Whole Genome Methylation Scanning Based on phi29 Polymerase Amplification
R. Brooks1, R. J. Rose1, M. B. Sheahan1, and S. Kurdyukov1,2*
1Australian Research Council Centre of Excellence for Integrative Legume Research, School of Environmental and Life Sciences, The University of Newcastle, Callaghan, New South Wales, 2308, Australia; fax: +61-2-4921-5472; E-mail: sergey.kurdyukov@sydney.edu.au; robert.brooks@uon.edu.au; ray.rose@newcastle.edu.au; michael.sheahan@newcastle.edu.au2Present address: Kolling Institute of Medical Research, Kolling Building, Royal North Shore Hospital, St. Leonards, New South Wales, 2065, Australia
* To whom correspondence should be addressed.
Received November 29, 2010; Revision received February 2, 2011
Identifying differences in DNA methylation is critical to understanding how epigenetics influences gene expression during processes such as development. Here, we propose a method that employs a single, methylation-sensitive restriction endonuclease of choice, to produce discrete pools of methylated and unmethylated DNA from the same sample. A pool of restriction fragments representing unmethylated regions of the genome is first obtained by digestion with a methylation-sensitive endonuclease. The restriction-digested DNA is then concatamerized in the presence of stuffer-adaptor DNA, which prevents interference from originally unmethylated DNA by blocking the ends of the restriction fragments. The concatamerized DNA is amplified by phi29 polymerase to remove methylation marks, and again digested with the same endonuclease to produce a pool of DNA fragments representing methylated portions of the genome. The two pools of DNA fragments thus obtained can be analyzed by end-sequencing or hybridization to a genomic array. In this report we detail a proof of concept experiment that demonstrates the feasibility of our method.
KEY WORDS: whole genome methylation, phi29 polymerase, whole genome amplification, next-generation sequencingDOI: 10.1134/S0006297911090021
Profiling differences in DNA methylation across a genome is relevant to understanding how epigenetic effects influence cell- and tissue-specific gene expression and processes such as cancer [1, 2]. Use of isoschizomers that possess differences in methylation-sensitivity, such as HpaII and MspI, to probe differences in methylation is well documented. With this approach, restriction fragments released after digestion by HpaII are representative of unmethylated regions of the genome, whereas an absence of HpaII digestion is diagnostic for DNA methylation. Combined with next-generation sequencing, this strategy was recently used to profile whole genome methylation using a method dubbed as Methyl-Seq [3]. However, methylation features are rarely unambiguous, and as such, a certain proportion of DNA molecules at a particular locus will produce fragments after digestion with methylation-sensitive endonucleases, even for generally methylated regions, as has been shown with particular epigenetic features [4]. While elegant solutions to the digital measurement of methylation frequency exist, such as MethylC-Seq [5] or BS-Seq [6], which employ bisulfite conversion [7] coupled to next-generation sequencing (e.g. Illumina Genome Analyzer) — their cost, especially for large genomes, may be prohibitive for some researchers [8].
Our solution to this problem is to use a single, methylation-sensitive restriction endonuclease of choice to derive two discrete pools of genomic fragments from the same sample — one containing unmethylated loci and the other containing methylated loci. The abundance of each fragment in these pools, and genomic location, could be determined through end sequencing. It would also be possible to use the fragments so obtained for hybridization to a whole genome microarray or tiling array. Our method is advantageous, as it can reduce the amount of sequencing required by methods such as MethylC-Seq, while also being applicable to species without sequenced genomes. The method we detail here should thus provide the ability to scan any genome at different levels of granularity (depending on the digestion frequency of the restriction endonuclease used) to identify and quantify the prevalence of novel methylated loci.
Our method exploits phi29 polymerase, an enzyme demonstrating unparalleled processivity and an ability to amplify long fragments of DNA devoid of methylation, which are thus suitable for digestion by methylation-sensitive restriction endonucleases [9]. Coupled to the ability of phi29 to amplify a fragmented genome following concatamerization with short adaptors, coined as stuffer DNA by Shoaib and coauthors [10], these features form the basis for a method that permits the quantitative analysis of methylation features in whole genomes. In our approach, DNA representing unmethylated regions of the genome is first digested with a methylation-sensitive restriction endonuclease. Next, methylation marks from methylated regions are removed by amplification with phi29 polymerase. Amplified DNA is then digested with the same endonuclease releasing originally methylated fragments. Interference from originally unmethylated DNA is avoided by blocking the ends of the restriction fragments with specifically-designed, stuffer-adaptor DNA. Thus, two pools of DNA are obtained that could be analyzed by end-sequencing or hybridization to a genomic array. In this report, we detail a proof of concept experiment that demonstrates the feasibility of our conceptual approach using an artificially constructed pseudogenome.
MATERIALS AND METHODS
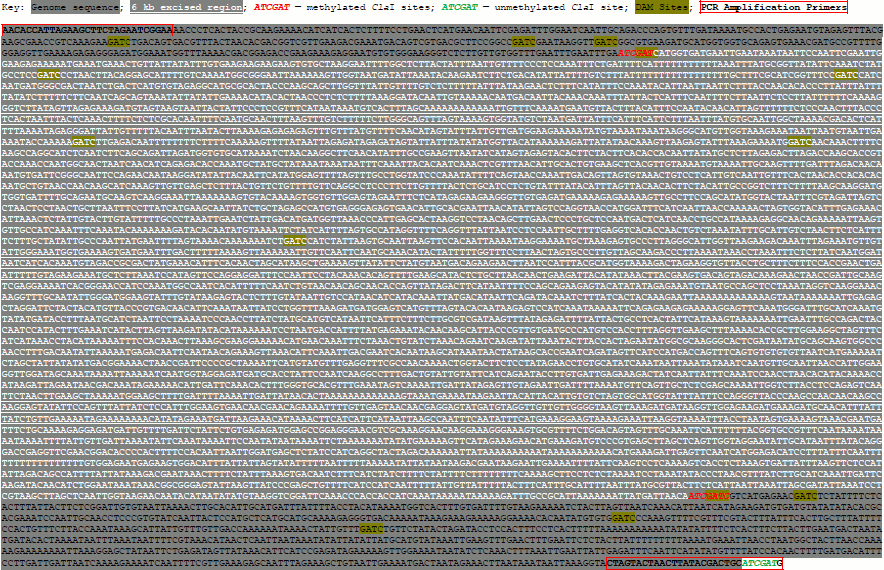
Synthesis of a pseudogenome. We synthesized a pseudogenome by ligating a mixture of digested λ-DNA and linearized plasmid to simulate unmethylated and methylated fractions of a genome, respectively. To create the plasmid, we cloned a 7.3 kb Medicago truncatula (cultivar Jemalong) genome fragment corresponding to bases 36,490,391-36,497,718 of chromosome 4 (see Supplement (Fig. S1) on site of Biochemistry (Moscow) journal http://protein.bio.msu.ru/biokhimiya). The genomic fragment was amplified using the primers 5′-AACACCATTAGAAGCTTCTAGAATCGGAA-3′ and 5′-CATCGATGCAGTCGTATAAGTTAGTACTAG-3′ using the Expand Long Template PCR System (Roche, Germany). The genome fragment contained two ClaI sites, while the second primer introduced a third ClaI site. The resulting PCR amplicon was cloned by TA cloning into the pGEM-T vector (Promega, USA). Unmethylated λ-DNA (20 µg; Promega; D1521) and the plasmid (40 µg) were digested by 20 units of ClaI (New England Biolabs, USA) in 100 µl reactions for 2 and 16 h at 37°C, respectively. The ClaI was inactivated at 65°C for 20 min. The efficiency of digestion was checked by gel electrophoresis. Electrophoresis was performed using a 0.6% (w/v) agarose gel prepared in Tris-acetate-EDTA buffer (pH 8.2) and run at 100 V, 22°C. The λ-DNA (1 µg in a 5 µl volume) and linearized plasmid (2 µg in a 5 µl volume) were combined with 1.2 µl of 10× T4 DNA ligase buffer and ligated by adding 1 µl of concentrated T4 DNA ligase (2000 cohesive end units; New England Biolabs, USA) for 24 h at 14°C.
Stuffer preparation. Stuffer-adaptor DNA was prepared by digestion of a pCR8/GW/TOPO plasmid (Invitrogen, USA) with AciI, HpaII, and HpyCh4IV restriction endonucleases (New England Biolabs), which produced short (50-100 bp) fragments of DNA possessing ClaI-compatible CG overhangs.
Stuffer ligation. To introduce stuffer-adaptor DNA into the pseudogenome, 1.25 µl (0.5 µg) of the ClaI-linearized plasmid (0.14 pmol), 1.25 µl (0.25 µg) of digested λ-DNA (0.1 pmol), and 5.5 µl (0.6 µg) of stuffer-adaptor (~7 pmol) were ligated with T4 ligase as described above.
Phi29 amplification and digestion. The stuffer-pseudogenome ligation mixture (1 µl) was diluted to 5 µl with water and used for phi29 amplification. The DNA was denatured at 95°C for 3 min and immediately chilled on ice. The amplification reaction was performed in a total volume of 20 µl using the REPLI-g DNA polymerase and buffer from the QuantiTect Whole Transcriptome Kit (Qiagen, Germany) at 30°C for 2 h and the reaction terminated at 65°C for 10 min. It would be possible to amplify using phi29 from other suppliers. In such cases, the reaction should be supplied with deoxyribonucleotide triphosphates (1-4 mM) and random hexamers (5-50 µM). Addition of pyrophosphatase and the use of modified (exonuclease-resistant) random hexamers could potentially increase yields significantly [11]. In our hands, the reaction yielded approximately 20 µg of the amplified DNA. After amplification, the reaction mixture was diluted to 100 µl with ClaI buffer and digested with ClaI as described above.
RESULTS AND DISCUSSION
We used a mixture of λ-DNA and a plasmid containing a 7.3 kb M. truncatula genome fragment to simulate unmethylated and methylated fractions of a genome, respectively. We chose to use a pseudogenome to more clearly illustrate how our procedure operates, but we believe our method has utility to genuine genomes. Although genuine genomes are longer and will exhibit more complex methylation profiles, we believe that our simple pseudogenome accurately reflects how the basic tenets of our procedure would operate on a genuine genome.
To produce the pseudogenome, we digested both the λ-DNA and plasmid with the methylation-sensitive endonuclease ClaI and then ligated the products of these digestion reactions (Fig. 1; see color insert). In this pseudogenome, the ClaI-digested λ-DNA fragments represent completely unmethylated genomic DNA (Fig. 1, blue regions; Fig. 2, lane 2) whereas the digested plasmid represents a region of methylated genomic DNA (~6 kb; see Supplement, Fig. S1) bounded by the remnants of an unmethylated ClaI site (Fig. 1, black region; Fig. 2, lane 3). Note that although the plasmid had three ClaI sites, two sites within the genome fragment were selected such that they would be protected by bacterial DNA adenine methylation (ClaI sites in a GATC context) and hence ClaI digestion resulted in plasmid linearization. We favored the use of in vivo adenine methylation over in vitro CpG methylation as it is more efficient.
Fig. 1. Procedural summary for generating pools of fragments corresponding to unmethylated and methylated regions of the genome. Black regions represent methylated DNA, blue regions unmethylated DNA, and red regions the stuffer-adaptor DNA. Green boxes, methylation-sensitive endonuclease (e.g. ClaI) sites. Starred, methylated endonuclease sites.
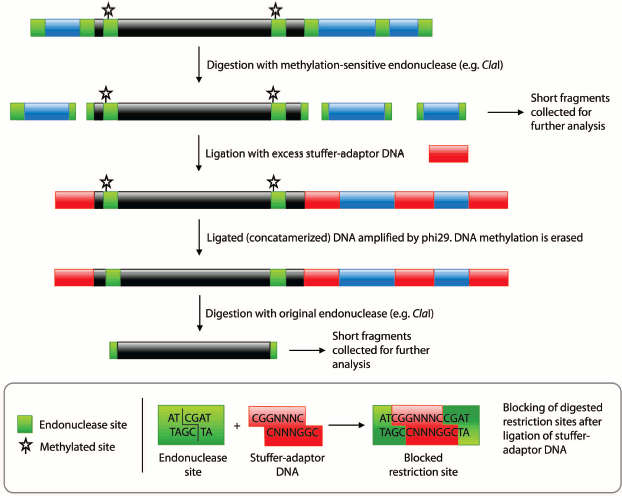
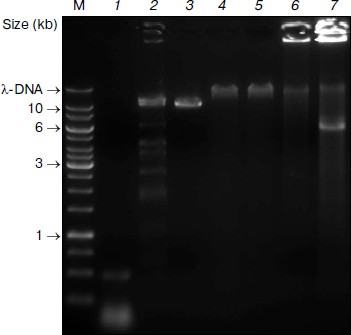
Subsequent digestion of the pseudogenome by ClaI, which results in restriction fragments possessing CG overhangs, produced two types of restriction fragment — predominantly short fragments arising from unmethylated loci and longer fragments from methylated, and therefore protected, loci. Although a range of fragment sizes would result from digestion of a complex genome, the use of a frequent cutting endonuclease would produce a majority of small (<2 kb) fragments. At this point in the procedure, the short fragments representative of unmethylated loci would be saved for further analysis by sequencing or hybridization to a genomic array (Fig. 1). We envisage that the loci contained in the short fragment pool would enable a scan of most unmethylated loci in the genome. It would, however, be possible to sequence the entire pool of restriction fragments is so desired.Fig. 2. Pseudogenome experiment. Products electrophoresed on a 0.6% agarose gel. M, 1 kb Fermentas GeneRuler DNA Ladder mixed with λ-DNA (uppermost band); 1) stuffer-adaptor DNA; 2) ClaI digested λ-DNA; 3) ClaI linearized plasmid (~10.3 kb); 4) ligation of ClaI digested λ-DNA, ClaI linearized plasmid and stuffer-adaptor DNA; 5) ClaI digest of ligation product shown in lane 4; 6) ligated DNA shown in lane 4 after phi29 amplification; 7) ClaI digestion of phi29-amplified DNA shown in lane 6.
To release fragments of methylated DNA, the digested pseudogenome was first concatamerized with an excess of stuffer-adaptor DNA (with ClaI-compatible ends) before being amplified with phi29 polymerase (Fig. 1). The phosphorylated CG overhangs of the ClaI-digested pseudogenome served to facilitate ligation of stuffer-adaptor DNA, in turn enabling amplification of the concatamerized pseudogenome by phi29. Ligation of stuffer-adaptor DNA into the ClaI-digested pseudogenome acts to block the re-digestion of unmethylated ClaI sites, while phi29 amplification removes methylation marks from originally methylated ClaI sites. Hence, after digestion of the phi29-amplified DNA by ClaI, fragments originally protected by methylation are released and can be saved for further analysis (Fig. 1).
We prepared the stuffer-adaptor DNA by digesting a pCR8/GW/TOPO plasmid, resulting in predominantly short (~50-100 bp) fragments of DNA with ClaI-compatible overhangs (Fig. 2, lane 1). By ligating the ClaI-digested pseudogenome with an excess of stuffer-adaptor DNA (1 : 30 molar ratio) we obtained high molecular weight products of around 50 kb (Fig. 2, lane 4) which were resistant to ClaI digestion, indicating that the stuffer-adaptor DNA efficiently blocked unmethylated ClaI sites from re-digestion (Fig. 2, lane 5). Indeed, even when we loaded 10-fold more stuffer-adaptor ligated pseudogenome treated with ClaI we saw no evidence of ClaI digestion (data not shown). Although our results clearly suggest that there was no re-ligation of unmethylated pseudogenome restriction fragments following ligation of stuffer-adaptor at a 1 : 30 molar ratio, it would be feasible to increase the ratio of stuffer-adaptor DNA to genome fragments, thus decreasing the potential for unwanted re-ligation even further.
We suggest that the prolonged ligation we used resulted not only in the concatamerization of restriction fragments but also in the self-ligation of concatamers. Consequently, rolling circle amplification by phi29 was possible, ensuring that long fragments of DNA (>40 kb) were generated during subsequent amplification and thereby attenuating the loss of restriction sites.
After the concatamerization reaction, the mixture was subject to phi29 amplification. The highly viscous nature of the phi29 solution following amplification and the fact that most DNA remained immobilized in the loading well of the 0.6% agarose gel we used supports the high molecular weight of the DNA obtained (Fig. 2, lane 6). Subsequent digestion of the phi29-amplified DNA with ClaI led to the excision of a 6-kb fragment corresponding to the region between the originally methylated ClaI sites of the plasmid (Fig. 2, lane 7). Thus, although the original unmethylated fragments are carried through the procedure, their ClaI sites are rendered indigestible, thereby negating contamination of the methylated DNA pool.
Our method should therefore be of use in performing scans for differential methylation across an entire genome. Moreover, coupled to next-generation end-sequencing technology, a digital readout of the prevalence of methylation marks at a particular locus can be ascertained — of particular value to complex tissues such as plant callus or to comparisons of different organs or developmental stages.
Although we have used ClaI here, by modifying the sequence of the stuffer-adaptor DNA, any methylation-sensitive endonuclease could be used. Thus, the granularity of genome methylation scans can be adjusted to suit the research agenda, with rare-cutting endonucleases producing a coarser scan at reduced costs with increasing frequencies of cutting producing finer maps at greater cost. Finally, our method circumvents potential difficulties in aligning read sequences from bisulfate-treated samples (which ultimately convert unmethylated C to T) to a genome. This is particularly pertinent in plants where perhaps close to half of methylation marks exist not only in a CpG context, but also in CHG and CHH contexts (where H refers to A, C, or T) [5].
This work was supported by an Australian Research Council Centre of Excellence grant (CEO348212) to the University of Newcastle Node of the Centre of Excellence for Integrative Legume Research to RJR and an Australian Research Council Australian Postdoctoral Fellowship (DP0770679) to MBS.
REFERENCES
1.Li, E., Bestor, T. H., and Jaenisch, R. (1992)
Cell, 69, 915-926.
2.Rhee, I., Bachman, K. E., Park, B. H., Jair, K. W.,
Yen, R. W., Schuebel, K. E., Cui, H., Feinberg, A. P., Lengauer, C.,
Kinzler, K. W., Baylin, S. B., and Vogelstein, B. (2002) Nature,
416, 552-556.
3.Brunner, A. L., Johnson, D. S., Kim, S. W.,
Valouev, A., Reddy, T. E., Neff, N. F., Anton, E., Medina, C., Nguyen,
L., Chiao, E., Oyolu, C. B., Schroth, G. P., Absher, D. M., Baker, J.
C., and Myers, R. M. (2009) Genome Res., 19,
1044-1056.
4.Soppe, W. J. J., Jacobsen, S. E., Alonso-Blanco,
C., Jackson, J. P., Kakutani, T., Koornneef, M., and Peeters, A. J. M.
(2000) Mol. Cell, 6, 791-802.
5.Lister, R., O’Malley, R. C., Tonti-Filippini,
J., Gregory, B. D., Berry, C. C., Millar, A. H., and Ecker, J. R.
(2008) Cell, 133, 523-536.
6.Cokus, S. J., Feng, S., Zhang, X., Chen, Z.,
Merriman, B., Haudenschild, C. D., Pradhan, S., Nelson, S. F.,
Pellegrini, M., and Jacobsen, S. E. (2008) Nature, 452,
215-219.
7.Frommer, M., McDonald, L. E., Millar, D. S.,
Collis, C. M., Watt, F., Grigg, G. W., Molloy, P. L., and Paul, C. L.
(1992) Proc. Natl. Acad. Sci. USA, 89, 1827-1831.
8.Lister, R., and Ecker, J. R. (2009) Genome
Res., 19, 959-966.
9.Adachi, E., Shimamura, K., Wakamatsu, S., and
Kodama, H. (2004) Plant Cell Rep., 23, 144-147.
10.Shoaib, M., Baconnais, S., Mechold, U., Le Cam,
E., Lipinski, M., and Ogryzko, V. (2008) BMC Genomics, 9,
415.
11.Dean, F. B., Nelson, J. R., Giesler, T. L., and
Lasken, R. S. (2001) Genome Res., 11, 1095-1099.
Fig. S1. 7.3-kb Genome Fragment from Chromosome 4 of Medicago truncatula