special hazard for cells intensively consuming oxygen—cardiomyocytes and neurocytes [154-156]. Possible sources for ROS are polymorphonuclear leukocytes, the xanthine-oxidase system, mitochondria, arachidonic acid metabolism, etc. A reduced NO production due to endothelium dysfunction and its inactivation by superoxide promotes the enhancement of damaging action of ROS. The role of ROS and the state of antioxidant enzymes on myocardial ischemia including angioplasty [167-170] was estimated by the CL method in papers [157-166]. NO production was measured with the ozone CL method [171, 172] as oxidation products (nitrite and nitrate) [173].
During investigations of cerebral ischemia, CL of blood plasma, isolated phagocytes, and cerebral tissue homogenates were measured on ischemia and the following reoxygenation, as well as the phagocyte-produced ROS level, the effect of ischemia duration on the extent of damage, and the role of antioxidants were estimated [174-177].
Endothelium Dysfunction
Vascular endothelium in the norm performs many metabolic and signal functions, in particular it regulates vasodilatation and vasoconstriction processes. Endothelium location in the interface between blood and tissues makes it very vulnerable for free radicals that cause damage to cells, bringing about the state of endothelium dysfunction and development of angiopathy, atherosclerosis, etc. [178]. The CL method enables estimation of NO and superoxide generation by endothelium properly, and the action of neutrophil reactive oxygen species on the state of vessels. The level of superoxide and more rarely that of NO is mainly determined by lucigenin CL [143]. Human [143] and animal [179-181] tissue homogenates and isolated vessels and also endothelial cell cultures of human umbilical vein [182, 183] and coronary microvessels [184] are used as objects. The authors studied the effect of acute inflammation on vascular endothelium [183, 185]. The role of oxidative stress in atherosclerosis pathogenesis is also shown in papers [186-188].
Central Nervous System Diseases
Recent investigations have proved the participation of ROS and other free radicals in the pathogenesis of different central nervous system diseases [189, 190], though it is not yet clear whether they are a cause or a concomitant phenomenon of these pathological processes. Oxidative stress is especially of great importance in the development of neurodegenerative diseases such as Alzheimer’s disease [191-193] and brain damage on epileptic seizures [140, 194]. The CL method was applied for measurements of the level of ROS produced by blood neutrophils or immediately the CL of cerebral tissue homogenates, and the state of antioxidant systems (Cu-Zn SOD, catalase, glutathione system, total blood antioxidant potentials, etc.) [195, 196] was also estimated.
Rheumatoid Arthritis
Rheumatoid arthritis is a systemic chronic inflammatory disease of immune genesis during which a long-term inflammation causes a stationary neutrophil activity and tissue damage by neutrophil-produced ROS. More than 150 papers on this subject are cited in the PubMed database, in which ROS products, first of all superoxide radical, are studied by measuring the CL of isolated neutrophils of the peripheral blood and synovial fluid of patients [197-201], CL-response of donor neutrophils after incubation with the blood serum and synovial fluid of patients [201-206], CL of isolated blood monocytes [207], isolated mitochondria [208], and fibroblast culture [209]. The action on neutrophils of some mediators and substances, in particular, hydroxyapatite [210, 211], phospholipase A2 [212], platelet-activating factor [213], oxygen [214], substance P [215], etc. on CL was investigated. Clinical behavior [216] and therapeutic effectiveness [217, 218] were estimated by the intensity of CL of polymorphonuclear leukocytes.
Oncological Diseases
Oncological diseases are supposed to be a result of dysfunction of certain genes. From this viewpoint, every factor that interacts with DNA and modifies it is a carcinogen. ROS, such as hydroxyl radical, are able to damage directly mitochondrial and nuclear DNA and, consequently, possess a mutagenic, carcinogenic action [219]. The role of free oxygen and nitrogen radicals is not confined to direct damage to DNA; they can impair apoptosis processes, upset redox balance, affect regulation of cell signaling, etc. [220]. The chemiluminescence method has been used to estimate the effect of chemotherapy on the proliferation and activation of tumor cells [221], to study the prooxidant and oxidant activity parameters of malignant tumors in comparison with benign tumors [222, 223], and to investigate metastasis mechanisms [224].
CONCLUSION
Free radicals are important participants of regulatory processes in living cells and simultaneously the cause of damage to cell structures and a trigger that activates a cascade of cell self-destruction reactions, apoptosis. Their leading role in the development of practically all diseases of the elderly and aging of the organism is universally recognized and is the object of numerous investigations. However, time-average (stationary) concentration of these active particles in living cell is very small, and their direct detection with ordinary biochemical methods is practically impossible. The chemiluminescence method is not based on the analysis of substances but on the measurement of the rates of luminescence-accompanied reactions, as it is these reactions that are characteristic of free radicals.
The most known chemiluminescent reactions in biological systems are intrinsic (ultraweak) luminescence during chain lipid oxidation, reactions of luminol with ROS (hydroxyl radical and superoxide) and organic radicals, reactions of lucigenin and a number of luciferin derivatives with superoxide radical, and all of them involve the interaction of two radicals at a certain stage which allows a generated molecule to accumulate an energy sufficient for the emission of a photon by the end product. It could seem that the reactions of active molecules with hydrogen peroxide, for example, the reaction of hypochlorite with luminol in the presence of H2O2, and some other reactions not mentioned in this review turn out to be an exception to this rule. It should not, however, be neglected that the bulk in our cells is formed as the result of a SOD-catalyzed reaction of interaction between two superoxide radicals since it is superoxide that is the main natural radical produced by cell membranes and mitochondria. H2O2 in this case is an accumulator of the radical interaction energy, which is a considerable constituent of the excited-state energy of the final reaction product.
About 80 years have passed since Gurvich discovered mitogenetic radiation, and this year it is 50 years since the date of publication of our first article on the investigation of ultraweak luminescence in biological systems [6]. Since that time, sensitive and easy-to-use CL-recording devices have been developed, substances that activate chemiluminescence in cell systems and solutions have been suggested, the methods of kinetics and mathematical modeling of CL reactions have been elaborated, and the mechanism of most of them has been discovered. The CL method has become one of the basic methods of investigation of free-radical processes in scientific and clinical studies. It will no doubt be widely used in the future.
This work was supported by projects 08-04-01074-a and 09-04-07075-d from the Russian Foundation for Basic Research.
REFERENCES
1.Gurvich, A. G. (1934) Mitogenetic Radiation
[in Russian], Gosmedizdat.
2.Gurvich, A. G., and Gurvich, L. D. (1945)
Mitogenetic Radiation [in Russian], Izd-vo Narkomzdrava
SSSR.
3.Colli, L., and Facchini, U. (1954) Nuovo
Cimento, 12, 150.
4.Colli, L., Facchini, U., Guidotti, G.,
Lonati, R. D., Arsenigo, M., and Sommariva, O. (1955)
Experientia, 11, 479-481.
5.Colli, L., Facchini, U., and Rossi, A. (1954)
Nuovo Cimento, 11, 255.
6.Vladimirov, Yu. A., and Litvin, F. F. (1959)
Biofizika, 4, 601-605.
7.Tarusov, B. N., Polivoda, A. N., and Zhuravlev, A.
I. (1961) Biofizika, 6, 490.
8.Tarusov, B. N., Polivoda, A. N., and Zhuravlev, A.
I. (1961) Radiobiologiya, 1, 150.
9.Tarusov, B. N., Polivoda, A. N., Zhuravlev, A. I.,
and Sekamova, E. N. (1962) Tsitologiya, 4, 696.
10.Vladimirov, Yu. A., and L’vova, O. F.
(1964) Biofizika, 9, 506-507.
11.Vladimirov, Yu. A. (1966) Ultraweak
Luminescences on Biochemical Reactions [in Russian], Nauka, Moscow,
p. 126.
12.Vladimirov, Yu. A., L’vova, O. F., and
Cheremisina, Z. P. (1966) Biokhimiya, 31, 507-514.
13.Vladimirov, Yu. A., Suslova, T. B., and
Cheremisina, Z. P. (1968) Biokhimiya, 33, 720-723.
14.Boveris, A., Cadenas, E., and Chance, B. (1981)
Fed. Proc., 40, 195-198.
15.Allen, R. C., Stjernholm, R. L., and Steele, R.
H. (1972) Biochem. Biophys. Res. Commun.,
47, 679.
16.Stjernholm, R. L., Allen, R. C., Steele, R. H.,
Waring, W. W., and Harris, J. A. (1973) Infect. Immun.,
7, 313-314.
17.Allen, R. C., and Loose, L. D. (1976)
Biochem. Biophys. Res. Commun., 69,
245-252.
18.Vladimirov, Yu. A., Azizova, O. A., Deev, A. I.,
Kozlov, A. V., Osipov, A. N., and Roshchupkin, D. I. (1992) Free
Radicals in Living Systems, in Advances in Science and
Technology, Ser. Biophysics [in Russian], Vol. 29, VINITI, Moscow,
p. 250.
19.Vladimirov, Yu. A. (1999) Soros. Obrazovat.
Zh., 25-32.
20.Babior, B. M. (1984) J. Clin. Invest.,
73, 599-601.
21.Babior, B. M. (1992) Adv. Enzymol. Relat.
Areas Mol. Biol., 65, 49-95.
22.Turrens, J. F. (1997) Biosci. Rep.,
17, 3-8.
23.Vladimirov, Yu. A. (1998) Vestn. Ros. Akad.
Med. Nauk, 7, 43-51.
24.Liochev, S. L. (1996) Free Radic. Res.,
25, 369-384.
25.Liochev, S. I., and Fridovich, I. (1994) Free
Radic. Biol. Med., 16, 29-33.
26.Osipov, A. N., Yakutova, E. Sh., and Vladimirov,
Yu. A. (1993) Biofizika, 38, 390-396.
27.Halliwell, B., and Gutteridge, J. M. C. (1999)
Free Radicals in Biology and Medicine, Oxford University
Press, Oxford, p. 783.
28.Halliwell, B., and Gutteridge, J. M. C. (1999)
Free Radicals in Biology and Medicine, Oxford University
Press, Oxford-New York, p. 936.
29.Men’shchikova, E. B., Zenkov, N. K., and
Lankin, V. Z. (2008) Oxidative Stress. Pathological States and
Diseases [in Russian], ARTA, Novosibirsk, p. 284.
30.Osipov, A. N., Savov, V. M., Yakh’yaev, A.
V., Zubarev, V. E., Azizova, O. A., Kagan, V. E., and Vladimirov, Yu.
A. (1984) Biofizika, 29, 533-336.
31.Azizova, O. A., Osipov, A. N., Savov, V. M.,
Yakh’yaev, A. V., Zubarev, V. E., Kagan, V. E., and Vladimirov,
Yu. A. (1985) Biofizika, 30, 36-39.
32.Tarusov, B. N., Ivanov, I. I., and Petrusevich,
Yu. M. (1967) Ultraweak Luminescence of Biological Systems [in
Russian], Moscow University Publishers, Moscow.
33.Vladimirov, Yu. A., and Archakov, A. I. (1972)
Lipid Peroxidation in Biological Membranes [in Russian], Nauka,
Moscow.
34.Boveris, A., Cadenas, E., Reiter, R.,
Filipkowski, M., Nakase, Y., and Chance, B. (1980) Proc. Natl.
Acad. Sci. USA, 77, 347-351.
35.Cadenas, E., Arad, I. D., Fisher, A. B., Boveris,
A., and Chance, B. (1980) Biochem. J., 192,
303-309.
36.Cadenas, E., Boveris, A., and Chance, B. (1980)
Biochem. J., 188, 577-583.
37.Cadenas, E., Boveris, A., and Chance, B. (1980)
Biochem. J., 186, 659-567.
38.Cadenas, E., Boveris, A., and Chance, B. (1980)
FEBS Lett., 112, 285-288.
39.Cadenas, E., Boveris, A., and Chance, B. (1980)
Biochem. J., 187, 131-140.
40.Cadenas, E., Ginsberg, M., Rabe, U., and Sies, H.
(1984) Biochem. J., 223, 755-759.
41.Cadenas, E., Varsavsky, A. I., Boveris, A.,
and Chance, B. (1981) Biochem. J., 198, 645-654.
42.Kakinuma, K., Cadenas, E., Boveris, A., and
Chance, B. (1979) FEBS Lett., 102, 38-42.
43.Cadenas, E., Varsavsky, A. I., Boveris, A.,
and Chance, B. (1980) FEBS Lett., 113, 141-144.
44.Cadenas, E., Arad, I. D., Boveris, A., Fisher, A.
B., and Chance, B. (1980) FEBS Lett., 111, 413-418.
45.Boveris, A., Cadenas, E., Reiter, R.,
Filipkowski, M., Nakase, Y., and Chance, B. (1980) Proc. Natl. Acad.
Sci. USA, 77, 347-351.
46.Slawinska, D., and Slawinski, J. (1987)
Pigment Cell Res., 1, 171-175.
47.Vladimirov, Y. A. (1994) Free Radicals in
the Environment, Medicine and Toxicology (Nohl, H., et al.,
eds.) Richelieu Press, London, pp. 345-373.
48.Vladimirov, Y. A. (1996) Free Radicals. A
Practical Approach (Punchard, F. J., ed.) Oxford University Press,
Oxford-New York-Tokyo, pp. 65-82.
49.Vladimirov, Yu. A., and Potapenko, A. Ya. (2006)
Physical-Chemical Principles of Photobiological Processes [in
Russian], Drofa, Moscow.
50.Vladimirov, Y. A., and Sherstnev, M. P. (1991)
Soviet Medical Reviews. Sec. B. Physicochemical Aspects of
Medicine, Vol. 2, Pt. 5 (Lopukhin, Y. M., ed.) Harwood Academic
Publishers GmbH, London, pp. 1-43.
51.Vladimirov, Yu. A., and Sherstnev, M. P. (1989)
in Advances in Science and Technology. Ser. Biophysics [in
Russian], Vol. 24.
52.Vladimirov, Y. A., Sharov, V. S., Driomina,
E. S., Reznitchenko, A. V., and Gashev, S. B. (1995) Free Rad.
Biol. Med., 18, 739-745.
53.Barnard, M. L., Robertson, B., Watts, B. P., Jr.,
and Turrens, J. F. (1997) Am. J. Physiol., 272,
L262-L267.
54.Turrens, J. F., Giulivi, C., Pinus, C., Roldan,
E., Lavagno, C., and Boveris, A. (1988) Basic Life Sci.,
49, 239-242.
55.Turrens, J. F., Giulivi, C., Pinus, C. R.,
Lavagno, C., and Boveris, A. (1988) Free Rad. Biol. Med.,
5, 319-323.
56.Vladimirov, Yu. A., Aksentsev, S. L., and Olenev,
V. I. (1965) Biofizika, 10, 614-618.
57.Vladimirov, Yu. A., Suslova, T. B., and Olenev,
V. I. (1969) Biofizika, 14, 836-845.
58.Vladimirov, Yu. A., Olenev, V. I.,
Suslova, T. B., and Cheremisina, Z. P. (1980) Adv. Lipid
Res., 17, 173-249.
59.Belikova, N. A., Vladimirov, Yu. A., Osipov, A.
N., Kapralov, A. A., Tyurin, V. A., Potapovich, M. V., Basova, L. V.,
Peterson, J., Kurnikov, I. V., and Kagan, V. E. (2006)
Biochemistry, 45, 4998-5009.
60.Kapralov, A. A., Kurnikov, I. V., Vlasova,
I. I., Belikova, N. A., Tyurin, V. A., Basova, L. V., Zhao,
Q., Tyurina, Y. Y., Jiang, J., Bayir, H., Vladimirov, Yu. A., and
Kagan, V. E. (2007) Biochemistry, 46, 14232-14244.
61.Vladimirov, Yu. A., Proskurnina, E. V., Izmailov,
D. Yu., Novikov, A. A., Brusnichkin, A. V., Osipov, A. N., and Kagan,
V. E. (2006) Biochemistry (Moscow), 71, 998-1005.
62.Vladimirov, Yu. A., Proskurnina, E. V., Izmailov,
D. Yu., Novikov, A. A., Brusnichkin, A. V., Osipov, A. N., and Kagan,
V. E. (2006) Biochemistry (Moscow), 71, 989-997.
63.Vladimirov, Yu. A., Korchagina, M. V., and
Olenev, V. I. (1971) Biofizika, 16, 953.
64.Vasil’ev, R. F. (1965) Opt.
Spektroskop., 18, 236-244.
65.Vasil’ev, R. F., and Rusina, I. F. (1964)
Dokl. AN SSSR, 156, 1402-1405.
66.Vasil’ev, R. F. (1998) J. Biolum.
Chemilum., 13, 69-74.
67.Vladimirov, Yu. A. (1986) Free
Radicals, Aging, and Degenerative Diseases
(Johnson, J. E., Jr., et al., eds.) Allan R. Liss, Inc., New York, pp.
141-195.
68.Vladimirov, Yu. A. (1987)
Physicochemical Aspects of Medicine. Reviews. Soviet
Medical Reviews, Sec. B. (Lopukhin, Y. M., ed.) Harwood
Academic Publishers GmbH, London, pp. 51-127.
69.Gukasov, V. M., Sergeev, P. V., Seifullo, R. D.,
and Vladimirov, Y. A. (1974) Byul. Eksp. Biol. Med., 11,
54-56.
70.Vladimirov, Yu. A., and Petrenko, Yu. M. (1976)
Biofizika, 21, 424-427.
71.Rubene, D. Ya., Sharov, V. S., Olenev, V. I.,
Tirzit Dubur, G. Ya., and Vladimirov, Yu. A. (1981) Zh. Fiz.
Khim., 15, 511-512.
72.Vladimirov, Yu. A., Boldyrev, A. A., Deev, A. I.,
Severin, S. E., and Li, H. I. (1988) Biofizika, 33,
140-145.
73.Klebanov, G. I., Babenkova, I. V., Teselkin, Yu.
O., Komarov, O. S., and Vladimirov, Yu. A. (1988) Lab. Delo,
No. 5, 59-62.
74.Li, H. I., Vladimirov, Yu. A., and Deev, A. I.
(1990) Biofizika, 35, 82-85.
75.Epanchitseva, O. M., Parfenov, E. A., Smirnov, L.
D., and Vladimirov, Yu. A. (1991) Byul. Eksp. Biol. Med.,
112, 358-360.
76.Vladimirov, Yu. A. (1996) Natural and
Synthetic Antioxidants (Packer, L., ed.) ISNA, NY, pp. 125-241.
77.Olenev, V. I., and Vladimirov, Yu. A. (1973)
Stud. Biophys., 38, 131-138.
78.Sharov, V. S., Driomina, E. S., and
Vladimirov, Yu. A. (1996) J. Biolum.
Chemilum., 11, 91-98.
79.Vladimirov, Yu. A., Gutenev, P. I., and
Kuznetsov, P. I. (1973) Biofizika, 18, 1024-1030.
80.Vladimirov, Yu. A., Tafel’shtein, E. E.,
and Kozlov, Yu. P. (1969) DAN SSSR, 188, 1163-1165.
81.Vladimirov, Y. A., Sergeev, P. V., Seifullo, R.
D., and Rudnev, Yu. N. (1973) Mol. Biol. (Moscow), 7,
247-253.
82.Gukasov, V. M., Sergeev, P. V., Seifulla, R. D.,
and Vladimirov, Yu. A. (1974) Byul. Eksp. Biol. Med., 78,
54-57.
83.Vladimirov, Yu. A., and L’vova, O. F.
(1965) Cell Biophysics (Frank, G. M., ed.) [in Russian], Nauka,
Moscow, pp. 74-83.
84.L’vova, O. F., and Vladimirov, Yu. A.
(1964) Symp. “Free-Radical Processes in Biological
Systems”, June 2-5, Abstracts, p. 34.
85.L’vova, O. F., and Vladimirov, Yu. A.
(1966) Trudy Mosk. Obshch. Ispytatelei Prirody, 16,
214-217.
86.Pollet, E., Martinez, J. A., Metha, B., Watts, B.
P., Jr., and Turrens, J. F. (1998) Arch. Biochem. Biophys.,
349, 74-80.
87.Watts, B. P., Jr., Barnard, M., and Turrens, J.
F. (1995) Arch. Biochem. Biophys., 317, 324-330.
88.Turrens, J. F. (1994) Int. Conf. on Clinical
Chemiluminescence, Berlin, April 25-28, 1994, Humboldt University
Publishers, Berlin.
89.Klebanov, G. I., Vladimirov, Yu. A., Benov, L.
Ts., and Ribarov, S. R. (1988) Byul. Eksp. Biol. Med.,
105, 674-676.
90.Vladimirov, Yu, A., Ribarov, S. R., Bochev, P.
G., Benov, L. C., and Klebanov, G. I. (1990) Gen. Physiol.
Biophys., 9, 45-54.
91.Boullier, A., Bird, D. A., Chang, M. K., Dennis,
E. A., Friedman, P., Gillotre-Taylor, K., Horkko, S., Palinski, W.,
Quehenberger, O., Shaw, P., Steinberg, D., Terpstra, V., and
Witztum, J. L. (2001) Ann. N. Y. Acad. Sci., 947,
214-222; discussion 22-23.
92.Kita, T., Kume, N., Minami, M., Hayashida,
K., Murayama, T., Sano, H., Moriwaki, H., Kataoka, H., Nishi, E.,
Horiuchi, H., Arai, H., and Yokode, M. (2001) Ann. N. Y. Acad.
Sci., 947, 199-205; discussion 05-06.
93.Nakajima, K., Nakano, T., and Tanaka, A. (2006)
Clin. Chim. Acta, 367, 36-47.
94.Nilsson, J., Nordin Fredrikson, G., Schiopu, A.,
Shah, P. K., Jansson, B., and Carlsson, R. (2007) Curr. Pharm.
Des., 13, 1021-1030.
95.Babior, B. M. (1995) Curr. Opin. Hematol.,
2, 55-60.
96.Klebanov, G. I., and Vladimirov, Yu. A. (1999)
Uspekhi Sovrem. Biol., 119, 461-474.
97.Roshchupkin, D. I., Belakina, N. S., and Murina,
M. A. (2006) Biofizika, 51, 99-107.
98.Merenyi, G., Lind, J., and Eriksen, T. E. (1990)
J. Biolum. Chemilum., 5, 53-56.
99.Krasowska, A., Rosiak, D., Szkapiak, K.,
Oswiecimska, M., Witek, S., and Lukaszewicz, M. (2001) Cell Mol.
Biol. Lett., 6, 71-81.
100.Ritov, V. B., Goldman, R., Stoyanovsky, D.
A., Menshikova, E. V., and Kagan, V. E. (1995) Arch. Biochem.
Biophys., 321, 140-152.
101.Tyurina, Y. Y., Tyurin, V. A., Yalowich, J. C.,
Quinn, P. J., Claycamp, H. G., Schor, N. F., Pitt, B. R., and Kagan, V.
E. (1995) Toxicol. Appl. Pharmacol., 131, 277-288.
102.Maulik, G., Maulik, N., Bhandari, V., Kagan, V.
E., Pakrashi, S., and Das, D. K. (1997) Free Radic. Res.,
27, 221-228.
103.Kagan, V. E., Tsuchiya, M.,
Serbinova, E., Packer, L., and Sies, H. (1993)
Biochem. Pharmacol., 45, 393-400.
104.Lissi, E., Salim-Hanna, M., Pascual, C., and
del Castillo, M. D. (1995) Free Radic. Biol. Med., 18,
153-158.
105.Dotan, Y., Lichtenberg, D., and Pinchuk, I.
(2004) Progr. Lipid Res., 43, 200-227.
106.Cormier, M. J., and Prichard, P. M. (1968)
J. Biol. Chem., 243, 4706-4714.
107.Lambeir, A. M., Markey, C. M., Dunford, H.
B., and Marnett, L. J. (1985) J. Biol. Chem., 260,
14894-14896.
108.Stepanov, G., Gnedenko, O., Mol’nar, A.,
Ivanov, A., Vladimirov, Y., and Osipov, A. (2009) FEBS
Lett., 583, 97-100.
109.Demin, E. M., Proskurnina, E. V., and
Vladimirov, Yu. A. (2008) Vestn. Mosk. Univ. Ser. 2,
Khim., 354-359.
110.Vladimirov, Yu. A., Proskurnina, E. V., Demin,
E. M., Matveeva, N. S., Lubitskiy, O. B., Novikov, A. A., Izmailov, D.
Yu., Osipov, A. N., Tikhonov, V. P., and Kagan, V. E. (2009)
Biochemistry (Moscow), 74, 301-307.
111.Osipov, A. N., Stepanov, G. O., Vladimirov, Yu.
A., Kozlov, A. V., and Kagan, V. E. (2006) Biochemistry
(Moscow), 71, 1128-1132.
112.Arnhold, J., Mueller, S., Arnold, K., and
Sonntag, K. (1993) J. Biolum. Chemilum., 8, 307-313.
113.Mueller, S., and Arnhold, J. (1995) J.
Biolum. Chemilum., 10, 229-237.
114.Murina, M. A., Belakina, N. S., and
Roshchupkin, D. I. (2004) Biofizika, 49, 1099-1105.
115.Murina, M. A., Roshchupkin, D. I., Belakina, N.
S., Filippov, S. V., and Khalilov, E. M. (2005) Biofizika,
50, 1100-1104.
116.Arnhold, J. (2004) Biochemistry
(Moscow), 69, 4-9.
117.Haqqani, A. S., Sandhu, J. K., and
Birnboim, H. C. (1999) Anal. Biochem., 273,
126-132.
118.Greenlee, L., Fridovich, I., and Handler, P.
(1962) Biochemistry, 1, 779-783.
119.Storch, J., and Ferber, E. (1988) Anal.
Biochem., 169, 262-267.
120.Wilson, M. E., Trush, M. A., van Dyke, K.,
and Neal, W. (1978) FEBS Lett., 94, 387-390.
121.Allen, R. C. (1986) Meth. Enzymol.,
133, 449-493.
122.Irani, K., Xia, Y., Zweier, J. L.,
Sollott, S. J., Der, C. J., Fearon, E. R., Sundaresan, M.,
Finkel, T., and Goldschmidt-Clermont, P. J. (1997)
Science, 275, 1649-1652.
123.Mohazzab, K. M., Kaminski, P. M., and Wolin, M.
S. (1994) Am. J. Physiol., 266, H2568-H2572.
124.Bhunia, A. K., Han, H., Snowden, A., and
Chatterjee, S. (1997) J. Biol. Chem., 272,
15642-15649.
125.Rembish, S. J., Yang, Y., and Trush, M. A.
(1994) Res. Commun. Mol. Pathol. Pharmacol., 85,
115-129.
126.Rembish, S. J., and Trush, M. A. (1994) Free
Radic. Biol. Med., 17, 117-126.
127.Faulkner, K., and Fridovich, I. (1993) Free
Radic. Biol. Med., 15, 447-451.
128.Liochev, S. I., and Fridovich, I. (1997)
Arch. Biochem. Biophys., 337, 115-120.
129.Li, Y., Zhu, H., Kuppusamy, P., Roubaud, V.,
Zweier, J. L., and Trush, M. A. (1998) J. Biol. Chem.,
273, 2015-2023.
130.Liochev, S. I., and Fridovich, I. (1998)
Free Radic. Biol. Med., 25, 926-928.
131.Afanas’ev, I. B., Ostrachovitch, E. A.,
and Korkina, L. G. (1999) Arch. Biochem. Biophys.,
366, 267-274.
132.Kowaltowski, A. J., and Vercesi, A. E. (1999)
Free Radic. Biol. Med., 26, 463-471.
133.Skulachev, V. P. (1998) FEBS Lett.,
423, 275-280.
134.Turrens, J. F., Alexandre, A., and
Lehninger, A. L. (1985) Arch. Biochem. Biophys.,
237, 408-414.
135.Vinogradov, A. D., and Grivennikova, V. G.
(2005) Biochemistry (Moscow), 70, 120-127.
136.Li, Y., and Trush, M. A. (1998)
Biochem. Biophys. Res. Commun., 253, 295-299.
137.Kervinen, M., Patsi, J., Finel, M., and
Hassinen, I. E. (2004) Anal. Biochem., 324, 45-51.
138.Matveeva, N. S., Lubitskiy, O. B., Osipov, A.
N., and Vladimirov, Yu. A. (2007) Biofizika, 52,
1120-1127.
139.Imada, I., Sato, E. F., Miyamoto, M., Ichimori,
Y., Minamiyama, Y., Konaka, R., and Inoue, M. (1999) Anal.
Biochem., 271, 53-58.
140.Patel, M., Li, Q. Y., Chang, L. Y., Crapo, J.,
and Liang, L. P. (2005) J. Neurochem., 92, 123-131.
141.Nakano, M. (1998) Cell Mol.
Neurobiol., 18, 565-579.
142.Tampo, Y., Tsukamoto, M., and Yonaha, M.
(1998) FEBS Lett., 430, 348-352.
143.Guzik, T. J., and Channon, K. M. (2005)
Meth. Mol. Med., 108, 73-89.
144.Brien, J. F., McLaughlin, B. E.,
Nakatsu, K., and Marks, G. S. (1991) J. Pharmacol.
Meth., 25, 19-27.
145.Hart, C. M., Kleinhenz, D. J., Dikalov, S.
I., Boulden, B. M., and Dudley, S. C., Jr. (2005) Meth.
Enzymol., 396, 502-514.
146.Dodeigne, C., Thunus, L., and Lejeune, R.
(2000) Talanta, 51, 415-439.
147.Monig, H., and Konermann, G. (1993)
Strahlenther Onkol., 169, 418-426.
148.Vladimirov, Y. A., Arroyo, A., Taylor, J. M.,
Tyurina, Y. Y., Matsura, T., Tyurin, V. A., and Kagan, V. E. (2000)
Arch. Biochem. Biophys., 384, 154-162.
149.Sharov, V. S., Briviba, K., and Sies, H. (1996)
Free Radic. Biol. Med., 21, 833-843.
150.Driomina, E., Polnikov, I., Sharov, V.,
Azizova, O., and Vladimirov, Y. (1994) Free Radic. Res.,
20, 279-288.
151.Sharov, V. S., Driomina, E. S., and Vladimirov,
Yu. A. (1995) Biofizika, 40, 428-433.
152.Kagan, V. E., Tyurin, V. A., Jiang, J.,
Tyurina, Y. Y., Ritov, V. B., Amoscato, A. A., Osipov, A. N., Belikova,
N. A., Kapralov, A. A., Kini, V. V., Vlasova, I. I., Zhao, Q.,
Zou, M., Di, P., Svistunenko, D. A., Kurnikov, I. V., and
Borisenko, G. G. (2005) Nature Chem. Biol., 1,
223-232.
153.Zenkov, N. K., Lankin, V. Z., and
Men’shchikova, E. B. (2001) Oxidative Stress [in Russian],
MAIK “Nauka/Interperiodika”, Moscow, p. 343.
154.Neuzil, J., Rayner, B. S., Lowe, H. C., and
Witting, P. K. (2005) Redox Rep., 10, 187-197.
155.Siesjo, B. K., and Siesjo, P. (1996) Eur. J.
Anaesthesiol., 13, 247-268.
156.Sivonova, M., Kaplan, P., Durackova, Z.,
Dobrota, D., Drgova, A., Tatarkova, Z., Pavlikova, M.,
Halasova, E., and Lehotsky, J. (2008) Cell Mol.
Neurobiol., 28, 431-441.
157.Prasad, K., Lee, P., Mantha, S. V., Kalra, J.,
Prasad, M., and Gupta, J. B. (1992) Mol. Cell Biochem.,
115, 49-58.
158.Bassenge, E., Sommer, O., Schwemmer, M.,
and Bunger, R. (2000) Am. J. Physiol. Heart Circ. Physiol.,
279, H2431-H2438.
159.Brovkovich, V. M., Nikitchenko, I. U., and
Lemeshko, V. V. (1987) Byul. Eksp. Biol. Med., 104,
546-548.
160.Wahi, S., Kaul, N., Ganguly, N. K., Varma,
S., Sharma, B. K., and Wahi, P. L. (1991) Can. J. Cardiol.,
7, 229-233.
161.Hoshida, S., Kuzuya, T., Fuji, H., Oe, H.,
Hori, M., Kamada, T., and Tada, M. (1993) Cardiovasc. Res.,
27, 377-383.
162.Klebanov, G. I., Kreinina, M. V., Pozin, V. M.,
Skuratovskaia, S. G., and Pocheptsova, G. A. (1988) Byul. Eksp.
Biol. Med., 106, 297-299.
163.Klebanov, G. I., Kreinina, M. V., Chukaeva, I.
I., Barbarash, O. L., Korochkin, I. M., and Vladimirov, Iu. A. (1990)
Byul. Eksp. Biol. Med., 109, 334-336.
164.Medvedeva, L. V., Popova, T. N.,
Artyukhov, V. G., Matasova, L. V., and Pinheiro de
Carvalho, M. A. (2002) Biochemistry (Moscow),
67, 696-705.
165.Safonova, O. A., Popova, T. N.,
Matasova, L. V., and Artiukhov, V. G. (2005) Biomed.
Khim., 51, 311-320.
166.Syrkin, A. L., Barsel, V. A., Alliluev, I. G.,
Kogan, I. G., Aksiutina, M. S., Eliubaeva, I. D., Nesmeianova, M.
A., and Drozdov, D. V. (1996) Klin. Med. (Moscow), 74,
24-27.
167.Baj, Z., Kowalski, J., Kantorski, J., Pokoca,
L., Kosmider, M., Pawlicki, L., and Tchorzewski, H. (1994)
Atherosclerosis, 106, 159-168.
168.Giannitsis, E., Tettenborn, I.,
Schmucker, G., Mitusch, R., Wiegand, U., Potratz, J.,
Sheikhzadeh, A., and Stierle, U. (1997) Int. J. Cardiol.,
61, 229-237.
169.Siminiak, T., O’Gorman, D. J.,
Shahi, M., Hackett, D., and Sheridan, D. J. (1995) Br. Heart
J., 74, 625-630.
170.Kowalski, J., Kosmider, M., Pasnik, J., Zeman,
K., Baj, Z., Janiszewska-Drobinska, B., and Czekalska, R. (1999)
Fundam. Clin. Pharmacol., 13, 237-242.
171.Fukuda, H., Sawa, Y., Kadoba, K., Taniguchi,
K., Shimazaki, Y., and Matsuda, H. (1995) Circulation,
92, II413-II416.
172.Stevens, R. M., Jahania, M. S., Stivers,
J. E., Mentzer, R. M., Jr., and Lasley, R. D. (2002) Ann. Thorac.
Surg., 73, 1261-1266.
173.Akiyama, K., Kimura, A., Suzuki, H., Takeyama,
Y., Gluckman, T. L., Terhakopian, A., Katagiri, T., Suh, K. Y., Roseto,
J., and Bing, R. J. (1998) J. Am. Coll. Cardiol., 32,
373-379.
174.Alexandrova, M. L., Bochev, P. G., Markova, V.
I., Bechev, B. G., Popova, M. A., Danovska, M. P., and
Simeonova, V. K. (2001) Luminescence, 16,
357-365.
175.Frassetto, S. S., Schetinger, M. R., Webber,
A., Sarkis, J. J., and Netto, C. A. (1999) Braz. J. Med. Biol.
Res., 32, 1295-1302.
176.Yamaguchi, S., Ogata, H., Hamaguchi, S.,
and Kitajima, T. (1998) Can. J. Anaesth., 45,
226-232.
177.Xie, H., Ray, P. E., and Short, B. L. (2005)
Stroke, 36, 1047-1052.
178.Roitberg, G. E. (ed.) (2007) Metabolic
Syndrome [in Russian], MEDpress-inform, Moscow, p. 224.
179.Florian, M., Freiman, A., and Magder, S. (2004)
Steroids, 69, 779-787.
180.Kougias, P., Chai, H., Lin, P. H.,
Lumsden, A. B., Yao, Q., and Chen, C. (2005) J. Vasc.
Surg., 41, 691-698.
181.Napoli, C., Aldini, G., Wallace, J. L., de
Nigris, F., Maffei, R., Abete, P., Bonaduce, D., Condorelli, G., Rengo,
F., Sica, V., D’Armiento, F. P., Mignogna, C., de Rosa, G.,
Condorelli, M., Lerman, L. O., and Ignarro, L. J. (2002)
Proc. Natl. Acad. Sci. USA, 99, 1689-1694.
182.Jacobi, J., Kristal, B., Chezar, J., Shaul, S.
M., and Sela, S. (2005) Free Radic. Biol. Med., 39,
1238-1248.
183.Niwa, Y., Sakane, T., Shingu, M., and
Yokoyama, M. M. (1983) J. Clin. Immunol., 3,
228-240.
184.Justice, J. M., Tanner, M. A., and Myers, P. R.
(2000) J. Cell. Physiol., 182, 359-365.
185.Zhu, L., Castranova, V., and He, P. (2005)
Am. J. Physiol. Heart Circ. Physiol., 288,
H1331-H1338.
186.Gamkrelidze, M., Mamamtavrishvili, N.,
Bejitashvili, N., Sanikidze, T., and Ratiani, L. (2008) Georgian
Med. News, 54-57.
187.Zalba, G., Beloqui, O., San Jose, G., Moreno,
M. U., Fortuno, A., and Diez, J. (2005) Arterioscler. Thromb. Vasc.
Biol., 25, 1452-1457.
188.Prasad, K., and Lee, P. (2003) J.
Cardiovasc. Pharmacol. Ther., 8, 61-69.
189.Radwanska-Wala, B., Buszman, E., and Druzba, D.
(2008) Wiad. Lek., 61, 67-73.
190.Sayre, L. M., Perry, G., and Smith, M. A.
(2008) Chem. Res. Toxicol., 21, 172-188.
191.Shibata, N., and Kobayashi, M. (2008) Brain
Nerve, 60, 157-170.
192.Repetto, M. G., Reides, C. G., Evelson,
P., Kohan, S., de Lustig, E. S., and Llesuy, S. F. (1999) Eur. J.
Clin. Invest., 29, 643-649.
193.Licastro, F., Morini, M. C., Davis, L. J.,
Malpassi, P., Cucinotta, D., Parente, R., Melotti, C., and
Savorani, G. (1994) J. Neuroimmunol., 51, 21-26.
194.Layton, M. E., and Pazdernik, T. L. (1999)
J. Mol. Neurosci., 13, 63-68.
195.Famulari, A. L., Marschoff, E. R., Llesuy, S.
F., Kohan, S., Serra, J. A., Dominguez, R. O., Repetto, M.,
Reides, C., and Sacerdote de Lustig, E. (1996) J. Neurol. Sci.,
141, 69-78.
196.Gatto, E. M., Carreras, M. C.,
Pargament, G. A., Riobo, N. A., Reides, C., Repetto, M.,
Fernandez Pardal, M. M., Llesuy, S., and Poderoso, J. J. (1996) Mov.
Disord., 11, 261-267.
197.Arnhold, J., Sonntag, K., Sauer, H.,
Hantzschel, H., and Arnold, K. (1994) J. Biolum. Chemilum.,
9, 79-86.
198.Bostan, M., Brasoveanu, L. I.,
Livescu, A., Manda, G., Neagu, M., and Iordachescu, D. (2001)
J. Cell Mol. Med., 5, 188-194.
199.Crouch, S. P., Crocker, I. P., and
Fletcher, J. (1995) J. Immunol., 155, 5436-5443.
200.Marhoffer, W., Stein, M., and Federlin, K.
(1993) Immun. Infekt., 21 (Suppl. 1), 15-16.
201.Bender, J. G., van Epps, D. E., Searles,
R., and Williams, R. C., Jr. (1986) Inflammation, 10,
443-453.
202.Gale, R., Bertouch, J. V., Bradley, J., and
Roberts-Thomson, P. J. (1983) Ann. Rheum. Dis., 42,
158-162.
203.Nurcombe, H. L., Bucknall, R. C., and Edwards,
S. W. (1991) Ann. Rheum. Dis., 50, 147-153.
204.Gale, R., Bertouch, J. V., Gordon, T. P.,
Bradley, J., and Roberts-Thomson, P. J. (1984) Ann. Rheum. Dis.,
43, 34-39.
205.D’Onofrio, C., Maly, F. E., Fischer,
H., and Maas, D. (1984) Klin. Wochenschr., 62,
710-716.
206.Bohm, U., Meretey, K., Sesztak, M., Hodinka,
L., Koo, E., and Zahumenszky, Z. (1988) Z. Rheumatol.,
47, 161-165.
207.Meske, S., Maly, F. E., Estefan, M., and
Muller, W. (1985) Z. Rheumatol., 44, 41-45.
208.Miesel, R., Murphy, M. P., and Kroger, H.
(1996) Free Radic. Res., 25, 161-169.
209.Meier, B., Radeke, H. H., Selle, S.,
Raspe, H. H., Sies, H., Resch, K., and Habermehl, G. G.
(1990) Free Radic. Res. Commun., 8, 149-160.
210.Hyvonen, P. M., and Kowolik, M. J. (1991) J.
Clin. Lab. Immunol., 35, 47-55.
211.Hyvonen, P. M., and Kowolik, M. J. (1993) J.
Clin. Lab. Immunol., 40, 69-76.
212.Bostan, M., Galatiuc, C., Hirt, M., Constantin,
M. C., Brasoveanu, L. I., and Iordachescu, D. (2003) J. Cell Mol.
Med., 7, 57-66.
213.Chou, C. T., Shaio, M. F., Chang, F. Y., and
Chang, M. L. (1991) Zhonghua Min Guo Wei Sheng Wu Ji Mian Yi Xue Za
Zhi (Chin. J. Microbiol. Immunol.), 24, 281-288.
214.Edwards, S. W., Hallett, M. B., and
Campbell, A. K. (1984) Biochem. J., 217,
851-854.
215.Hafstrom, I., Gyllenhammar, H., Palmblad, J.,
and Ringertz, B. (1989) J. Rheumatol., 16, 1033-1037.
216.Leirisalo-Repo, M., Paimela, L., Koskimies, S.,
and Repo, H. (1993) Inflammation, 17, 427-442.
217.Magaro, M., Altomonte, L., Zoli, A., Mirone,
L., De Sole, P., Di Mario, G., Lippa, S., and Oradei, A. (1988) Ann.
Rheum. Dis., 47, 793-796.
218.Magaro, M., Zoli, A., Altomonte, L., Mirone,
L., De Sole, P., Di Mario, G., and De Leo, E. (1992) Ann. Rheum.
Dis., 51, 877-880.
219.Halliwell, B. (2007) Biochem. J.,
401, 1-11.
220.Valko, M., Rhodes, C. J., Moncol, J., Izakovic,
M., and Mazur, M. (2006) Chem. Biol. Interact., 160,
1-40.
221.Chen, C., Liu, F. K., Qi, X. P., and Li, J. S.
(2003) World J. Gastroenterol., 9, 242-245.
222.Antonov, A. R., Babyshkina, Yu. G., and
Kiselev, A. B. (2004) Vestnik Nov. Med. Tekhnol., 24.
223.Tullgren, O., Giscombe, R., Holm, G.,
Johansson, B., Mellstedt, H., and Bjorkholm, M. (1991) Clin. Exp.
Immunol., 85, 436-440.
224.Shaughnessy, S. G., Buchanan, M. R., Turple,
S., Richardson, M., and Orr, F. W. (1989) Am. J. Pathol.,
134, 787-796.
225.Bilenko, M. V., Vladimirov, Iu. A., Pavlova, S.
A., Thu Thuy, N. T., and Hai Yen, T. T. (2008) Biomed. Khim.,
54, 445-453.