REVIEW: Modular Nanotransporters of Anticancer Drugs Conferring Cell Specificity and Higher Efficiency
A. S. Sobolev1,2
1Institute of Gene Biology, Russian Academy of Sciences, ul. Vavilova 34/5, 119334 Moscow, Russia; E-mail: sobolev@igb.ac.ru2Biological Faculty, Lomonosov Moscow State University, 119191 Moscow, Russia
Received December 3, 2008; Revision received June 18, 2009
This review deals with artificial modular nanotransporters (MNT) of polypeptide nature for drug delivery into target cells and then into a specified cell compartment like the nucleus. The developed approach is based on the use of intracellular transport processes characteristic of practically all cells, including cancer cells. The first MNT module ligand carries out a double function: specific recognition of a cancer target cell and penetration into the cell via receptor-mediated endocytosis. The movement of the MNT within the cell along this path specifies the need to supply the MNT with an endosomolytic module making it possible to leave the endocytotic pathway before getting into lysosomes in order to have time for interaction with importins. For this purpose, a polypeptide fragment able to make defects in membranes only at the pH of endosomes is used as the second module. Delivery into the cell nucleus is provided by the third module containing an amino acid sequence of nuclear localization, “recognized” by importins located in the hyaloplasm. And finally, the fourth module, a carrier for joining the transported drug, is incorporated into the MNT. Depending on the type of ligand module, MNT for different target cell types have been produced. Each module retains its activity within the MNT, ligand modules bind target receptors with high affinity, while the module with the nuclear localization sequence binds importins. The endosomolytic module forms pores in lipid membranes through which MNT are able to leave acidifying cell compartments (endosomes). Modules within MNT can be replaced or transposed, which makes it possible to use them for delivery of different drugs into different target cells and their compartments. It was shown that photosensitizers and radionuclides used for cancer therapy acquire pronounced cell specificity as well as the 10-1000-fold higher efficiency resulting from their delivery into the most vulnerable compartment – the cell nucleus.
KEY WORDS: modular drug nanotransporters, drug delivery, photosensitizers, alpha-emitters, ErbB1 over-expression, intracellular transport, cancer therapyDOI: 10.1134/S0006297909130094
Abbreviations: APE, alpha-particle emitters; 211At-SAGMB, N-succinimidyl-3-[211At]astato-4-guanidinomethylbenzoate; DTox, endosomolytic module based on diphtheria toxin translocation domain; EGF, epidermal growth factor; HMP, hemoglobin-like protein of Escherichia coli; MSH, α-melanocyte-stimulating hormone; MNT, modular nanotransporters; NLS, nuclear localization sequence (amino acid); PS, photosensitizers.
It is desirable to determine just which drugs are reasonable to deliver
into a prescribed cell compartment using modular nanotransporters (MNT)
before starting their description and characteristics of the latter. At
least two groups of drugs can be considered. The first group consists
of substances that are able to exhibit their action only after
appearing in a certain cell compartment (like DNA in the cell nucleus).
The second group might include antitumor drugs able to carry out
cytotoxic action in any part of a cell, but the cell compartment most
sensitive to their action can be found. It can be characterized as the
compartment into which a minimal dose of this cytotoxic agent will
cause cell death. Examples of this group are photosensitizers (PS) used
for photodynamic therapy of a number of diseases, especially for tumor
diseases, and radionuclides emitting short-range particles (such as
alpha-particle emitters (APE)) used for endotherapy of malignant tumors
or Auger1 electron emitters suggested for the same
purpose.
1 Electron emission emerging during an atomic transition from an excited state to a lower energy state not accompanied by photon emission. It is known concerning PS that: (i) active oxygen forms (singlet oxygen, hydroxyl radical, and some other free radicals) are the primary cytotoxic factor of PS; (ii) the cell nucleus is the cell compartment most sensitive to damaging effect of reactive oxygen species; (iii) PS are localized in different cell compartments with the exception of cell nucleus; (iv) in the case of systemic introduction, blood proteins bind PS, which probably define greater uptake of PS by cells than the physicochemical properties of the PS; (v) photo-physical properties of PS within their complexes with blood proteins can differ from the properties of the free PS. Previous reviews, including those by the author and his colleagues [1, 2], dealt with the bases of these theses. It is known already for over 50 years that the cell nucleus is the most sensitive cell compartment for alpha particles [3]. As for Auger electron emitting radionuclides, they are practically outside the cell nucleus [4]. These conclusions suggest the expediency of minimization of random PS interactions with blood components and giving to PS and APE the ability to penetrate into the target cell nuclei.
ADDRESSED DELIVERY OF PHOTOSENSITIZERS INTO THE NUCLEI OF TARGET
CELLS
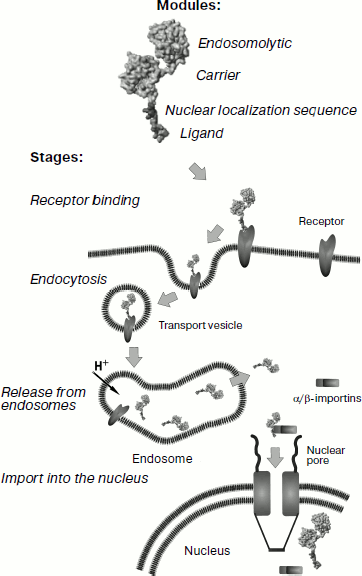
It is possible to deliver PS or APE into the cell nucleus by creation of special transporters with properties, prescribed in advance, which could provide for the target cell “recognition”, absorption by this cell of transporters with PS or APE, and their following penetration into the cell nucleus. To achieve this, we elaborated modular transporters (for review see [1]) having (1) an internalized ligand module providing for the target cell “recognition” and subsequent receptor-mediated transporter endocytosis; (2) an endosomolytic module allowing the transporter to leave endosomes; (3) a module with the nuclear localization sequence (NLS) via which it interacts with importins, the cytosol proteins providing for active transport into the nucleus, and (4) a carrier module for joining other modules into the unit as well as for the delivered drug. Terms “module” and “modular” are used here in their direct sense, because the design of such transporters should suggest the possibility of transporter switching to different target cell types and even to different cell compartments, which can be achieved more rapidly and efficiently if the transporter components are easily replaceable/rearrangeable. The necessity of several, not less than four, components is defined by the following reasons. First, it is possible to confer cell specificity to the transporter simultaneously with the ability to penetrate into the target cell if it has a component that highly specifically binds the internalized and endocytosis-specific receptors. It is important that the used receptors are overexpressed on target cells and are poorly represented (in an ideal case they are absent) on closely located normal cells. Second, it is possible to achieve specific intranuclear delivery if the transporter has NLS. Third, the above-mentioned importins are cytosol proteins, whereas the transporter that entered the cell via receptor-mediated endocytosis is included in endocytotic vesicles (endosomes, etc.) and therefore it is separated from importins. This means that the transporter cannot interact with them, and it needs an additional component to provide for transporter release from endocytotic vesicles. Fourth, all components or modules should be combined in one moiety, the transporter, and it should be possible to join transported drugs to it; the carrier module is intended for this purpose.
In the beginning, we tried to prove in principal the possibility of solving this problem using polypeptide conjugates containing the above-mentioned modules and created by the cross-linking of the modules by bifunctional reagents (see in more detail [5-9]). It appeared that these molecular constructs, having the prescribed set of modules, really provided for the specific delivery of PS into target cells, internalization in the latter, release from intracellular vesicles, and delivery into the nucleus. Individual modules within the constructs retained their functions and served for the main task – achievement of the high efficiency and cell specificity of the PS. Note, the internalized PS exhibited higher cytotoxicity compared to the same PS localized on the cell surface [10-12], whereas the same PS that got into the nucleus via the construct was more efficient than the internalized PS [5, 6]; free PS exhibited the least efficiency in photodynamic cell damage. Thus, the conclusion concerning the cell nucleus hypersensitivity to the photodynamic effect of PS was confirmed.
It is important to know to what extent different transport constructs are realized technologically; multicomponent transporters, obtained by cross-linking using special reagents (see above), could scarcely become widely used because their production is labor-consuming and expensive. However, these transporters designed by us at the first stage did not pretend to possible practical use: their task was to check the correctness of the approach as such. However, for the aims of possible practical application we have designed recombinant modular nanotransporters (MNT; the scheme of their structure and stages of penetration into a cell is shown in Fig. 1) containing (1) α-melanocyte-stimulating hormone (MSH) or epidermal growth factor (EGF) as internalized ligand modules binding to melanocortin-1 receptors, overexpressed on the human and mouse melanoma cells, or by ErbB1 receptors, overexpressed on the cells of head and neck cancer, cancer of esophagus, (urinary) bladder, and some others; (2) optimized NLS of SV40 virus large T-antigen; (3) the hemoglobin-like protein (HMP) of E. coli as a carrier module, and (4) translocation domain of diphtheria toxin as the endosomolytic module (DTox) [13-15]. MNT with the purity of 90-98% were obtained. Later MNT with different ligand modules such as interleukin-3 (targets of this MNT were the acute myeloid leukemia cells with overexpression of interleukin-3 receptors) and somatostatin (for neuroblastoma cells with overexpression of somatostatin receptors) were designed [16].
At the next step it was necessary to check to what extent modules incorporated in MNT retained their functions required for achievement of the main goal – specific intracellular drug delivery to target cells.Fig. 1. Scheme of modular MNT structure and stages of its penetration into the target cell. The first MNT module, ligand, performs a double function: specific “recognition” of cancer target cell and penetration into this cell via receptor-mediated endocytosis. The second, endosomolytic module allows MNT to “go off” the endocytotic pathway before getting into lysosomes in order to allow the nanotransporter to interact with importins. For this purpose, a polypeptide fragment able to make defects in membranes at pH specific for endosomes was used as the second module. Delivery into the cell nucleus is provided by the third module, containing NLS, “recognized” by importins present in the hyaloplasm. The fourth MNT module is the carrier module for attachment of the transported drug.
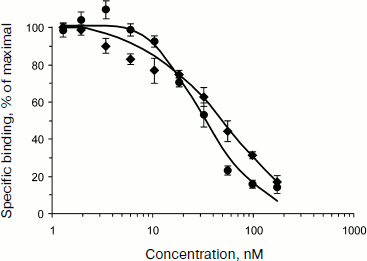
Functional activity of EGF-containing MNT was estimated [15] by binding by EGF receptors on human A431 epidermoid carcinoma cells overexpressing these receptors [17], while for MSH-containing MNT this estimation was carried out on mouse melanoma B16-F1 cells with overexpressed melanocortin-1 receptors [13]. Dissociation constants (Kd) for MNT complexes like HMP-NLS-DTox-EGF and DTox-HMP-NLS-EGF with EGF receptors (Fig. 2) were 40 and 29 nM, respectively, which is close to the Kd for 125I-labeled EGF (Fig. 2). In the 13-amino acid oligopeptide MSH, affinity to melanocortin receptors was somewhat lower after its incorporation into MSH-containing MNT (approximately 2·10–8 M) [13].
The fate of MNT bound to internalized receptors is predetermined by processes of receptor-mediated endocytosis: in particular, MNT should appear in endosomes, closed membrane vesicles with gradually acidified content, and the MNT should actively leave them in order to enter the cytosol in which importins, able to provide for MNT delivery to the cell nucleus after binding to NLS, are localized. The release from endosomes should be carried out by the endosomolytic module DTox, the task of which was creation of defects in membranes on the side characterized by weakly acidic medium (like that inside endosomes).Fig. 2. Competition of two MNT—DTox-HMP-NLS-EGF (closed circle) and HMP-NLS-DTox-EGF (closed rhomb)—with 125I-labeled DTox-HMP-EGF (20 nM) for binding to ErbB1 receptors on human epidermoid carcinoma A431 cells [15]. The labeled truncated MNT variant, 125I-labeled DTox-HMP-EGF, has Kd = 110 nM and 3.1·106 specific binding sites per A431 cell.
The ability of polypeptides to make pores in membranes can be estimated by the release of dye from preloaded liposomes [18]. The MNT under study caused dye release in two pH intervals. The first was between pH 5.5 and 6.5 that was close to pH in endosomes and was dependent on the DTox module [13, 15], because it itself made pores in this pH interval [13]. The second was revealed in a more acidic region with pH maximum from 3.0 to 4.0 and it was possible to attribute it to an effect of HMP.
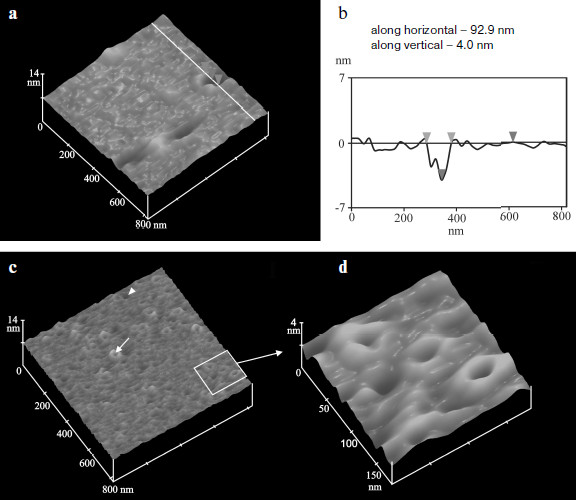
Defects in membranes created by MNT and found in experiments on liposomes were characterized electrochemically and by atomic-force microscopy [15, 19, 20]. Studying of conductivity of planar bilayer lipid membranes after addition of MNT at pH 5.5 revealed the emergence of ion channels with conductivity about 2-5 nS, whereas MNT without the endosomolytic module did not exhibit such effect. No channels emerged in response to the full-sized MNT at neutral pH 7.0. In 5-15 min after medium acidification to pH 5.5, atomic-force microscopy reveals circular structures of 30-50 nm diameter in supported lipid bilayer (egg lecithin) in the presence of MNT (Fig. 3). In 40-60 min pores of 50-200 nm diameter and of depth equal to bilayer thickness could be detected. No such alterations were found at pH 7.0. It was possible to show that the emergence of fluctuating pores was caused by the effect of two membrane-active domains—DTox and HMP. The diameter of these pores (50-200 nm) significantly exceeds the dimensions of MNT, and owing to this the MNT molecules not bound to bilayer are probably able to leave endosomes and reach their destination. It is interesting that the endosomolytic module DTox, included in different MNT regions, nevertheless caused formation of identical defects in lipid membranes [15], which suggests its ability to function in different polypeptide contexts; this supposition agrees with results of Nizard et al. [21].
It was found by biospecific atomic-force microscopy that elevations, observed on bilayer and forming circular structures, are often seen near fluctuating pores and are formed by MNT molecules. Biospecificity to MNT of the cantilever tip of the atomic-force microscope was achieved by its modification using affinity-purified rabbit antibodies to MNT. Such tip was used to obtain the curves of dependence of strength of the tip interaction with lipid bilayer and with MNT on the latter from the distance between the tip and these objects. It was shown that the average strength of the MNT–antibody specific interaction was 192 ± 23 pN, which is characteristic of specific antigen–antibody interactions [22].Fig. 3. Atomic-force microscopy of defects in lipid bilayers caused by MNT DTox-HMP-NLS-EFG at pH 5.5 [15]. a) Large pores and their cross-section (b) by perpendicular plane along white line with characteristics of width (between light gray signs) and depth (between gray signs); c) small defects and their enlarged image (d). One small defect surrounded by a bolster is shown by a white arrow, and one large fluctuating pore is designated by a white sign.
It is known that in the case of scanning by a tip modified by antibodies (or by antigen) an increase in apparent height of scanned objects by 1-2 nm due to the antigen–antibody interactions is observed under the tapping mode conditions [23]. Scanning the MNT-containing lecithin bilayers by unmodified probe at pH 5.5 revealed three types if particles with mean heights 1.1, 2.2, and 4.4 nm. Three types of particles were also observed during scanning under the tapping mode conditions by the tip modified with antibodies to MNT. However, in this case their apparent heights were significantly higher by 1-2 nm: 2.0 (p < 0.001), 4.3 (p < 0.05), and 7.0 nm. This means that all these particles were formed by MNT molecules and, according to their mean height, they are MNT molecules incorporated in the bilayer, adsorbed on bilayer surface, and forming aggregates there. Thus, the full-sized MNT, i.e. MNT containing all four modules including the endosomolytic module DTox, in the case of medium acidification to pH 5.5 can form in bilayers pores bordered by MNT and having dimensions sufficient for MNT release through them.
Finally, functional activity of the endosomolytic module was confirmed on the cellular level. Measuring pH of intracellular MNT microenvironment by image ratio video microscopy [13] was used to reveal the ability of MNT with incorporated DTox to leave the acidified endocytotic compartments. In experiments on live cells of Claudmann mouse melanoma S91 (clone M3), the truncated MNT variant without endosomolytic module was detected in vesicles with weakly-acidic and acidic content, whereas the full-sized MNT (with DTox module) was present in the neutral microenvironment.
Surface plasmon resonance was used to characterize the interaction of NLS within MNT with the α/β-importin dimer [15]: affinity constants of the studied MNT to the importin dimer appeared to be very close to the constant of free polypeptide with the same NLS [24], which suggests that this module is fully functional [25].
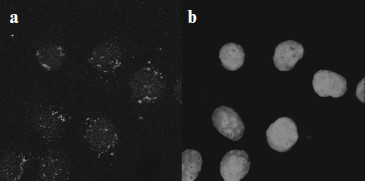
Intracellular localization of full-sized MNT appeared to be almost completely intranuclear [13, 15] (Fig. 4 shows as an example localization of DTox-HMP-NLS-EGF in cells of human epidermoid carcinoma A431).
Finally, the covalent PS (bacteriochlorin p) attachment to MNT via spacer 1,5-diaminopentane had no effect on production of active oxygen forms by this PS, which was possible to show using spin traps for hydroxyl radicals and singlet oxygen [15].Fig. 4. Intracellular localization of DTox-HMP-NLS-EGF in human epidermoid carcinoma A431 cells [15]. a) Immunocytochemical detection of DTox-HMP-NLS-EGF; b) the same group of cells as in (a) with DNA stained by intercalating dye ToPro-3.
Thus, all modules within MNT retained their inherent functions which made it possible to achieve the main goal – MNT delivery into the target cell nucleus; besides the PS drug covalently attached to MNT did not lose the ability to generate an active participant of all this type cytotoxic drugs – reactive oxygen species.
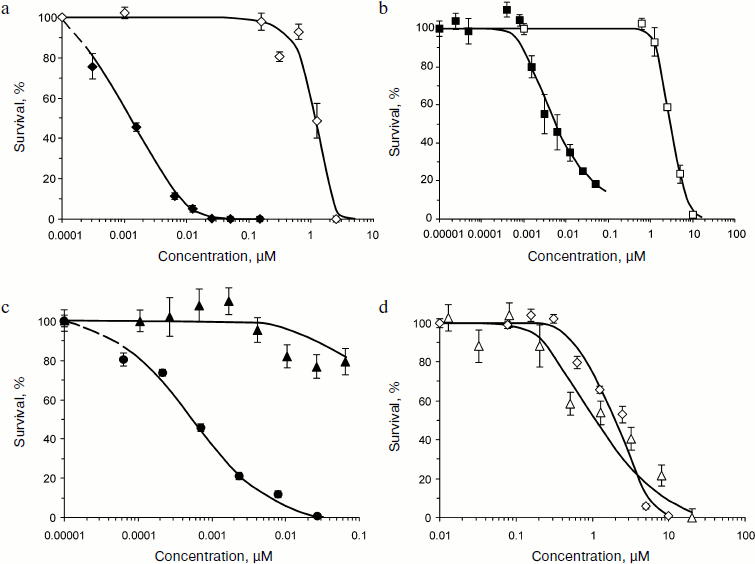
The complete functional activity of modules within MNT made it possible to use MNT for cell-specific intranuclear delivery of antitumor drugs. The pronounced, over 1000-3000 times enhancement of cytotoxic effect of PS chlorin e6 and bacteriochlorin p, delivered by MNT into cell nuclei compared to the effect of free PS (Fig. 5) was revealed in experiments [15] on human epidermoid carcinoma A431 cells overexpressing ErbB1 receptors; the estimation was based on the EC50 ratio, i.e. on the PS concentrations providing for half-maximal effect. Moreover, MNT conferred cell specificity to PS: chlorin e6 practically equally affected both target cells (A431) and “non-target” cells expressing a low amount of ErbB1 receptors (NIH 3T3 cells), whereas in MNT-PS conjugate was inefficient for NIH 3T3 cells within the range of concentrations killing target A431 cells (Fig. 5). Similar results [13] were obtained using MSH-containing MNT on mouse melanoma B16-F1 cells overexpressing receptors for MSH [26-29]. Cytotoxic effect of PS bacteriochlorin p conjugated with MNT (bacteriochlorin p-DTox-HMP-NLS-EGF) was characterized by EC50 = 22 nM, whereas free bacteriochlorin p had EC50 = 4990 nM, 230 times higher. Bacteriochlorin p-DTox-HMP-NLS-EGF was not toxic for normal mouse fibroblasts C3H/10T1/2 and NIH 3T3 that did not overexpress melanocortin-1 receptors. The authors believe that differences in efficiency of EGF- and MSH-containing MNT may be caused by different amounts of overexpressed receptors on the cells: ~104 per cell of B16-F1 melanoma and >106 per cell of A431epidermoid carcinoma. The truncated variant of MNT (bacteriochlorin p-HMP-NLS-MSH) devoid of endosomolytic module was 5.3 times less active than the full-sized MNT, while MNT without NLS-containing module was even less cytotoxic. The comparison of efficiencies of the full-sized MNT and its truncated variants devoid of any modules indicates that the presence of all the modules in MNT is necessary for its maximal efficiency.
In vivo experiments [15] were carried out on mouse model of skin melanoma, a tumor that is non-optimal for photodynamic therapy because it almost completely absorbs light due to very intensive melanin pigmentation. However, a significant MNT effect obtained in experiments with bacteriochlorin p on melanoma cells in vitro as well as spectral peculiarities of this PS (absorption maximum at 761 nm, i.e. in the region of deeper light penetration into tissues compared to most PS) were arguments in favor of such experiments. The MSH-containing MNT administered intravenously to C57/black mice with syngeneic melanoma B16-F1 (inoculated subcutaneously) already 3 h later began to accumulate in tumor cells and their nuclei. Bacteriochlorin p in itself was incapable of influencing either the rate of tumor growth or an average survival of mice with melanoma, whereas the same doses of this PS introduced by the same scheme but transported by MNT reliably inhibited tumor growth and increased average survival of tumor-carrying mice.Fig. 5. Photocytotoxicity and cell specificity of photosensitizers transported by MNT compared to free photosensitizers [15]. a) Chlorin e6-HMP-NLS-DTox-EGF (closed rhomb) and free chlorin e6 (open rhomb); b) bacteriochlorin p-HMP-NLS-DTox-EGF (closed square) and free bacteriochlorin p (open squares); c) chlorin e6-DTox-HMP-NLS-EGF on target cells A431 (closed circle) and on “non-target” cells NIH 3T3 (closed triangle); d) free chlorin e6 on target cells A431 (open rhomb) and on “non-target” cells NIH 3T3 (open triangle). Mean ± standard error.
ADDRESSED DELIVERY OF ALPHA-PARTICLE EMITTERS INTO TARGET CELL
NUCLEI
Results obtained with PS point to promising use of nanotransporters for delivery of different drugs like radionuclides emitting alpha-particles. In our first experiments with APE [30] we used modular polypeptide conjugates obtained by module joining by bifunctional cross-linking reagents (their application for PS delivery is described above); they exhibited the expected efficiency: the radioactivity dose necessary for damage of 63% cells (A0) by alpha-emitter 211At delivery into the nuclei of human hepatoma PLC/PRF/5 cells decreased by one order of magnitude.
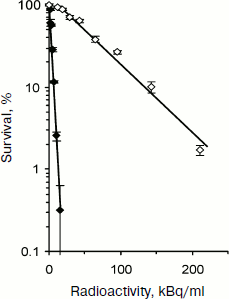
Recently [31, 32] we applied recombinant MNT (DTox-HMP-NLS-EGF) labeled by 211At using N-succinimidyl-3-[211At]astato-4-guanidinomethylbenzoate (211At-SAGMB) [33] for directed damage of cancer cells with ErbB1 receptor overexpression: human epidermoid carcinoma A431 and two lines of human glioblastoma D247 MG and U87MG.wtEGFR. It was shown in preliminary experiments that MNT labeling does not change its properties. Experiments on determination of cell survival (clonogenic ability) clearly showed more pronounced cytotoxicity of 211At-SAGMB-MNT compared to free 211At on all three lines of cancer cells. For D247 MG glioblastoma cells (Fig. 6) the A0 values for 211At-SAGMB-MNT and free 211At were equal to 3.8 and 69.0 kBq/ml, respectively. In other words, the delivery of 211At into target cell nuclei increased its cytotoxicity by 18.2 times. For two other lines A431 and U87MG.wtEGFR the increase in cytotoxicity was 14.5 and 8.3 times, respectively. In the case of prolonged incubation of glioblastoma D247 MG cells with 211At-SAGMB-MNT or free 211At (control), when about 90% radionuclide decayed, comparison of obtained A0 values and following quantitative calculations indicated the involvement of recoil nuclei formed during 211At alpha-decay in the 211At-SAGMB-MNT effect [32].
Modular MNT developed by us make it possible to confer cell specificity and high efficiency to a number of antitumor drugs because they were constructed of modules with prescribed properties that provided for “recognition” of a necessary target cell and its following directed transport into the cell nucleus. MNT modules constituting either fragments of various natural polypeptides (NLS-containing and endosomolytic modules) or originally whole molecules (ligand modules, module-carrier) are functional within a single chimeric artificial MNT molecule. Target cell “recognition” by transporters along with penetration into them is achieved due to the MNT ligand module, capable of binding with high affinity to internalized receptors, overexpressed on target cells (but not on surrounding “non-target” cells). Thus, highly specific ligand binding to receptor provides for cell specificity and following receptor-mediated MNT endocytosis. The MNT release from endosomes, necessary for MNT protection against degradation in lysosomes and final entering the nucleus, is carried out by the endosomolytic module. Specific intracellular delivery is carried out by the module with an appropriate amino acid sequence, and in the case of delivery into the nucleus – with NLS. Finally, the carrier module provides for joining the MNT modules and attachment of delivered drug. The modular principle of MNT design makes possible module replacement or change of their arrangement within the MNT upon changing the task: a change in the target cell type, a change in intracellular target compartment, etc. We believe that the MNT described here can be considered as new pharmacological agents of wide use.Fig. 6. Survival (estimation by colony formation) of glioblastoma D247 MG cells at different added radioactivity of astatine-211 in the form of 211At-SAGMB-MNT (closed signs) or free astatide-211 (open signs) (according to [32]).
This work was supported by grants of the Netherlands Organization for Scientific Research (NOW) (grant No. 047.017.025), CRDF (grant No. RUB-02-2663-MO-05) and of the National Institutes of Health of the USA NIH/NINDS (grant No. NS20023).
REFERENCES
1.Sobolev, A. S., Jans, D. A., and Rosenkranz, A. A.
(2000) Prog. Biophys. Mol. Biol., 73, 51-90.
2.Castano, A. P., Demidova, T. N., and Hamblin, M. R.
(2004) Photodiagnosis Photodynam. Ther., 1, 279-293.
3.Hall, E. J. (1994) Radiobiology for the
Radiologist, 4th Edn., J. B. Lippincott, Philadelphia, p. 376.
4.Boswell, C. A., and Brechbiel, M. W. (2005)
J. Nucl. Med., 46, 1946-1947.
5.Akhlynina, T. V., Jans, D. A., Rosenkranz, A. A.,
Statsyuk, N. V., Balashova, I. Y., Toth, G., Pavo, I., Rubin, A. B.,
and Sobolev, A. S. (1997) J. Biol. Chem., 272,
20328-20331.
6.Akhlynina, T. V., Jans, D. A., Statsyuk, N. V.,
Balashova, I. Y., Toth, G., Pavo, I., Rosenkranz, A. A., Naroditsky, B.
S., and Sobolev, A. S. (1999) Int. J. Cancer, 81,
734-740.
7.Chopp, M., Dereski, M. O., Madigan, L., Jiang, F.,
and Logie, B. (1996) Radiat. Res., 146, 461-465.
8.Rosenkranz, A. A., Jans, D. A., and Sobolev, A. S.
(2000) Immunol. Cell Biol., 78, 452-464.
9.Sobolev, A. S., Akhlynina, T. V., Rosenkrants, A.
A., and Jans, D. A. (2002) US Patent No. 6,500,800.
10.Akhlynina, T. V., Gulak, P. V., Serebryakova, N.
V., Rosenkranz, A. A., and Sobolev, A. S. (1990) Byul. Eksp. Biol.
Med., 109, 150-152.
11.Akhlynina, T. V., Rosenkranz, A. A., Jans, D. A.,
Gulak, P. V., Serebryakova, N. V., and Sobolev A. S. (1993)
Photochem. Photobiol., 58, 45-48.
12.Hamblin, M. R., Miller, J. L., and Ortel, B.
(2000) Photochem. Photobiol., 72, 533-540.
13.Rosenkranz, A. A., Lunin, V. G., Gulak, P. V.,
Sergienko, O. V., Shumiantseva, M. A., Voronina, O. L., Gilyazova, D.
G., John, A. P., Kofner, A. A., Mironov, A. F., Jans, D. A., and
Sobolev, A. S. (2003) FASEB J., 17, 1121-1123.
14.Rosenkranz, A. A., Lunin, V. G., Sergienko, O.
V., Gilyazova, D. G., Voronina, O. L., Jans, D. A., Kofner, A. A.,
Shumiantseva, M. A., Mironov, A. F., and Sobolev, A. S. (2003)
Genetika, 39, 259-268.
15.Gilyazova, D. G., Rosenkranz, A. A., Gulak, P.
V., Lunin, V. G., Sergienko, O. V., Khramtsov, Y. V., Timofeyev, K. N.,
Grin, M. A., Mironov, A. F., Rubin, A. B., Georgiev, G. P., and
Sobolev, A. S. (2006) Cancer Res., 61, 10534-10540.
16.Sobolev, A. S. (2008) BioEssays,
30, 278-287.
17.Lokeshwar, V. B., Huang, S. S., and Huang, J. S.
(1989) J. Biol. Chem., 264, 19318-19326.
18.Nir, S., and Nieva, J. L. (2000) Progr. Lipid
Res., 39, 181-206.
19.Khramtsov, Y. V., Rokitskaya, T. I., Rosenkranz,
A. A., Gnuchev, N. V., Antonenko, Y. N., and Sobolev, A. S. (2008)
J. Contr. Release, 128, 241-247.
20.Rosenkranz, A. A., Khramtsov, Y. V., Trusov, G.
A., Gnuchev, N. V., and Sobolev, A. S. (2008) Doklady AN (Biokhim.
Biofiz. Mol. Biol.), 421, 835-837.
21.Nizard, P., Chenal, A., Beaumelle, B., Fourcade,
A., and Gillet, D. (2001) Protein Eng., 14,
439-446.
22.Hinterdorfer, P., Baumgartner, W., Gruber, H. J.,
Schilcher, K., and Schilder, H. (1996) Proc. Natl. Acad. Sci.
USA, 93, 3477-3481.
23.Raab, A., Han, W., Badt, D., Smith-Gill, S. J.,
Lindsay, S. M., Schindler, H., and Hinterdorfer, P. (1999) Nat.
Biotechnol., 17, 901-905.
24.Catimel, B., Teh, T., Fontes, M. R., Jennings, I.
G., Jans, D. A., Howlett, G. J., Nice, E. C., and Kobe, B. (2001) J.
Biol. Chem., 276, 34189-34198.
25.Hodel, M. R., Corbett, A. H., and Hodel, A. E.
(2001) J. Biol. Chem., 276, 1317-1325.
26.Jiang, J., Sharma, S. D., Fink, J. L., Hadley, M.
E., and Hruby, V. J. (1996) Exp. Dermatol., 5,
325-333.
27.Funasaka, Y., Sato, H., Chakraborty, A. K.,
Ohashi, A., Chrousos, G. P., and Ichihashi, M. (1999) J. Invest.
Dermatol. Symp. Proc., 4, 105-109.
28.Wikberg, J. E., Muceniece, R., Mandrika, I.,
Prusis, P., Lindblom, J., Post, C., and Skottner, A. (2000)
Pharmacol. Res., 42, 393-420.
29.Salazar-Onfray, F., Lopez, M., Lundqvist, A.,
Aguirre, A., Escobar, A., Serrano, A., Korenblit, C., Petersson, M.,
Chhajlani, V., Larsson, O., and Kiessling, R. (2002) Br. J.
Cancer, 87, 414-422.
30.Rosenkranz, A. A., Nabatnikov, P. A., Aliev, R.
A., Jans, D. A., and Sobolev, A. S. (2002) Mol. Med. (Moscow),
2, 47-55.
31.Rosenkranz, A., Vaidyanathan, G., Pozzi, O.,
Lunin, V., Khramtsov, Y., Zalutsky, M., and Sobolev, A. (2007) J.
Nucl. Med., 48, 80P.
32.Rosenkranz, A. A., Vaidyanathan, G., Pozzi, O.
R., Lunin, V. G., Zalutsky, M. R., and Sobolev, A. S. (2008) Int. J.
Rad. Oncol. Biol. Phys., 72, 193-200.
33.Vaidyanathan, G., Affleck, D. J., Bigner, D. D.,
and Zalutsky, M. R. (2003) Nucl. Med. Biol., 30,
351-359.