Biophysical Characterization of a Recombinant Leucyl Aminopeptidase from Bacillus kaustophilus
Meng-Chun Chi1, Hui-Ping Chang1, Gu-Gang Chang2, Tzu-Fan Wang3, Hsien-Bin Huang3, and Long-Liu Lin1*
1Department of Applied Chemistry, National Chiayi University, 300 University Road, Chiayi, Taiwan; fax: +886(5)271-7901; E-mail: llin@mail.ncyu.edu.tw2Department of Life Sciences and Institute of Genome Science, National Yang-Ming University, Taipei, Taiwan
3Department of Life Sciences and Institute of Molecular Biology, National Chung Cheng University, Chiayi, Taiwan
* To whom correspondence should be addressed.
Received August 26, 2009; Revision received January 26, 2010
The biophysical properties of Bacillus kaustophilus leucyl aminopeptidase (BkLAP) were examined in terms of analytical ultracentrifugation, fluorescence spectroscopy, and circular dichroism. By using the analytical ultracentrifuge, we demonstrated that tetrameric BkLAP exists as the major form in solution at protein concentration of 1.5 mg/ml at pH 8.0. The native enzyme started to unfold beyond ~1 M GdnHCl and reached an unfolded intermediate with [GdnHCl]1/2 at 1.8 M. Thermal unfolding of BkLAP was found to be highly irreversible and led to a marked formation of aggregates.
KEY WORDS: Bacillus kaustophilus, leucyl aminopeptidase, analytical ultracentrifuge, thermal unfolding, chemical denaturationDOI: 10.1134/S0006297910050159
Abbreviations: BkLAP, B. kaustophilus LAP; CD, circular dichroism; GdnHCl, guanidine hydrochloride; LAP, leucyl aminopeptidase; L-Leu-p-NA, L-leucine-p-nitroanilide; Ni2+-NTA, nickel nitrilotriacetate; p-NA, p-nitroaniline; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Leucyl aminopeptidases (LAPs; EC 3.4.11.1) are members of the M17 family
metallopeptidases that catalyze the removal of amino acids from the
N-terminus of proteins, peptides, and synthetic substrates. The enzyme
is widely distributed in organisms from bacteria to humans with the
best characterized being those from Bos taurus, Escherichia
coli, and Lycopersicon esculentum [1, 2]. X-Ray crystal structures of the bovine lens and
E. coli enzymes have shown that they exist as homohexamers
comprising two trimers stacked on top of one another [3, 4]. In many cases, each monomer
binds two zinc ions and folds into two α/β-type
quasi-spherical globular domains to generate a comma-like shape,
although the E. coli pepA-encoded aminopeptidase presumably
binds manganese through residues that are absolutely conserved and
located at the C-terminal catalytic domain [4].
The biological roles of LAPs are not completely understood, and might be complex and species-specific. Escherichia coli PepA serves as an aminopeptidase [5] and a DNA-binding protein that mediates both site-specific recombination and transcriptional control [6]. LAP-A of tomato is induced in response to wounding, tissue-damaging insects, and exogenous jasmonic acid [7]. Moreover, the human homolog is implicated in the processing of peptides released from the proteasome [8]. Besides the uncertainty of their biological roles, there has been rather little experimental work done on protein unfolding of the M17 LAPs. Protein unfolding reactions have been shown to proceed through a variety of mechanisms [9]. The simplest mechanism is that of a two state transition in which a reaction that proceeds directly from folded to unfolded state without the occurrence of any detectable intermediate. However, many studies have demonstrated that folding and unfolding processes of several proteins involve multiple steps that are associated with one or more intermediates [10-12]. Investigation of unfolding pathways of proteins is an intriguing scientific challenge, and a wide variety of biophysical methods, such as fluorescence and circular dichroism (CD), have been employed for this purpose. The typical CD spectra of proteins originating in the near- and far-UV region form the basis of CD applications to protein structure [13].
Earlier, a gene encoding Bacillus kaustophilus LAP (BkLAP) was cloned and overexpressed in E. coli [14]. Site-directed mutagenesis studies demonstrated that the conserved Asn345, Thr346, Ala348, Gly350, Leu352, and Asn435 residues play important roles in the catalytic reaction of the enzyme [15-17]. In this investigation, we have demonstrated the quaternary structure and the thermal and chemical unfolding of BkLAP, a member of the M17 family enzyme. These biophysical studies can be expected to provide insights into the molecular structure and the stability of the enzyme.
MATERIALS AND METHODS
Materials. Growth medium components were purchased from Difco Laboratories (USA). The expression vector pQE-LAP used was constructed as previously described [14]. Escherichia coli M15 and nickel nitrilotriacetate (Ni2+-NTA) resin were obtained from Qiagen Inc. (USA). LAP activity was determined with L-leucine-p-nitroanilide (L-Leu-p-NA) and p-nitroaniline (p-NA), which were acquired from Sigma-Aldrich Fine Chemicals (USA).
Purification of His6-tagged BkLAP. Escherichia coli M15 (pQE-LAP) was cultivated overnight with shaking in Luria–Bertani (LB) medium (10 g/liter bacto-tryptone, 5 g/liter yeast extract, and 5 g/liter NaCl) containing ampicillin (100 mg/liter) and kanamycin (25 mg/liter) at 37°C. The culture was transferred to fresh LB medium and incubated at 28°C. Four hours after the transfer, 1 mM isopropyl-β-D-thiogalactopyranoside was added and the cultivation was continued for 12 h. Cells were collected by centrifugation, resuspended in 20 mM Tris-HCl buffer (pH 8.0) containing 5 mM imidazole and 0.5 M NaCl, and disrupted by sonication at 4°C. Cellular debris was removed by centrifugation and the supernatant was subjected to His6-tagged BkLAP purification using a Ni2+-NTA column. Sequential washing and eluting procedures were performed according to the protocol of Qiagen to obtain the purified enzyme.
Electrophoresis and activity staining. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed using the Laemmli buffer system [18]. Prior to electrophoresis, the samples were heated at 100°C for 5 min in dissociating buffer containing 2% SDS and 5% 2-mercaptoethanol. After electrophoresis, the gels were stained with 0.25% Coomassie Brilliant Blue R-250 dissolved in 50% methanol–10% acetic acid and destained in a solution of 30% methanol and 10% acetic acid. Protein markers were phosphorylase b (97.4 kDa), bovine serum albumin (66.3 kDa), ovalbumin (45.0 kDa), carbonic anhydrase (31.0 kDa), and trypsin inhibitor (21.5 kDa).
The native protein was fractioned on a 10% nondenaturing polyacrylamide gel and electrophoresis was done at 4°C at constant voltage of 100 V for 3 h. The LAP activity band was detected by incubating the gel at 55°C in 50 ml of 100 mM phosphate buffer (pH 5.8) containing 0.4% L-leucyl-2-naphthylamide (Sigma-Aldrich) and 1% Fast garnet salt GBC (Sigma-Aldrich). A dark blue band was visualized within 30 min.
Analytical ultracentrifugation. Sedimentation velocity was measured in a Beckman-Coulter XL-A analytical ultracentrifuge. Sample (370 µl) and buffer (400 µl) solutions were loaded into the double sector centerpiece separately, and built up in a Beckman An-50Ti rotor. Experiments were performed at 20°C and rotor speed of 42,000 rpm. Protein sample was monitored by UV absorbance at 280 nm in a continuous mode with a time interval of 8 min and a step size of 0.003 cm. Multiple scans at different time points were fitted to a continuous size distribution model by the program SEDFTT [19]. All size distributions were solved at a confidence level of P = 0.95, a best-fitted average anhydrous frictional ration (f/fo), and a resolution N of 200 sedimentation coefficients between 0.1 and 20 S.
Unfolding of BkLAP in guanidine hydrochloride. The purified BkLAP was unfolded with different concentrations of GdnHCl in 20 mM Tris-HCl buffer (pH 8.0) at 30°C for 10 min. The unfolding of the enzyme was monitored by fluorescence and the loss of enzymatic activity.
Fluorescence spectra were recorded in a Hitachi F-7000 fluorescence spectrophotometer at 30°C, and all spectra were corrected for buffer absorption. The excitation wavelength was set at 280 nm and the fluorescence emission spectrum was scanned from 300 to 400 nm. The maximal peak of the fluorescence spectrum and the change in fluorescence intensity were used to monitor the unfolding of the enzyme.
Circular dichroism spectroscopy. CD spectra were measured using a JASCO J-815 spectropolarimeter equipped with a PTC-423S/15 Peltier-type cell holder for temperature control. Thermal denaturation transitions were tracked by examining the ellipticity changes at a wavelength of 220 nm. Heating rates varied from 0.5 to 4°C/min, and 1 cm pathlength cells were used. Mean residue ellipticity [θ] is expressed in the unit of degrees cm2·dmol–1, which is defined as [θ] = M·θ/(100·c·l), where M represents the average molecular weight of amino acids, θ is the observed ellipticity in degrees, c is the concentration in residue moles per liter, and l is the pathlength in centimeters.
The kinetics of secondary structure changes upon thermal denaturation of BkLAP were followed by changes in ellipticity at 220 nm. Cells of 1 cm pathlength were filled up to 98% of their total volumes with 20 mM Tris-HCl buffer (pH 8.0). After temperature equilibration, the necessary amount of concentrated BkLAP solution was added to complete the cell volume. Samples were vigorously stirred to promote rapid mixing and temperature equilibration. Under these conditions, the dead time of experiments was less than 10 sec. Unfolding data were fitted to a biphasic decay equation, θt = θf + A1·exp(–ku1t) + A2·exp(–ku2t), where θt is the ellipticity measured at time t, θf is the final ellipticity value, A1 and A2 represent the amplitudes of each phase, and ku1 and ku2 are the unfolding rate constants for each phase.
RESULTS AND DISCUSSION
The M17 LAPs are bilobed protomers that contain a larger C-terminal catalytic domain and a smaller, more variable N-terminal domain. The primary structure of BkLAP comprises 497 amino acid residues corresponding to a molecular mass of 53,785 Da. To determine the biophysical properties of BkLAP, the enzyme in the crude extract of E. coli M15 (pQE-LAP) was purified to near homogeneity using Ni2+-NTA resin. The molecular mass of the purified enzyme was 54 kDa (Fig. 1a) and the purification procedure resulted in a protein yield of ~45%.
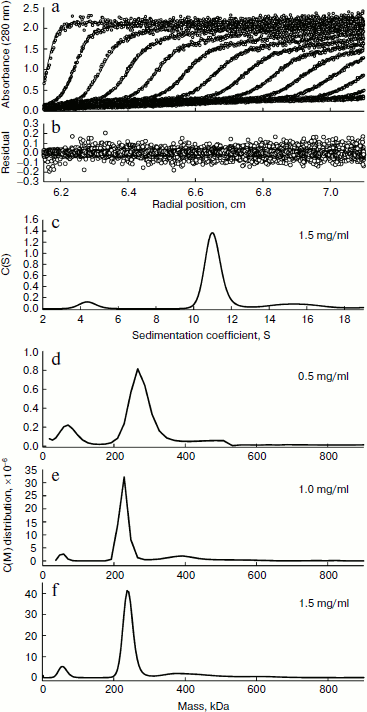
The quaternary structure of BkLAP was examined by analytical ultracentrifugation. Most of the BkLAP molecules sediment at 11.1 S (Fig. 2c) corresponding to a species with molar mass of 231.4 kDa (Fig. 2d), which is in agreement with the molecular weight calculated from amino acid sequence of tetramer (215.2 kDa). There are two small peaks at 4.4 and 15.4 S, respectively, corresponding to 57.8 kDa (monomer) and 410.1 kDa (octamer), respectively. The excellent matching of the experimental data points and the fitted curve (Fig. 2a), the homogeneous bitmap picture (Fig. 2b), and the randomly distributed residual values (data not shown) all indicated that a highly reliable model for the sedimentation velocity experiments was obtained and the analytical ultracentrifugation was an excellent biophysical probe for assessing the quaternary structure of the enzyme.Fig. 1. a) Analysis of the purified BkLAP by SDS-PAGE. b, c) Non-denaturing PAGE of the purified enzyme. The protein sample was electrophoresed on 10% polyacrylamide gel and visualized by Coomassie brilliant blue staining (b) and activity staining (c).
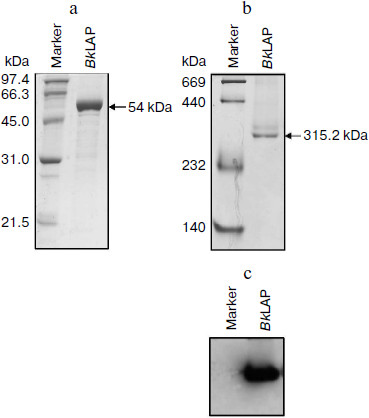
The self-association of proteins plays an important role in the catalytic activity of a variety of enzymes, such as cytidine 5′-triphosphate synthase from E. coli [20], dihydrofolate reductase from Thermotoga maritima [21], and γ-cyclodextrin-specific cyclodextrinase from Bacillus clarikii [22]. X-Ray crystal structures of bovine lens and E. coli LAPs have shown these enzymes exist as homohexamers and fold into two α/β-type quasi-spherical globular domains [3, 4]. Similar to bovine lens and E. coli enzymes, the tomato wound-induced LAP-A was determined to be a hexameric enzyme using a native gel assay [23], and the multimeric structure is critical for the catalytic activity [24]. Native-PAGE analysis also shows that the molecular size of a M17 family LAP from Schizosaccharomyces pombe appears to be a homohexameric protein [25]. As shown in Figs. 1b and 1c, the molecular size of native BkLAP was 315.2 kDa as estimated from the native gel, reflecting that the enzyme exists as a hexamer, and it had LAP activity (Fig. 1c). However, an accurate molecular size cannot be determined on native gel due to factors affecting the mobility of proteins in native gels include size, shape, and intrinsic charge [26]. Additionally, the AUC data clearly indicate that the tetramer is a predominant form of BkLAP at protein concentrations up to 1.5 mg/ml (Fig. 2f). Protein oligomerization is reported as a concentration-dependent event and increases logarithmically with increasing concentration [27]. Accordingly, many proteins were reported to have an interconvertible mixture of monomers and oligomers [20, 28, 29]. Although bovine lens and E. coli LAPs have been described as hexamers in the past, it is still not clear whether the M17 family metallopeptidases form active tetramers or hexamers, or what the physiological role of a possible oligomerization state is. At present our findings cannot directly address these issues, but we report for the first time that BkLAP, a M17 family enzyme, exists in solution as a tetramer.Fig. 2. Sedimentation velocity experiments of the purified BkLAP. a) A typical trace of absorbance at 280 nm of the enzyme during the sedimentation velocity experiment. The symbols are experimental data and the lines are computer-generated results by fitting the experimental data to the Lamm equation using the SEDFIT program. b) The residuals of the model fitting of the data in panel (a). In panels (a) and (c), the protein concentration was 1.5 mg/ml in 50 mM Tris-HCl buffer (pH 8.0). c) The continuous sedimentation coefficient distribution of the enzyme at high protein concentration. d-f) The continuous molar mass distribution of the enzyme at different protein concentrations.
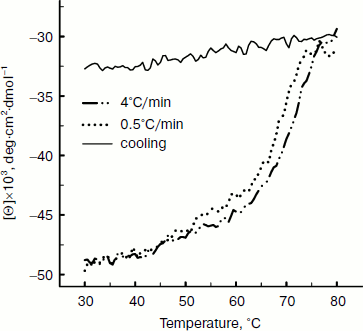
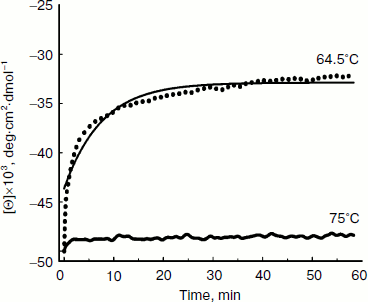
Thermal denaturation of BkLAP was followed by monitoring the ellipticity at 220 nm at constant heating rates. After the thermal denaturation transitions went to completion, the protein solutions were cooled to 30°C at the same scan speed. Figure 3 shows the transition curves obtained with BkLAP solutions at heating rates of 0.5 and 4°C/min and the curve observed when cooling the sample at a scan speed of 4°C/min. It is clear that the transition at 0.5°C/min appeared at lower temperature than that of 4°C/min. This resembles a common result in transitions under kinetic control [30-32]. Additionally, the lower heating rate resulted in larger changes in the far-UV CD transition, probably indicating aggregation of the enzyme. In all cases, aggregation of the protein samples was confirmed by light scattering at wavelengths higher than 300 nm (data not shown). Also, it can be seen that the secondary structure was not recovered in either case. To further explore if the unfolding reaction was reversible or not, the sample was heated for 1 min at 75°C (Fig. 4). Under these conditions, the secondary structure of the native protein disappeared as revealed by CD signal. The native secondary structure of the polypeptide was not recovered immediately after the sample was cooled to 30°C, and the secondary structure of the native polypeptide was not recovered (data not shown). This indicates that the thermal denaturation of BkLAP is highly irreversible even at the very early stages of the reaction. The time course of BkLAP denaturation at 64.5°C was also examined by changes in far-UV CD at 220 nm. As shown in Fig. 4, a double exponential decay curve fitted well to the experimental data. This might indicate the presence of at least one kinetic intermediate.
Fig. 3. Heating and cooling profiles of BkLAP in Tris-HCl buffer (pH 8.0). Transitions were obtained by recording the ellipticity of the sample at 220 nm. Protein concentration was 1.0 mg/ml.
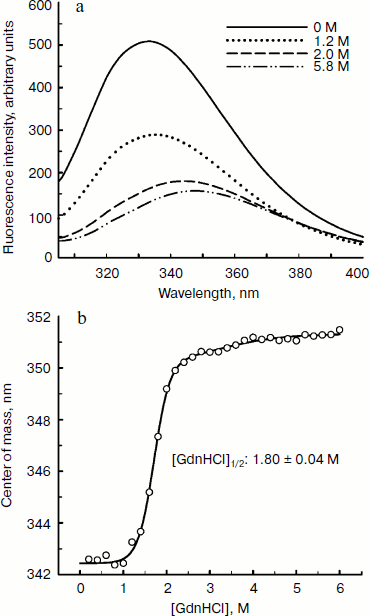
Since aromatic amino acid residues are sensitive to the polarity of their immediate environment, changes to the center of mass (λcm, which represents the average energy of the emission spectrum) will reflect conformational changes induced in the protein. Figure 5 shows that BkLAP exhibited maximum emission at approximately 334 nm in the absence of GdnHCl and suffered typical alterations to the spectra as a function of GdnHCl concentration (Fig. 5a). Low concentration of GdnHCl (~1 M) induced only a small red shift in the spectral center of mass, whereas the tryptophan residues in the protein are highly exposed to the buffer environment at values above 3 M, allowing us to consider the protein completely unfolded. Fitting the data shown in Fig. 5b resulted in a [GdnHCl]1/2 value of 1.80 ± 0.04 M.Fig. 4. Kinetics of thermal denaturation of BkLAP in Tris-HCl buffer (pH 8.0) at two different temperatures followed by CD spectroscopy at 220 nm. Protein concentration was 1.0 mg/ml.
In conclusion, the analytical ultracentrifugation studies of BkLAP can elucidate the inherent quaternary structure of the M17 family enzyme. Although further work is required to construct the N-terminal and C-terminal domain-truncated enzyme for the evaluation of their roles in thermal and GdnHCl-induced unfolding, the current work contributes to a better understanding of the biophysical properties of the M17 family enzyme, an aspect which has been largely neglected in the past and which is fundamental to a fuller appreciation of the mechanism of their activities at the molecular level.Fig. 5. Fluorescence spectroscopy of BkLAP. a) The effect of GdnHCl-induced unfolding on the tertiary structure of the enzyme. The protein sample was exposed to 0-5.8 M GdnHCl at 25°C in 50 mM Tris-HCl buffer (pH 8.0). b) Spectral center of mass as a function of GdnHCl concentration.
Financial support (NSC 95-2313-B-415-2-MY3 and NSC 97-2628-B-415-0-MY3) provided by the National Science Council of Taiwan is gratefully acknowledged.
REFERENCES
1.Taylor, A. (1993) FASEB J., 7,
290-298.
2.Matsui, M., Fowler, J. H., and Walling, L. L.
(2006) Biol. Chem., 387, 1535-1544.
3.Burley, S. K., David, P. R., Taylor, A., and
Lipscomb, W. (1990) Proc. Natl. Acad. Sci. USA, 87,
6878-6882.
4.Strater, N., Sherrat, D. J., and Collons, S. D.
(1999) EMBO J., 18, 4513-4522.
5.Vogt, V. M. (1970) J. Biol. Chem.,
245, 4760-4769.
6.Charlier, D., Kholti, A., Huysveld, N., Gigot, D.,
Maes, D., Thia-Toong, T. L., and Glansdorff, N. (2000) J. Mol.
Biol., 302, 411-426.
7.Chao, W. S., Gu, Y. Q., Pautot, V., Bray, E. A.,
and Walling, L. L. (1999) Plant Physiol., 120,
979-992.
8.Beninga, J., Rock, K. L., and Goldberg, A. L.
(1998) J. Biol. Chem., 273, 18534-18542.
9.Daggett, M., and Fersht, A. R. (2003) Trends
Biochem. Sci., 28, 18-25.
10.Nolting, B., Golbik, R., and Fersht, A. R. (1995)
Proc. Natl. Acad. Sci. USA, 92, 10668-10672.
11.Plaxco, K. W., and Dobson, C. M. (1996) Curr.
Opin. Struct. Biol., 6, 630-636.
12.Panda, K. W., Gorovits, B. M., and Horowitz, P.
M. (2000) J. Biol. Chem., 275, 63-70.
13.Screerama, N., and Woody, K. W. (2004) Meth.
Enzymol., 383, 318-351.
14.Lin, L. L., Hsu, W. H., Wu, C. P., Chi, M. C.,
Chou, W. M., and Hu, H. Y. (2004) Extremophiles, 8,
79-87.
15.Chi, M. C., Huang, H. B., Liu, J. S., Wang, W.
C., Liang, W. C., and Lin, L. L. (2006) FEMS Microbiol. Lett.,
260, 156-161.
16.Chi, M. C., Liu, J. S., Wang, W. C., Lin, L. L.,
and Huang, H. B. (2008) Biochimie, 90, 811-819.
17.Chi, M. C., Ong, P. L, Hsu, W. H., Chen, Y. H.,
Huang, H. B., and Lin, L. L. (2008) Int. J. Biol. Macromol.,
43, 481-487.
18.Laemmli, U. K. (1970) Nature, 227,
680-685.
19.Brown, P. H., and Schuck, P. (2006) Biophys.
J., 90, 4651-4661.
20.Lunn, F. A., MacLeod, T. J., and Bearne, S. L.
(2008) Biochem. J., 412, 113-121.
21.Loveridge, E. J., Rodriguez, R. J., Swanwick, R.
S., and Allemann, R. K. (2009) Biochemistry, 48,
5822-5933.
22.Nakagawa, Y., Saburi, W., Takada, M., Hatada, Y.,
and Horikoshi, K. (2008) Biochim. Biophys. Acta, 1784,
2004-2011.
23.Gu, Y. Q., Pautot, V., Holzer, F. M., and
Walling, L. L. (1996) Plant Physiol., 110, 1257-1266.
24.Gu, Y. Q., Holzer, F. M., and Walling, L. L.
(1999) Eur. J. Biochem., 263, 726-735.
25.Herrera-Camacho, I., Rosas-Murrieta, N. H.,
Rolo-Domingguez, A., Millan, L., Reyes-Leyva, J., Santos-Lopez, G., and
Suarez-Rendueles, P. (2007) FEBS J., 274, 6228-6240.
26.Gallagher, S. R. (2001) in Curr. Protoc.
Protein Sci., Chap. 6, Unit 6.5, John Wiley & Sons, New
York.
27.Shriver, J. W., and Edmondson, S. P. (2009)
Meth. Mol. Biol., 490, 57-82.
28.Gruber, C. W., Cemazar, M., Mechler, A., Martin,
L. L., and Crail, D. J. (2009) Peptide Sci., 92,
35-43.
29.Dengra-Pozo, J., Martínez-Rodriguez, S.,
Contreras, L. M., Prieto, J., Andujar-Sanchez, M., Clemente-Jimenez, J.
M., Las Heras-Vazquez, F. J., Rodriguez-Vico, F., and Neira, J. L.
(2009) Biopolymers, 91, 757-772.
30.Freire, E., van Osdol, W. W., Mayorga, O. L., and
Sanchez-Ruiz, J. M. (1990) Annu. Rev. Biophys. Biophys. Chem.,
19, 159-188.
31.Sanchez-Ruiz, J. M. (1992) Biophys. J.,
61, 921-935.
32.Tello-Solis, S. R., and Hernandez-Arana, A.
(1995) Biochem. J., 311, 969-974.