Effects of C-Terminal Truncation on Autocatalytic Processing of Bacillus licheniformis γ-Glutamyl Transpeptidase
Hui-Ping Chang1, Wan-Chi Liang1, Rui-Cin Lyu1, Meng-Chun Chi1, Tzu-Fan Wang2, Kuo-Liang Su3, Hui-Chih Hung3, and Long-Liu Lin1*
1Department of Applied Chemistry, National Chiayi University, 300 Syuefu Road, Chiayi County 60004, Taiwan; fax: +886-5-271-7901; E-mail: llin@mail.ncyu.edu.tw2Department of Life Sciences and Institute of Molecular Biology, National Chung Cheng University, 168 University Road, Minhsiung Township, Chiayi County 62102, Taiwan
3Institute of Genomics and Bioinformatics, National Chung Hsing University, 250 Kuokuang Road, Taichung County 400, Taiwan
* To whom correspondence should be addressed.
Received January 18, 2010; Revision received April 30, 2010
The role of the C-terminal region of Bacillus licheniformis γ-glutamyl transpeptidase (BlGGT) was investigated by deletion analysis. Seven C-terminally truncated BlGGTs lacking 581-585, 577-585, 576-585, 566-585, 558-585, 523-585, and 479-585 amino acids, respectively, were generated by site-directed mutagenesis. Deletion of the last nine amino acids had no appreciable effect on the autocatalytic processing of the enzyme, and the engineered protein was active towards the synthetic substrate L-γ-glutamyl-p-nitroanilide. However, a further deletion to Val576 impaired the autocatalytic processing. In vitro maturation experiments showed that the truncated BlGGT precursors, pro-Δ(576-585), pro-Δ(566-585), and pro-Δ(558-585), could partially precede a time-dependent autocatalytic process to generate the L- and S-subunits, and these proteins showed a dramatic decrease in catalytic activity with respect to the wild-type enzyme. The parental enzyme (BlGGT-4aa) and BlGGT were unfolded biphasically by guanidine hydrochloride (GdnCl), but Δ(577-585), Δ(576-585), Δ(566-585), Δ(558-585), Δ(523-585), and Δ(479-585) followed a monophasic unfolding process and showed a sequential reduction in the GdnCl concentration corresponding to half effect and ΔG0 for the unfolding. BlGGT-4aa and BlGGT sedimented at ~4.85 S and had a heterodimeric structure of approximately 65.23 kDa in solution, and this structure was conserved in all of the truncated proteins. The frictional ratio (f/fo) of BlGGT-4aa, BlGGT, Δ(581-585), and Δ(577-585) was 1.58, 1.57, 1.46, and 1.39, respectively, whereas the remaining enzymes existed exclusively as precursor form with a ratio of less than 1.18. Taken together, these results provide direct evidence for the functional role of the C-terminal region in the autocatalytic processing of BlGGT.
KEY WORDS: Bacillus licheniformis, γ-glutamyl transpeptidase, C-terminal truncation, autocatalytic processing, analytical ultracentrifugationDOI: 10.1134/S0006297910070151
Abbreviations: AEW, average emission wavelength; BlGGT, EcGGT, and HpGGT, Bacillus licheniformis, Escherichia coli, and Helicobacter pylori γ-glutamyl transpeptidases; CD, circular dichroism; IPTG, isopropyl-β-D-thiogalactopyranoside; L-γ-Glu-p-NA, L-γ-glutamyl-p-nitroanilide; Ntn, N-terminal nucleophile; p-NA, p-nitroaniline; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
γ-Glutamyl transpeptidase (GGT; EC 2.3.2.2) is a member of the
N-terminal nucleophile (Ntn) hydrolase superfamily [1] and catalyzes the transfer of the γ-glutamyl
group of glutathione and related compounds to an acceptor that can be
an amino acid, a peptide, or water [2]. The
inactive precursor of Ntn hydrolases must undergo intramolecular
autocatalytic processing to generate the mature active enzyme. The new
N-terminal residue, typically a serine, threonine, or cysteine, acts as
a nucleophile for both autocatalytic processing and catalytic reaction.
GGTs are generally considered to be involved in the metabolism of
glutathione and in salvaging of cysteine [3, 4]. Helicobacter pylori GGT (HpGGT) has
been shown to degrade extracellular glutathione and glutamine,
providing an advantage for the bacterium within its growth environment
[5]. Similarly, upregulation of human GGT in cancer
cells is generally known to help supplement these rapidly dividing
cells with essential amino acids for protein biosynthesis [6, 7].
Members of Ntn hydrolases, despite lacking any discernible sequence identity, share the same tertiary fold consisting of a four-layer αββα-structure with two antiparallel β-sheets between α-helical layers [8]. Recently, crystal structures of the bacterial GGTs (PDB code for GGT of Bacillus subtilis, Escherichia coli, and H. pylori is 2v36, 2dbu, and 2nqo, respectively) have indicated a heterodimeric arrangement (one large subunit + one small subunit) and contributed greatly to our understanding of the maturation and catalytic reaction of Ntn hydrolases. Escherichia coli GGT (EcGGT) becomes active only after cleavage of the peptide bond between Gln390 and Thr391 in its precursor protein [9]. The structure of the acyl-enzyme intermediate of EcGGT elucidates the role of Thr391 and the location of the donor substrate-binding site [10]. Structural evidence further confirms the autocatalytic processing of EcGGT [11], and the structure of HpGGT has provided more information on this mechanism [12, 13].
Earlier, the gene encoding Bacillus licheniformis GGT (BlGGT) was cloned and overexpressed in E. coli [14]. BlGGT is synthesized as a 61.259-kDa polypeptide precursor made up a large subunit of 374 residues and a small subunit of 187 residues (Swiss-Prot Q65KZ6). Recently, we constructed six N-terminally truncated mutants of BlGGT and found that removal of the signal peptide significantly affects the functional expression of the enzyme in recombinant E. coli [15]. Kinetic and mutagenesis studies of HpGGT have demonstrated that a Thr380-Thr398 dyad is critical for efficient cleavage of the γ-glutamyl peptide bond of glutathione [13]. The corresponding residues, Thr399 and Thr417, of BlGGT have been shown to be important for the enzymatic maturation and reaction [16]. To our knowledge, there has been no experimental work done to investigate effects of deletion of the C-terminal region of GGTs on enzyme maturation. In this study, one full-length BlGGT and seven truncated proteins lacking the last 5, 9, 10, 20, 28, 63, and 107 amino acids were constructed to gain insight into the role of this region. We have narrowed the critical amino acid residues to Val576, which is involved in autocatalytic processing and provides the correct conformation of the active site. These results reveal that deletion of more than nine amino acids from the C-terminus of the enzyme would lead to the expression of an inactive precursor, suggesting that the C-terminal region might play a crucial role in BlGGT maturation.
MATERIALS AND METHODS
Materials. A QuikChange II site-directed mutagenesis kit for mutagenic PCR amplification was obtained from Stratagene (USA). Ni2+-nitrilotriacetate (Ni2+-NTA) resin was acquired from Qiagen Inc. (USA). The GGT activity was determined with L-γ-glutamyl-p-nitroanilide (L-γ-Glu-p-NA), Gly-Gly, p-nitroaniline (p-NA), 5-L-glutamyl-2-naphthylamide, and Fast garnet GBC, which were purchased from Sigma-Aldrich Chemicals (USA). All other chemicals were commercial products of analytical or molecular biological grade.
Site-directed mutagenesis. The pQE-BlGGT plasmid [15], which contains a recombinant B. licheniformis ggt gene encoding in order the N-terminal hexahistidine tag, BlGGT, and four additional amino acid residues (Trp-Gly-Thr-Asn), was used as the DNA template for site-directed mutagenesis of codons 586, 581, 577, 576, 566, 558, 523, and 479 to stop codons to generate pQE-BlGGT(1-585), pQE-BlGGT(1-580), pQE-BlGGT(1-576), pQE-BlGGT(1-575), pQE-BlGGT(1-565), pQE-BlGGT(1-557), pQE-BlGGT(1-522), and pQE-BlGGT(1-478), respectively. Two overlapping complementary primers containing the desired nucleotide changes were designed for each mutation (Table 1). The mutagenic PCR reaction mixture consisted of 20 mM Tris-HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100, 0.1 mg/ml nuclease-free bovine serum albumin, 15 ng of template DNA, 0.5 mM dNTPs, 1 µM each of the complementary primers, and 3 units of Pfu DNA polymerase. Mutant DNA was generated with a thermocycling program of 2 min at 95°C and 16 cycles of 30 sec at 95°C, 60 sec at 55°C, and 12 min at 68°C on an Applied Biosystems thermal cycler. The amplified products were digested with 10 units of DpnI at 37°C for 1 h prior to their use for transformation of E. coli XL-1 blue cells. Mutations were confirmed by DNA sequencing, which was carried out with a dye terminator sequencing kit and an automatic DNA sequencer (Applied Biosystems, USA).
Table 1. Oligonucleotide primers used for
site-directed mutagenesis

Expression and purification of parental and truncated enzymes. For high-level expression of parental and mutant enzymes, E. coli M15 harboring pQE-BlGGT or each of the mutated plasmids was grown on LB medium supplemented with ampicillin (100 µg/ml) and kanamycin (25 µg/ml) to an optical density at 600 nm of approximately 0.8. Protein expression was induced by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. The cultivation was continued at 20°C for 12 h. Cells were harvested by centrifugation (4000g for 10 min at 4°C), and the cell pellets were stored at –20°C until required.
To purify His6-tagged enzymes, cell pellets were resuspended in binding buffer (5 mM imidazole, 500 mM NaCl, and 20 mM Tris-HCl, pH 7.9) and lysed by sonication (30-sec bursts and pulses for 5 min). The cell extract was obtained by centrifugation, and the soluble His6-tagged protein was bound to 2 ml of Ni2+-NTA resin by gentle mixing at 4°C for 30 min. Subsequently, the resin was loaded onto a column and washed with three volumes of 50 mM phosphate buffer (pH 7.9) containing 0.3 M NaCl and 20 mM imidazole, and the bound protein was eluted with 5 ml of 250 mM imidazole added to the washing buffer.
Gel electrophoresis, activity staining, and determination of protein concentration. Polyacrylamide gel electrophoresis (PAGE) was performed in a vertical mini-gel system (mini-Protean III system; Bio-Rad Laboratories, USA) with a 10% non-denaturing polyacrylamide gel. Electrophoresis was done at 4°C and a constant voltage of 100 V for 3 h. To detect the GGT activity, the gel was immersed into 1 mM 5-L-glutamyl-2-naphthylamide and 50 mM Gly-Gly in 100 mM Tris-HCl buffer (pH 10) and incubated at 40°C for 30 min. The gel was then transferred into a solution containing 0.05% Fast garnet GBC in 8% acetic acid until the red dark GGT band appeared.
Sodium dodecyl sulfate PAGE (SDS-PAGE) was carried out using the Laemmli buffer system [17]. The gels were stained with 0.25% Coomassie brilliant blue R-250 dissolved in 50% methanol–10% acetic acid and destained in a solution of 30% methanol and 10% acetic acid.
Protein concentrations were measured using the Bio-Rad protein assay reagent and bovine serum albumin as the standard.
Enzyme assays. GGT activity was routinely assayed at 40°C using L-γ-Glu-p-NA as substrate. The reaction mixture contained 1.25 mM L-γ-Glu-p-NA, 30 mM Gly-Gly, 1 mM MgCl2, 50 mM Tris-HCl buffer (pH 8.0), 20 µl enzyme solution at a suitable dilution, and enough distilled water to bring the final volume to 1 ml. After 10-min incubation, the reaction was quenched by the addition of 100 µl 3.5 N acetic acid. The formation of p-NA was recorded by monitoring the absorbance changes at 410 nm. One unit of GGT activity is defined as amount of enzyme that releases 1 µmol of p-NA per minute under the assay conditions.
Km and kcat values were estimated by measuring p-NA production in 1-ml reaction mixtures with various concentrations of the substrate (0.3-2.0 Km). Samples were incubated at 40°C for 10 min. The kinetic parameters were determined using Lineweaver–Burk plots.
Unfolding of recombinant proteins in GdnCl. Parental BlGGT or each of the C-terminally truncated enzymes was unfolded with different concentrations of GdnCl in 20 mM Tris-HCl buffer (pH 8.0) at room temperature. A 10-min incubation was employed in unfolding experiments. The unfolding of the enzyme was monitored by fluorescence and circular dichroism (CD).
Spectrofluorimetric and CD analyses. Fluorescence spectra of parental and mutant enzymes were monitored at 30°C in a Hitachi F-7000 fluorescence spectrophotometer with an excitation wavelength of 280 nm. All spectra were corrected for buffer absorption. The fluorescence emission spectra of protein samples with a concentration of 42 μM were recorded from 300 to 400 nm at a scanning speed of 240 nm/min. The maximal peak of the fluorescence spectrum and the change in fluorescence intensity were used in monitoring the unfolding processes of the enzyme. Both the red shift and the change in fluorescence intensity were analyzed together using the average emission wavelength (AEW) (λ) according to Eq. (1) [18]:
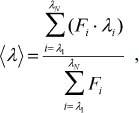 (1)
(1)in which Fi is the fluorescence intensity at the specific emission wavelength (λi).
Far UV-CD spectra of parental and mutant enzymes were acquired on a JASCO model J-815 spectropolarimeter (JASCO Inc., Japan) from 250 to 190 nm in cuvettes at 25°C using 1.0-nm bandwidth, 0.1-nm resolution, 0.1-cm pathlength, 1-sec response time, and 100-nm/min scanning speed. The photomultiplier voltage was always below 600 V in the analyzed region. Each scanning was repeated ten times, and the average is reported. Data were corrected for the buffer effect, and the results are expressed as molar ellipticity [Θ] in the units of deg⋅cm2⋅dmol–1 according to Eq. (2):
where l represents the light pathlength (cm), C is the molar concentration of protein (mol/liter), and Θ represents the observed ellipticity (mdeg). Thermal denaturation experiments were performed by monitoring the ellipticity at 222 nm. The temperature was increased with a heating rate of 2°C/min from 20 to 100°C, and the temperature at which half of the protein molecules were unfolded was recorded (Tm).
Analytical ultracentrifugation. Sedimentation velocity was measured in a Beckman-Coulter XL-A analytical ultracentrifuge with an An50Ti rotor at 20°C and 42,000 rpm in 12-mm double-sector Epon charcoal-filled centerpieces. The ultraviolet (UV) absorption of the cells at 280 nm was scanned in continuous mode with time interval of 8 min and step size of 0.003 cm. The partial specific volume of the enzyme, solvent density, and viscosity were calculated by the free software SEDNTERP (http://www.jphilo.mailway.com/). All samples were visually checked for clarity after ultracentrifugation to make sure that there was no precipitation due to unfolding of the protein. Multiple scans at different time points were analyzed with the SEDFIT program [19, 28]. The sedimentation velocity data were fitted using a two-dimensional distribution with respect to frictional ratio c(s, f/fo) according to the Lamm equation [19] (Eq. (3)):
with a(r,t) denoting the observed optical signal at radius r and time t; χ(s,D,r,t), the solution of the Lamm equation; and D(s, f/fo), the dependence of diffusion coefficient (D) on sedimentation coefficient (s) and frictional ratio (f/fo), where
with ω denoting angular velocity; k, Boltzmann constant; T, absolute temperature; v̄, enzyme partial specific volume; η, buffer viscosity; and ρ, buffer density.
All two-dimensional distributions were solved and normalized to a confidence level of p = 0.95 by maximum entropy and a resolution N of 200 with sedimentation coefficients between 0.1 and 20 S. The anhydrous friction ratio is from 1.0 to 2.0 or 3.5 at a resolution of 10.
Unfolding data analysis. Because unfolding/refolding of BlGGT is a reversible process, the unfolding data were treated with the following thermodynamic models by global fitting of the data. The two-state unfolding model (Scheme 1) was described by Eq. (6) [20]. Equations (7) and (8) were used to fit post-slope and both pre- and post-slope unfolding curves, respectively:
Scheme 1
where
The three-state unfolding model (Scheme 2) was described by Eq. (9) [21]:
Scheme 2
where
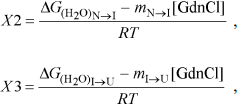
yobs is the observed biophysical signal; yN, yI, and yU are the calculated signals of the native, intermediate, and unfolded states, respectively. [GdnCl] is the GdnCl concentration, and ΔGN–I and ΔGI–U are the free energy changes for the N ↔ I and I ↔ U processes, respectively. The values mU, mN–I, and mI–U are the sensitivities of the respective unfolding states or process to denaturant concentration.
RESULTS AND DISCUSSION
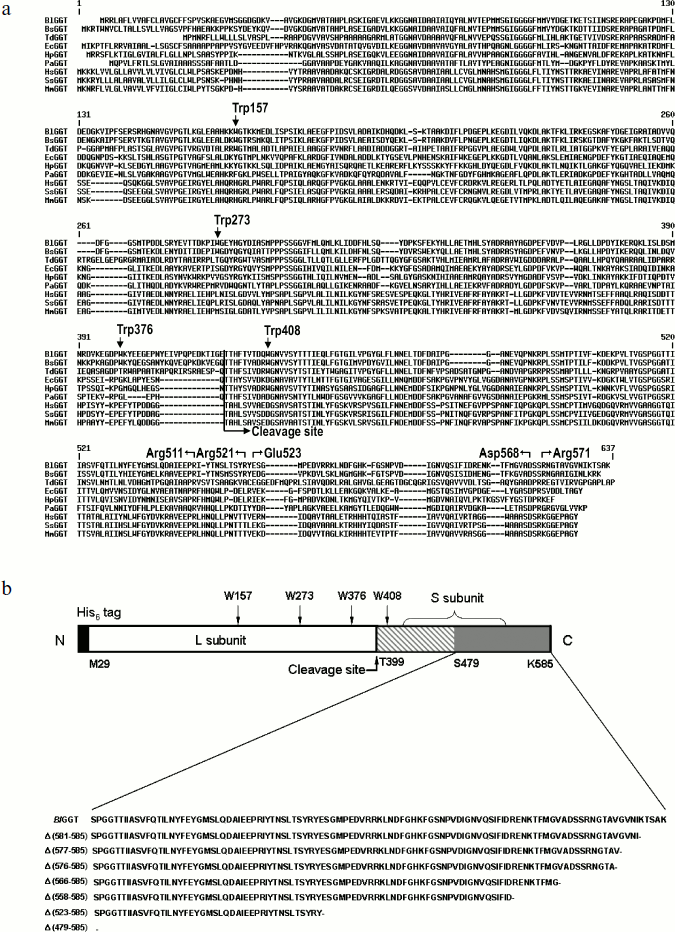
Sequence comparison and plasmid constructions. Figure 1a shows the sequence alignment of the full-length BlGGT. The alignment reveals a significant degree of primary sequence identity in the small subunits (Fig. 1). The small subunit spanning the BlGGT residues 399-585 had between 33 to 78% identity with B. subtilis, E. coli, H. pylori, and Pseudomonas aeruginosa GGTs, while identities with non-microbial enzymes (bovine, human, and mouse GGTs) ranged from 25 to 31%. In HpGGT, the mobile C-terminus has been shown to be positioned by several electrostatic interactions within the C-terminal region, and mutational studies have confirmed that these salt bridges play an important role in the autocatalytic processing and catalysis of the enzyme [22]. As shown in Fig. 1a, the alignment also revealed a strict conservation of residues implicated in the formation of salt bridges.
Recently, the overexpression plasmid pQE-BlGGT was constructed for the functional expression of BlGGT-4aa [15]. Based on pQE-BlGGT, eight recombinant plasmids were further constructed for IPTG-induced production of the full-length and C-terminally truncated BlGGTs. The molecular mass for the small unit of BlGGT, Δ(581-585), Δ(577-585), Δ(576-585), Δ(566-585), Δ(558-585), Δ(523-585), and Δ(479-585) was estimated to be 20517, 20001, 19618, 19519, 18560, 17596, 13694, and 8701 Da, respectively, whereas the large subunit of these proteins shares the same molecular size of ~44,729 Da. To have a comprehensive presentation, the schematic diagram of full-length BlGGT and partial amino acid sequences at the C-termini of these constructs are shown in Fig. 1b.Fig. 1. Sequence alignment of BlGGT and schematic presentation of full-length and truncated enzymes. a) A graphic representation of amino acid sequence alignment generated with the ExPAsy molecular server (Swiss Institute of Bioinformatics, Basel, Switzerland). The deduced amino acid sequence for B. licheniformis ATCC 14580 GGT (BlGGT; Swiss-Prot Q62WE3), Bacillus subtilis GGT (BsGGT; Swiss-Prot P54422), Thiobacillus denitrificans GGT (TdGGT; Swiss-Prot Q3SJ07), E. coli GGT (EcGGT; Swiss-Prot P18956), H. pylori GGT (HpGGT; Swiss-Prot O25743), Pseudomonas aeruginosa GGT (PaGGT; Swiss-Prot Q9I406), Homo sapiens GGT (HsGGT; Swiss-Prot P19440), Sus scrofa GGT (SsGGT; Swiss-Prot P20735), and Mus musculus GGT (MmGGT; Swiss-Prot Q60928) were used for the alignment. Gaps in aligned sequences (dashes) were introduced to maximize similarities. The vertical line shows the putative cleavage site. HpGGT residues involved in the formation of salt bridges are indicated above the sequence. b) Schematic presentation of the engineered enzymes. Positions of the tryptophanyl residues in BlGGT are indicated. The putative cleavage for autocatalytic processing of BlGGT is also marked. Every truncated protein has a histidine tag fused to its N terminus.
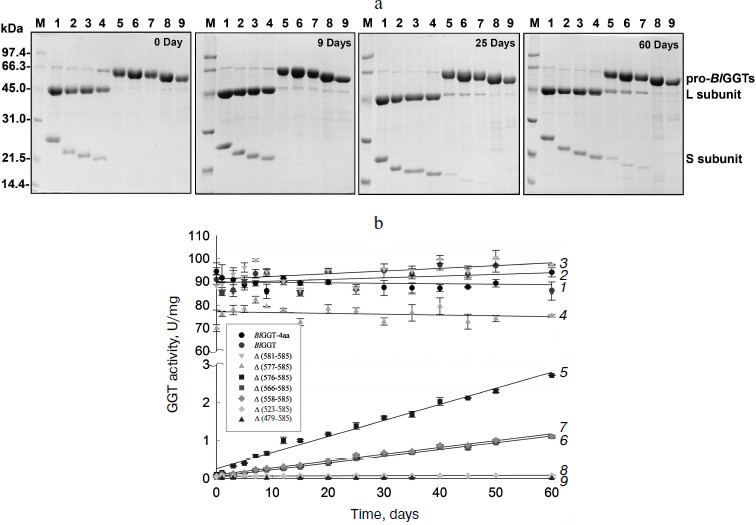
Purification and characterization of parental and C-terminally truncated enzymes. The expression of parental and truncated enzymes was evaluated by analyzing the whole-cell lysate preparations. BlGGT-4aa, BlGGT, Δ(581-585), and Δ(577-585) were expressed as three predominant bands with the molecular masses of approximately 65, 44.9, and 20-21.7 kDa, respectively, whereas the remaining mutant enzymes were present as a precursor form (data not shown). Importantly, all of the mutant proteins were expressed in the soluble fraction, suggesting that gross disruption of protein structure does not occur as a result of these deletions. The recombinant enzymes constructed in this study have a His-tag at the N-terminus, which does not have detrimental effects on BlGGT activity [14, 15]. To characterize these enzymes, the His6-tagged proteins were purified to near homogeneity using a Ni2+-NTA column (Fig. 2a). The one-step purification procedure resulted in a protein yield of 13-27% for the recombinant enzymes. The assay for GGT activity with the synthetic substrate, L-γ-Glu-p-NA, and the kinetic parameters of these proteins are summarized in Table 2. The Km and kcat values for BlGGT were 0.08 mM and 97 sec–1 (91 U/mg), respectively, which is comparable to those of BlGGT-4aa. A slight decrease in the enzyme activity was observed in Δ(577-585). Moreover, Δ(581-585) and Δ(577-585) showed comparable catalytic constant (kcat) and specificity constant (kcat/Km) values respective to the parental enzyme, but the GGT activity was not detected in Δ(576-585), Δ(566-585), Δ(558-585), Δ(523-585), and Δ(479-585). These results indicate that the local structural motifs in the C-terminus of BlGGT are critical for the proper function of the enzyme.
Table 2. GGT activity and kinetic parameters of parental and mutant enzymesFig. 2. Autocatalytic processing of pro-BlGGTs. Approximately 1 mg protein/ml in the elution buffer was incubated for 0, 9, 25, and 60 days. Aliquots of 100 µl were withdrawn at the indicated intervals for SDS-PAGE analysis (a) and enzymatic assays (b). Lanes: M, molecular mass maker; 1) BlGGT-4aa; 2) BlGGT; 3) Δ(581-585); 4) Δ(577-585); 5) Δ(576-585); 6) Δ(566-585); 7) Δ(558-585); 8) Δ(523-585); 9) Δ(479-585).
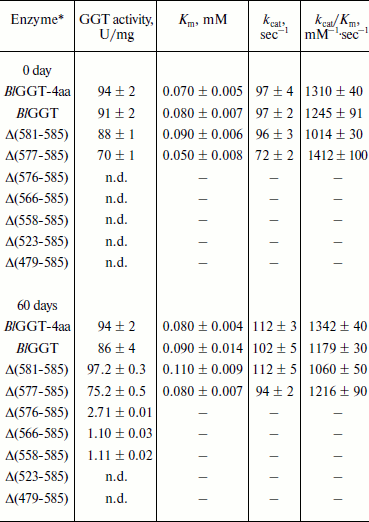
Note: n.d., not detected; –, not determined.
*Protein concentration of these samples was approximately 1 mg/ml.
In vitro processing of enzyme precursors. To investigate in vitro processing of the truncated enzymes, the purified proteins were incubated at 4°C for a period of time. At each sampling time, the amount of precursors was analyzed by SDS-PAGE, and the GGT activity was simultaneously assayed. As shown in Fig. 2a, BlGGT-4aa, BlGGT, Δ(581-585), and Δ(577-585) were predominantly as the mature form. A significant amount of the processed Δ(576-585), Δ(566-585), and Δ(558-585) was observed after 60-day incubation. GGT activity of the incubated Δ(576-585), Δ(566-585), and Δ(558-585) was 2.71 ± 0.01, 1.10 ± 0.03, and 1.11 ± 0.02 U/mg, respectively (Table 2). However, the remaining two proteins, Δ(523-585) and Δ(479-585), were actually detrimental to the autocatalytic capability. These results clearly indicate that the C-terminus of BlGGT participates in the autocatalytic processing, and residue Val576 is located at the boundary.
BlGGT must undergo autocatalytic processing before it has enzymatic activity, but the mechanism of activation has not been elucidated. A base is generally proposed to be crucial for the autocatalytic activation of Ntn-hydrolase precursors into L- and S-subunits. It has been suggested that a histidine residue in the vicinity of the processing site of Flavobacterium meningosepticum glycosyl asparaginase is essential for its maturation and acts as a base in this reaction [23]. However, X-ray crystallographic results demonstrated that the β-carboxyl group of the Asp residue just before the catalytic Thr residue, which is the nucleophile of Flavobacterium enzyme, acts as the base [24]. In the case of the S-subunit of 20 S proteasome, the residue just ahead the catalytic residue is the invariant Gly, which has no side chain to act as a base similar to glycosyl asparaginase. These findings therefore rule out the involvement of Asp in the autocatalytic processing. In contrast, Ditzel et al. [25] showed that a water molecule acts as the base at the active center for the processing of S-subunit precursor of yeast 20 S proteasome. Also, a water molecule was found to be the general base in cephalosporin acylase [26] and glutaryl 7-aminocephalosporanic acid acylase [27]. As shown in Fig. 2a, the purified Δ(576-585), Δ(566-585), and Δ(558-585) could slowly undergo autocatalytic processing in the elution buffer. In this regard, it is likely that a water molecule plays a critical role in the autocatalytic activation of BlGGT precursors into L- and S-subunits. These results also reflect the fact that the C-terminus of BlGGT plays a role in enzyme maturation, and C-terminal deletion of more than nine amino acids would lead to the expression of inactive precursors.
The C-terminus of HpGGT has been reported to play critical roles in autocatalytic processing and catalysis [22]. Although the indicated region shows low sequence conservation among GGT members, the disruption of conserved salt bridges in the C-terminal region of the 20-kDa subunit has been demonstrated to cause a significant reduction in autoprocessing and hydrolase activities of HpGGT. In the HpGGT structure, the carboxylate of Asp562 interacts with Arg564 as well as another conserved arginine residue, Arg502. Similarly, Arg564 forms a salt bridge with a conserved glutamate residue, Glu515. Given the involvement of these residues in the extensive hydrogen bond network and the remote location relative to the catalytic nucleophile, it was proposed that these four highly conserved residues are important for enzyme maturation [22]. For BlGGT, the corresponding pairs of salt bridges are Arg571/Asp568 and Glu523/Arg571. In the present constructions, four of the truncated enzymes, including Δ(566-585), Δ(558-585), Δ(523-585), and Δ(479-585), definitely abolish the proposed salt bridges. As noted by Williams et al. [22], disruption of the corresponding salt bridges in the small subunit of BlGGT might be responsible for the suppression of autocatalytic processing and enzymatic activity. However, the reasons for the loss of autocatalytic processing in Δ(576-585) remain to be explored.
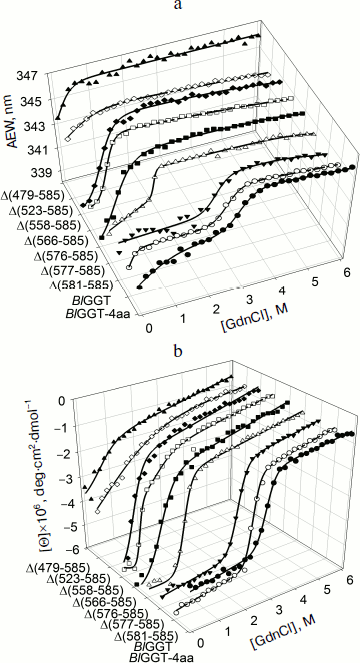
Unfolding of parental and C-terminally truncated enzymes at various concentrations of GdnCl. BlGGT has a total of four tryptophanyl residues (Trp157, Trp273, and Trp376 in L subunit and Trp408 in S subunit) (Fig. 1). When excited at 280 nm, the protein intrinsic emission fluorescence comes mostly from the tryptophanyl residues. Deletion at the C-terminus of BlGGT even for Δ(479-585) did not exclude these four tryptophanyl residues. Moreover, no precipitation was observed, and also the unfolding was completely reversible for BlGGTs at any GdnCl concentration (data not shown). Thus, the GdnCl-induced unfolding is very suitable for conformational stability studies of the enzyme. Unfolding of the enzyme at different concentrations of GdnCl is shown in Fig. 3. The average emission wavelength (AEW) that reports on the changes in both fluorescence wavelength and fluorescence intensity was used to calculate the thermodynamic parameters of the unfolding/refolding process (Table 3). By fitting the data with Eq. (9), BlGGT-4aa was seen to start to unfold at very low concentration of GdnCl and reach an unfolding intermediate with [GdnCl]0.5,N–I of 1.10·10–12 M. The second unfolding phase started at 1.23 M denaturant with [GdnCl]0.5,I–U of 2.874 M. The calculated free energy change (ΔG) for the N ↔ I and I ↔ U processes was 1.19·10–12 and 7 ± 1 kcal/mol, respectively. The stability parameters of BlGGT are comparable to those of BlGGT-4aa. However, the fluorescence signals of GdnCl-induced C-terminally truncated enzymes followed a monophasic process (Fig. 3a). Similar results were obtained with CD spectra (Fig. 3b). Δ(581-585), Δ(577-585), Δ(576-585), Δ(566-585), Δ(558-585), Δ(523-585), and Δ(479-585) showed [GdnCl]0.5,N–U of 2.92, 1.37, 0.44, 0.56, 0.63, 1.2·10–6 and 1.9·10–9 M, corresponding to free energy change of 8 ± 3, 16 ± 10, 0.8 ± 0.3, 2.2 ± 0.2, 1.5 ± 0.2, 1.5·10–6, and 4.8·10–9 kcal/mol, respectively, for the N ↔ U process. It is very likely that as more amino acid residues at the C-terminus of the enzyme were deleted, a lower concentration of GdnCl was required to start unfolding. This allows one to distinguish the conformation stabilities of the parental and truncated BlGGTs and assign the functional role of the C-terminus. Clearly, the significant changes in protein structure occur as a consequence of the deletions.
Table 3. Stability parameters of C-terminally truncated enzymesFig. 3. Unfolding curves of parental and truncated BlGGTs in different concentrations of GdnCl. The enzyme in 30 mM Tris-HCl buffer (pH 7.7) was incubated with various concentrations of GdnCl for 10 min. The enzyme solution was excited at 280 nm, and the emission was recorded at 25°C. a) Average emission wavelength (AEW) of the enzyme was used to analyze the unfolding data. b) CD changes at 222 nm versus GdnCl concentration. Solid symbols are experimental data and the smooth curves are computer-generated best-fit lines according to a two-state unfolding model (Eq. (6) or (7)).
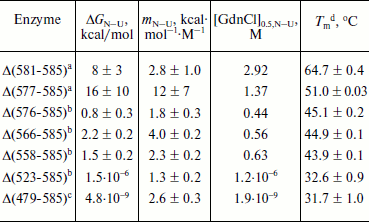
a Unfolding data were fitted to a two-state unfolding model according to Eq. (8).
b Unfolding data were fitted to a two-state unfolding model according to Eq. (7).
c Unfolding data were fitted to a two-state unfolding model according to Eq. (6).
d Melting temperature determined by CD spectropolarimetry.
To probe the structural integrity of parental and mutant enzymes, their thermal unfolding patterns were determined by following the changes in ellipticity at 222 nm (Table 3). The unfolding curves of the tested proteins showed single sigmoidal transitions. Midpoints of thermal transition, Tm, were calculated as 64.8 ± 0.3 and 64.7 ± 0.4°C for BlGGT and Δ(581-585), respectively. The Tm values for Δ(577-585), Δ(576-585), Δ(566-585), Δ(558-585), Δ(523-585), and Δ(479-585) were 12.9-32.2°C lower than that of BlGGT-4aa Tm (63.8 ± 0.2°C). These data clearly indicate that deletions of more than nine amino acids from the C-terminus have a profound effect on the thermal stability of the enzyme. Also, the results shown in Table 3 indicate the close correlation of inability to autoprocess with the unstable structure.
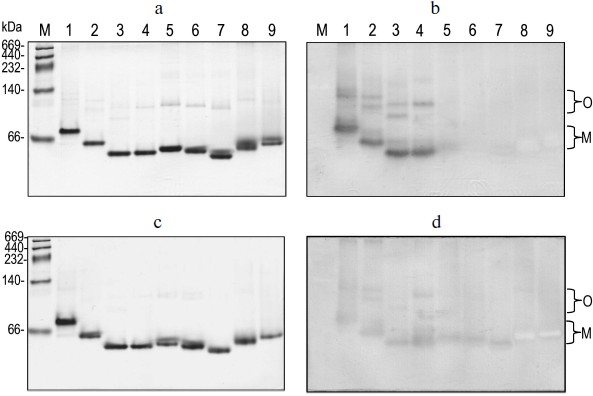
Correlation of quaternary structural changes of BlGGT with autocatalytic processing. To determine the oligomeric structure of the parental and mutant BlGGTs, the purified enzymes were subjected to native PAGE and the gel was stained with Coomassie brilliant blue R-250. As shown in Fig. 4a, one major band with a similar migration pattern to the recombinant BlGGT heterodimer was detected, ruling out a possible role of the His-tag in the polymerization process. Most importantly, higher polymers were not as prominent for these enzymes. Activity staining showed that the heterodimer and the higher order oligomers of BlGGT-4aa, BlGGT, Δ(581-585), and Δ(577-585) had GGT activity (Fig. 4b). Although Δ(576-585), Δ(566-585), Δ(558-585), Δ(523-585), and Δ(479-585) migrated at the positions corresponding to their heterodimers on the gel, these precursor enzymes displayed abnormal mobility. Actually, the truncations lead to a modification in the net charge and a decrease in the pI values (5.07-5.11 compared with 5.19 for BlGGT), which might cause an abnormal mobility of the mutant enzymes during native PAGE. After 60 days incubation, however, a significant amount of Δ(576-585), Δ(566-585) was processed and the mature enzymes also retained GGT activity (Figs. 4c and 4d).
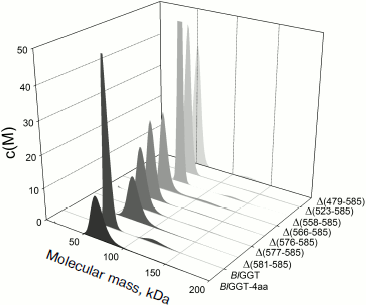
EcGGT and HpGGT were reported to have heterodimeric structures [10, 12]. The quaternary structures of parental and mutant BlGGTs were examined using analytical ultracentrifugation, and the sedimentation velocity data were analyzed using the continuous size distribution model using the SEDFIT program (Fig. 5). In these experiments no aggregation was observed at sedimentation coefficients of up to 20 S. Analytical ultracentrifugation provides very detailed conformational information about enzyme structural changes. The parental enzyme and BlGGT sedimented at ~4.85 S, and there was a peak between 4.82 and 5.60 S corresponding to the truncated enzymes (data not shown). An equivalent continuous distribution C(s,M) plot (Fig. 5) indicated that both BlGGT-4aa and BlGGT have a calculated molecular mass of approximately 65.23 kDa in buffer. Analytical ultracentrifugation analysis verified the expected decreases in molar masses of Δ(581-585) (76.0 kDa), Δ(577-585) (66.5 kDa), Δ(576-585) (60.1 kDa), Δ(566-585) (59.0 kDa), Δ(558-585) (62.0 kDa), Δ(523-585) (55.0 kDa), and Δ(479-585) (50.5 kDa). All these experimentally determined molar mass values are in agreement with the theoretical values calculated from the amino acid sequences. The parental BlGGT existed as a heterodimer, and almost all the truncated enzymes were heterodimeric in solution too (Fig. 5). This confirms that the active form of BlGGT is the heterodimeric one.Fig. 4. Staining of native polyacrylamide gel for GGT activity. Purified parental and mutant BlGGTs (100 µg of protein) were subjected to native PAGE followed by staining for GGT activity. The purified proteins are visualized by non-denaturing PAGE at 0 (a) and 60 days (c); activity staining at 0 (b) and 60 days (d). Lanes: M, molecular mass markers; 1) BlGGT-4aa; 2) BlGGT; 3) Δ(581-585); 4) Δ(577-585); 5) Δ(576-585); 6) Δ(566-585); 7) Δ(558-585); 8) Δ(523-585); 9) Δ(479-585). The positions of heterodimeric and oligomeric GGTs are indicated by M and O, respectively.
Significant mature form with a frictional ratio (f/fo) of 1.58, 1.57, 1.46, and 1.39 was detected with BlGGT-4aa, BlGGT, Δ(581-585), and Δ(577-585), respectively. The processed form was not detected in Δ(576-585), Δ(566-585), Δ(558-585), Δ(523-585), and Δ(479-585), in which the enzyme existed exclusively as precursor with f/fo of 1.18, 1.05, 1.23, 1.31, and 1.32. After autocatalytic processing, the conformational changes of BlGGTs were clearly demonstrated by the increase in frictional ratio of the enzyme upon maturation (data not shown). The truncated BlGGTs with deletions of more than nine amino acids from the C-terminus could not induce the autocatalytic process. Thus, the C-terminal region close to Val576 is essential for the autocatalytic processing of BlGGT.Fig. 5. Quaternary structure analysis of the full-length and C-terminally truncated BlGGTs. Continuous sedimentation of the protein in 20 mM Tris-HCl (pH 8.0) was monitored with the analytical ultracentrifuge. The sedimentation velocity data were analyzed with SEDFIT [28]. All enzyme preparations were at 0.4 mg/ml concentration.
In summary, we have studied the role of the C-terminal region of BlGGT by deletion analysis. The mutagenesis results revealed that deletion of up to nine amino acid residues from the C-terminal end of BlGGT definitely affects the autocatalytic activity of the enzyme. Although the sequence in the C-terminal region is not strictly conserved among GGTs, these enzymes show a high level of similarity in structure organization. Thus, the role of the C-terminal region proposed herein might also be true for other GGTs, and this research also provides a starting point for further studies of the quaternary structure of this family.
The authors acknowledge financial support (NSC 95-2313-B-415-2-MY3 and NSC 97-2628-B-415-0-MY3) from the National Science Council of Taiwan.
REFERENCES
1.Brannigan, J. A., Dodson, G., Duggleby, H. J.,
Moody, P. C., Smith, J. L., Tomchick, D. R., and Murzin, A. G. (2006)
Nature, 90, 4651-4661.
2.Tate, S. S., and Meister, A. (1981) Mol. Cell
Biochem., 39, 357-368.
3.Hanigan, M. H., and Ricketts, W. A. (1993)
Biochemistry, 32, 6302-6306.
4.Suzuki, H., Hashimoto, W., and Kumagai, H.
(1999) J. Mol. Catal. B: Enzym., 6, 175-184.
5.Shibayama, K., Wachino, J., Arakawa, Y., Saidijam,
M., Rutherford, N. G., and Henderson, P. J. (2007) Mol.
Microbiol., 64, 396-406.
6.Godwin, A. K., Meister, A., O’Dwyer, P. J.,
Huang, C. S., Hamilton, T. C., and Anderson, M. E. (1992) Proc.
Natl. Acad. Sci. USA, 89, 3070-3074.
7.Hanigan, M. H., Gallagher, B. C., Townsend, D. M.,
and Gabarra, V. (1999) Carcinogenesis, 20,
553-559.
8.Oinonen, C., and Rouvinen, J. (2000) Protein
Sci., 9, 2329-2337.
9.Suzuki, H., and Kumagai, H. (2002) J. Biol.
Chem., 277, 43536-43543.
10.Okada, T., Suzuki, H., Wada, K., Kumagai, H., and
Fukuyama, K. (2006) Proc. Natl. Acad. Sci. USA, 103,
6471-6476.
11.Okada, T., Suzuki, H., Wada, K., Kumagai, H., and
Fukuyama, K. (2007) J. Biol. Chem., 282,
2433-2439.
12.Boanca, G., Sand, A., and Barycki, J. J.
(2006) J. Biol. Chem., 281, 19029-19037.
13.Boanca, G., Sand, A., Okada, T., Suzuki, H.,
Kumagai, H., Fukuyama, K., and Barycki, J. J. (2007) J. Biol.
Chem., 282, 534-541.
14.Lin, L. L., Chou, P. R., Hua, Y. W., and Hsu, W.
H. (2006) Appl. Microbiol. Biotechnol., 73,
103-112.
15.Lin, L. L., Yang, L. Y., Hu, H. Y., and Lo, H. F.
(2008) Curr. Microbiol., 57, 603-608.
16.Lyu, R. C., Hu, H. Y., Kuo, L. Y., Lo, H. F.,
Ong, P. L., Chang, H. P., and Lin, L. L. (2009) Curr.
Microbiol., 59, 101-106.
17.Laemmli, U. K. (1970) Nature,
227, 680-685.
18.Royer, C. A., Mann, C. J., and Matthews, C. R.
(1995) Protein Sci., 2, 1844-1852.
19.Schuck, P. (2000) Biophys. J., 78,
1600-1619.
20.Pace, C. N. (1990) Trends Biotechnol.,
8, 93-98.
21.Morjana, N. A., McKeone, B. J., and Gilbert, H.
F. (1993) Proc. Natl. Acad. Sci. USA, 90, 2107-2111.
22.Williams, K., Cullati, S., Sand, A., Biterova, E.
I., and Barycki, J. J. (2009) Biochemistry, 48,
2459-2467.
23.Guan, C., Cui, T., Rao, V., Liao, W., Benner, J.,
Lin, C. L., and Comb, D. (1996) J. Biol. Chem.,
271, 1732-1737.
24.Xu, Q., Buckley, D., Guan, C., and Guo, H. C.
(1999) Cell, 98, 651-661.
25.Ditzel, L., Huber, R., Mann, K., Heinemeyer, W.,
Wolf, D. H., and Groll, M. (1998) J. Mol. Biol.,
279, 1187-1191.
26.Kim, Y., Kim, S., Earnest, T. N., and Hol, W. G.
(2002) J. Biol. Chem., 277, 2823-2829.
27.Kim, J. K., Yang, I. S., Rhee, S., Dauter, Z.,
Lee, Y. S., Park, S. S., and Kim, K. H. (2003) Biochemistry,
42, 4084-4093.
28.Brown, P. H., and Schuck, P. (2006) Biophys.
J., 90, 4651-4661.