REVIEW: What Is “Phenoptosis” and How to Fight It?
V. P. Skulachev
Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology and Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia; E-mail: skulach@genebee.msu.ru
Received May 11, 2012
Phenoptosis is the death of an organism programmed by its genome. Numerous examples of phenoptosis are described in prokaryotes, unicellular eukaryotes, and all kingdoms of multicellular eukaryotes (animals, plants, and fungi). There are very demonstrative cases of acute phenoptosis when actuation of a specific biochemical or behavioral program results in immediate death. Rapid (taking days) senescence of semelparous plants is described as phenoptosis controlled by already known genes and mediated by toxic phytohormones like abscisic acid. In soya, the death signal is transmitted from beans to leaves via xylem, inducing leaf fall and death of the plant. Mutations in two genes of Arabidopsis thaliana, required for the flowering and subsequent formation of seeds, prevent senescence, strongly prolonging the lifespan of this small semelparous grass that becomes a big bush with woody stem, and initiate substitution of vegetative for sexual reproduction. The death of pacific salmon immediately after spawning is surely programmed. In this case, numerous typical traits of aging, including amyloid plaques in the brain, appear on the time scale of days. There are some indications that slow aging of higher animals and humans is also programmed, being the final step of ontogenesis. It is assumed that stepwise decline of many physiological functions during such aging increases pressure of natural selection on organisms stimulating in this way biological evolution. As a working hypothesis, the biochemical mechanism of slow aging is proposed. It is assumed that mitochondria-generated reactive oxygen species (ROS) is a tool to stimulate apoptosis, an effect decreasing with age the cell number (cellularity) of organs and tissues. A group of SkQ-type substances composed of plastoquinone and a penetrating cation were synthesized to target an antioxidant into mitochondria and to prevent the age-linked rise of the mitochondrial ROS level. Such targeting is due to the fact that mitochondria are the only cellular organelles that are negatively charged compared to the cytosol. SkQs are shown to strongly decrease concentration of ROS in mitochondria, prolong lifespan of fungi, invertebrates, fish, and mammals, and retard appearance of numerous traits of aging. Clinical trials of SkQ1 (plastoquinonyl decyltriphenylphosphonium) have been successfully completed so that the Ministry of Health of the Russian Federation recommends drops of very dilute (0.25 µM) solution of this antioxidant as a medicine to treat the syndrome of dry eye, which was previously considered an incurable disease developing with age. These drops are already available in drugstores. Thus, SkQ1 is the first mitochondria-targeted drug employed in medical practice.
KEY WORDS: evolution, phenoptosis, programmed aging, mitochondria, antioxidants, therapy of dry eye syndromeDOI: 10.1134/S0006297912070012
Abbreviations: Δψ, transmembrane difference of electric potentials; BLM, bilayer planar phospholipid membrane; C12TPP, dodecyltriphenylphosphonium; MitoQ, ubiquinonyl decyltriphenylphosphonium; ROS, reactive oxygen species; SkQ, derivatives of plastoquinone and penetrating cations (Sk+); SkQ1, plastoquinonyl decyltriphenylphosphonium; SkQR1, plastoquinonyl decylrhodamine 19.
In memory of my friend Mikhail Leonovich Gasparov
I once asked my old friend, the famous philologist and metrician M. L. Gasparov: “Mikhail Leonovich! How would you call the programmed death of an organism if a similar type of phenomenon with respect to a single cell is called “apoptosis”? “Phenoptosis!” – answered the academician without any hesitation.
That happened back in 1997, and today, 15 years later, we are releasing the first (and hopefully not the last) issue of the journal with this word on the cover. If successful, the journal will be published as a series of special editions of Biochemistry (Moscow), in both English and Russian. Since 1990, the impact factor of Biochemistry (Moscow) has grown almost an order of magnitude, so that it has already become one of the most cited Russian journals, with the largest contribution to the citations being made by special thematic issues composed mainly of review articles by leading Russian and foreign scientists [1]. In January 2012, the editorial board of the journal decided to make some such issues periodic. The title of this issue is Biochemistry (Moscow). Phenoptosis (similar, for example, to American Journal of Physiology. Heart Circ. Physiol.). I would like to express my sincere gratitude to all those who helped me, the editor-compiler of this issue, to prepare for publication the first volume of the new edition (and I am especially grateful to those of them who also wrote articles for this issue): my deputies G. Libertini (Italy) and R. D. Ozrina (Russia), R. Lozier (USA, the editor of the English version of the journal), V. N. Anisimov (Russia), M. Blagosklonny (USA), L. A. Gavrilov (USA), T. Goldsmith (USA), D. B. Zorov (Russia), V. B. Ivanov (Russia), V. Longo (USA), K. Lewis (USA), V. N. Manskikh (Russia), A. V. Markov (Russia), J. J. Mitteldorf (USA), F. F. Severin (Russia), and M. V. Skulachev (Russia). Excellent work of the biochemist-translator A. Brzyska should also be noted.
Phenoptosis is defined as genetically programmed death of an organism [2-4]. As a rule, the death program is encoded in the genome of the dying organism, being a chain of biochemical events that ultimately cause its suicide. That is why it is first of all biochemists that study the phenomenon of phenoptosis. More rarely, death occurs as a result of behavioral responses coded in the genome of the dying individual, its sexual partner, or a close relative of the victim [2, 3, 5].
EXAMPLES OF PHENOPTOSIS
Prokaryotes. Examples of phenoptotic phenomena can be found in all the kingdoms of living organisms. Homologs of eukaryotic genes that in higher cells participate in apoptosis, i.e. cell suicide, have been found also in prokaryotes [6]. In bacteria there exist “long-lived toxin–short-lived antitoxin” systems, when the cell slowly synthesizes a protein potentially able to kill it. Such a “murder” does not occur as long as amino acids necessary for protein synthesis are in sufficient quantity, for another protein, an antitoxin forming an inactive complex with the toxin, is rapidly synthesized. The toxin is not only slowly synthesized, it is also slowly decomposed. At the same time, the antitoxin decays faster than the toxin. It is because of these relationships that the decrease in the level of free amino acids leads to the disappearance of antitoxin, whereas the amount of toxin is only slightly reduced. The toxin released from a complex with the antitoxin is activated, and it kills the bacteria. As a result, the concentration of bacteria decreases, and therefore the consumption of amino acids by these bacteria is also reduced. In the end, the level of amino acids in the few surviving bacteria increases, reaching the amount sufficient for protein synthesis, and there appears the possibility of maintenance of an antitoxin concentration at a level higher than that of the toxin [7, 8]. It seems to be quite essential that the lack of not only amino acids, but also respiratory substrates and oxygen, as well as the appearance of pollutants – inhibitors of transcription, translation, or metabolism, and other adverse factors inhibiting the biosynthesis of proteins, can trigger the phenoptotic “toxin–antitoxin” system as the last line of defense of a bacterial population against complete extinction [8].
Yeast as an example of unicellular eukaryotes. F. F. Severin and coworkers have shown that the long-known phenomenon of the toxicity of pheromone excess for yeast should be attributed to the phenomenon of phenoptosis [9-12]. Mating partners of these unicellular organisms, marked by the letters A and B, secrete short peptides (α and β, respectively), which are specifically bound by the receptors of the cells of the opposite sex, where they cause certain biochemical and morphological changes favorable for sexual reproduction. Yeasts possess no mechanism of active movement. They follow the laws of Brownian motion in the liquid medium and, in case of random collision, stick together for the time needed for DNA transfer from the donor cell to the recipient cell. If this process is unsuccessful, the cells cannot separate, but they continue secreting pheromones into the narrow intercellular gap. In this situation, the pheromones become the killers of cells with corresponding receptors (i.e. pheromone α kills the cells of A type and pheromone β kills the cells of B type). This effect was shown to be absolutely specific (i.e. pheromone α does not affect cells of B type, and pheromone β does not affect cells of A type). Excess of pheromone α, added to a suspension of A cells, triggers the following cascade of events:
– activation of a special protein kinase necessary for the biosynthesis of some proteins that cause a sharp increase of Ca2+ level in the cytosol;
– stimulation of respiration, increase in membrane potential, and powerful generation of reactive oxygen species (ROS) in mitochondria, the process being accompanied by fragmentation of elongated mitochondria into small spherical ones;
– subsequent decrease in potential, swelling of mitochondria, and the release of cytochrome c (apparently due to the rupture of the outer mitochondrial membrane) to cytosol.
The described cascade strongly resembles the one of mechanisms of animal cells’ apoptosis [8]. The biological significance of yeast phenoptosis caused by the pheromone excess can be attributed to the stimulation of transition of these unicellular organisms from vegetative to sexual reproduction. This effect is in turn a response to deteriorating external conditions. In fact, the pheromones discard the cells that were incapable of sexual reproduction that increases the diversity of offspring and, therefore, increases the chances of the emergence of new properties that could be useful for adaptation to the changing environment [11, 13]. (For other cases of apoptosis in yeast, see review [14]).
Plants. The rapid aging of semelparous plants that reproduce only once in their life is perhaps the most frequently described example of phenoptosis, the biochemical mechanism of which is to some extent understood. The well-known plant biochemist and gerontologist L. D. Nooden wrote: “Even before much was known about the biochemistry of senescence [of such plants], it was considered to be internally programmed, because it is specific and orderly in terms of when, where, and how it occurs. Although senescence does progress with age, it is not simply a passive aging process. It is controlled by internal and external signals, and it can be delayed or accelerated by altering these signals. In contrast, the process of aging is a passive accumulation of lesions with age, and this might best be represented by the gradual loss of viability of seeds during storage” [15].
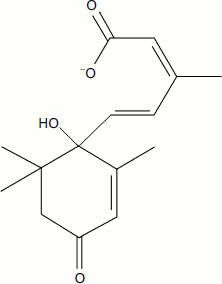
It is well known that rapid aging and death of soya can be prevented by the removal of soybean pods [16] or the ripening beans in them [17]. An interesting experiment was performed by Nooden and Murray [18]. They removed all but one pod cluster at the stage of bean ripening. In due time (after about 3 months), the plant did not die. It remained green, but the leaves on a branch with remaining pods turned yellow and withered. The death of these leaves could not be prevented by killing the phloem with a stream of hot steam directed to the branch between the leaf and the pod. The steam was supposed to prevent the transport of compounds from the leaves to the pod through the living tissue of the phloem, but not from the pod to the leaves through the xylem, which was already dead in the mature plant. This experiment proves the aging of soya to be induced by pods generating some deadly signal or producing a poisonous compound that kills the leaves [19]. This observation contradicts the statement of Kirkwood and Melov who recently published an article on the impossibility of the existence of an aging program. In their opinion, “There is a little evidence that semelparous (capable of single reproduction) organisms are actively destroyed once reproduction is completed” [20]. Certain evidence suggests the phytohormone abscisic acid plays the key role in triggering the process of soya aging [21] (Fig. 1). Nanomolar concentrations of this organic acid regulate the expression of enzymes that produce or destroy ROS in the leaves of this plant [22]. Aging of plants is known to be accompanied by a sharp increase in ROS level [22].
The aging of soya is a relatively fast process that takes about 10 days (the maximum lifespan of this plant is 90 days) [15]. Timely removal of ripening pods causes manifold increase in soya lifespan. Similar phenomena were described for other higher plants that reproduce only once, including wall cress (Arabidopsis thaliana), a classic plant model for biochemists, geneticists, and gerontologists. It was on this model that S. Melzer and coworkers, Belgian biologists from Ghent [23], could directly refute one more argument from the already mentioned article by Kirkwood and Melov: “Yet among the many gene mutations that have been discovered that affect the lifespan often increasing it significantly, none has yet been found that abolish aging all together” [20]. Melzer and coworkers recently reported in their article published in Nature Genetics [23] that mutations in two (out of more than 20,000) genes of A. thaliana, namely soc1 and ful, cause the plant to switch from sexual to vegetative reproduction. The mutant does not bloom, and, hence, it does not form seeds. It completely loses rapid aging induced by these seeds. The lifespan of an organism (usually less than 3 months) is multifold increased, becoming virtually endless like in case of most other plants that reproduce vegetatively. The plant develops cambium, secondary growth, woody stem, and many rhizomes. Wall cress turns into a large bush with bulky leaves (Fig. 2; see color insert). Floral meristem is transformed into vegetative meristem. The authors believe the present A. thaliana to have appeared initially as a vegetatively-propagated perennial plant, which originally competed with other bushes and trees and only later changed into a grass in the course of evolution (as happened with horsetails and ferns). This transition was accompanied by the appearance of sexual reproduction culminating in the formation of numerous very small seeds easily spread by wind over long distances. Seeds quickly sprout and, having fallen onto open ground, form the sprouts of grass, which grows without competing with slow-growing plants that have not yet had time to grow on this soil.Fig. 1. Abscisic acid, a phytohormone formed in seeds and causing withering and falling of leaves.
The present-day wall cress is a short-lived organism killed by its own seeds. Early death accelerates the change of generations and, therefore, the evolution of A. thaliana [19]. There is another feature that also accelerates evolution – the fact that the wall cress reproduces only once in a lifetime, so that the new portion of seeds is surely formed by another individual. This increases the diversity of the offspring and, therefore, increases the likelihood of the emergence of new traits. The very change from vegetative to sexual reproduction also accelerates evolution, for due to such a change the offspring genome is always a hybrid of the genomes of both parents. It is not a coincidence that in the case of yeast, the transition to sexual reproduction is one of the responses to deteriorating environmental conditions and, therefore, an attempt to search for novel traits that might help to survive in the new circumstances [11]. The transformation of the wall cress from a vegetatively reproducing plant to a flowering one seems to have happened relatively recently, and that is why the old (vegetative) program has remained in its genome as a backup. Arabidopsis thaliana may be seen as a precedent, when the inactivation of several genes completely prevents aging, and hence, the senescence-induced death [19]. And the wall cress does not seem to be a rare exception. According to Melzer and colleagues [23], “in angiosperms, the perennial woody habit is believed to be the ancestral condition from which annual herbaceons lineages have evolved several times independently. Conversely, evolution from annual herbaceons ancestors to perennial woody taxa has also repeatedly occurred. For example, in various annual herbaceons lineages, such as Sonchus and Echium, woody perennial species evolved on isolated islands from their continental annual ancestors [26-28]”.Fig. 2. A double mutant of Arabidopsis thaliana in genes soc1 and ful switches from sexual reproduction (by seeds) to vegetative reproduction (by rhizomes). It neither blooms nor forms seeds that kill the wild-type plants at the age of about 2.5 months. The plant changes from a small grass to a large shrub with a woody stem. a) Two-month-old plant of wild type (left) and an eight-month-old mutant (right); b) fourteen-month-old mutant (after Melzer et al. [23]).

The brevity of life of A. thaliana may provide this species with one more advantage. This plant appears in the open ground and dies so quickly that other plants simply have no time to develop enough to compete with the thale grass. So it manages to live its short life and produce the seeds in relatively comfortable conditions. It is striking that the case of A. thaliana quite literally confirms the thought of August Weismann, the first researcher of aging as a program, who said that highly organized organisms “carry in them the seeds of death” [24]. Among the perennials, there are examples of organisms that reproduce vegetatively for many years before switching to sexual reproduction and dying after seed maturation. Some bamboo species have a fixed lifespan that is determined by their flowering time. This time varies greatly: certain species flower at the age of 6 years, while others at 120 years. Agave and the Madagascan palm Ravenala madagascariensis, which bloom, respectively, on the 10th and 100th year, die right after the seeds ripen. Experiments have shown that the removal of the peduncle from the flowering agave results in a multifold prolonging of its lifespan [19, 24, 29].
Invertebrates. One might assume that senescence is programmed only in plants. However, the already discussed cases of phenoptosis in unicellular pro- and eukaryotes, as well as the examples of phenoptosis in animals that will be discussed below, indicate the general prevalence of this phenomenon.
The female of one octopus species, Octopus hummalincki, ceases to eat and dies of hunger after her young have hatched and she no longer needs to protect her laying from marine predators. If the so-called “optical glands” are removed from the female’s brain, she does not die in her due time and can propagate many times. Such an operation extends her lifespan several-fold [30]. The males of some squids die right after transferring their spermatophores to a female. The females die a bit later, after having laid the eggs [31]. The female of the praying mantis bites off the male’s head at the end of sexual intercourse, the phenomenon being cited for many years as an example of the most refined cruelty in the world of insects. However, later it was proved that the male mantis’ ejaculation becomes possible only after his decapitation [32]. Mayflies are born without a functional mouth, and their intestines are filled with air; this is why these insects die of starvation several days later, having mated with the males and laid many thousands of eggs [24].
Vertebrates. Some vertebrates also show examples of acute phenoptosis connected to reproduction. There are birds that lay eggs on barren cliffs above the Arctic Circle. Some of the species lay two eggs next to each other. The chick that hatches first splits the second egg and uses the contents to feed itself [29]. An interesting case of suicide is described in males of Australian marsupial mouse. They die a couple of weeks after rut. The death is caused by their own pheromones, which are originally produced to attract the females. The pheromone interacts with the receptors of the male vomeronasal organ, and in case of prolonged exposure, this blocks the control functions of the hippocampus in relation to the hypothalamus. As a result, the increased production of corticosteroids, adrenaline and noradrenaline, causes very strong stress, which results in a violation of salt metabolism leading to acute renal failure [33]. Significantly, the Australian mouse, as well as yeast (see above), uses pheromone as a phenoptotic tool, although the biochemical mechanisms of the functioning of this tool are completely different in these two cases.
All the examples listed in this section can be attributed to one way to accelerate evolution, i.e. the increase in the diversity of offspring. This diversity increases when an individual can become a father (or a mother) only once (we have already noted this fact when explaining the existence of plants reproducing only once in a lifetime).
The described rapid programmed aging of annual plants can be compared with progeria (accelerated aging) of Pacific salmon. Before spawning, this fish turns into a ridiculous humpbacked creature with a mouth not suitable for the taking of food. The fish dies soon after spawning. The death of the salmon resembles an accelerated movie, where the entire aging program is carried out within a couple of weeks; it starts with the decrease of immunity and ends with severe osteoporosis in the bones, sarcopenia, thinning of the skin, and the appearance of tumors [34]. According to the traditional explanation of this phenomenon, the accelerated aging of salmon is due to spending too much energy when moving against the current of the river to its headwaters, sometimes for a thousand kilometers. This explanation is wrong though, for it was shown that (i) aging and death do not occur if the gonads or adrenal glands are removed and (ii) progeria develops even if the spawning takes place in the headwaters of very short rivers that are only several hundred meters long [34]. It may be suggested that dead fish provide excellent food for river invertebrates, that are in turn eaten by salmon whitebait, explaining the biological significance of the discussed phenomenon. It is the transfer from the seawater to freshwater that seems to be the signal for actuation of progeria in this case. Austad believes salmon progeria and slow aging of higher vertebrates to be of completely different nature, that accelerated aging is a program while slow aging is the result of accumulation of occasional injuries [35]. But the discussed above similarity of many features of these two aging types contradicts this belief2.
2 T. Maldonaldo and colleagues [36-38] recently discovered peptides of amyloid plaques in the brain of spawning salmon. The same kind of plaques is present in the brain of old people with Alzheimer’s disease (for the discussion of similarity between the development of progeria and “normal” aging in humans, see [39]). The study of aging of different species of small African fish of genus Nothobranchius provides another argument against the position of Austad [40]. The lifespan of different species of this genus was shown vary up to five times depending on their habitat in the wild. For instance, N. furzeri, which lives in Zimbabwe in puddles formed during the rainy season and dried after this period, was shown to live for only 3 months, which corresponds to the length of the rainy season in this country. Nothobranchius rachovii and N. kuhntae from Mozambique, where the rainy season is four times longer, live for about 9.5 months, while N. guentheri from Zanzibar (a humid climate with two rainy seasons) for over 16 months. In the case of the shortest-lived species N. furzeri, growth and sexual maturation occur very rapidly, within the first month of life. Then the fish mates many times and lays eggs up to the end of the third month, till the puddle dries up. The next year fry will hatch from the eggs that have survived the drought. It seems to be quite remarkable that over the short period determined by the habitat conditions (the last two months of life), the fish manages to age, manifesting by the end of its life a whole set of signs of senescence (slowing of movements, loss of a “research”-type behavior in an open space, osteoporosis (gibbosity), accumulation of lipofuscin granules in liver, a sharp increase in β-galactosidase activity in skin fibroblasts, etc.).
In case of the longer-living species, this type of completion of growth and sexual maturation occur much later. Hence, aging biomarkers also start to increase later. It seems to be significant that these properties remain even in an aquarium, which means that they are determined genetically and are no longer dependent on the current habitat conditions. It looks like the fish of the short-lived species seek to age within that short lifespan given to them. It is also important that the aging program has similar traits even in such distant vertebrate classes as fish and mammals. This conservatism presents clear evidence of the ancientness of this program.
AGING OF HIGHER ANIMALS AND HUMANS AS A CASE OF SLOW
PHENOPTOSIS
Aging of an organism can be defined as a balanced decay of many physiological functions with age, leading to gradually increasing risk of death. The key question is: what is the cause of such a decay of body functions? Two main views on this problem have been competing in biology for a long time – an optimistic and a pessimistic one. The first approach believes aging to be the final stage of our ontogenetic program; this assumption implies the possibility of canceling aging by switching off this stage. The second approach considers aging to be an inevitable result of the functioning of a complex living system: the accumulation of errors and damage in its biomolecules, depletion of “life force”, operation of certain genes that used to be originally useful but became harmful with age, etc.
It is obvious that if the pessimistic hypothesis is true, then any attempts to fight the process of aging are doomed to failure: all of us are destined to get broken like an old car. One of the leading gerontologists of the XX century, Alex Comfort, stated though that it is difficult to believe a horse and a carriage age in the same way [41]. Nevertheless, the majority of gerontologists still oppose the theory of programmed aging. It is only very recently that certain data have been obtained providing direct support for the optimistic concept. These data allow us to understand how these age-activated biochemical mechanisms of the decay of body functions might have appeared in the course of evolution.
The dominance of “pessimists” began with Charles Darwin, who believed natural selection of individuals to be the basis of biological evolution. Within this paradigm, aging as a feature clearly counterproductive for an organism could not be selected as a result of struggle for survival. But that is how the philosopher Arthur Schopenhauer defined the role of an individual in the process of evolution in 1818, i.e. well before Darwin: “An individual is… not only exposed to destruction in a thousand ways from the most insignificant accidents but is even destined for this and is led towards it by nature herself, from the moment that the individual has served the maintenance of the species” [42].
And that is what Alfred Russell Wallace (who is first of all known due to the fact that he postulated the idea of natural selection at the same time as Darwin, but independently from him) wrote in one of his letters in the 1860-1870s: “When one or more individuals have provided a sufficient number of successors, they themselves, as consumers of nourishment in a constantly increasing degree, are an injury to those successors. Natural selection therefore weeds them out, and in many cases favor such races as die almost immediately after they left successors” (cited from [24]). Later (in 1881), this principle was independently formulated and developed by another great biologist, August Weismann [24]: “Worn-out individuals are not only valueless to the species, but they are even harmful, for they take the place of those which are sound… I consider death to be not a primary necessity, but that it has been secondarily acquired as an adaptation” (italicized by the author).
Weismann was immediately accused by his contemporaries of being anti-Darwinist, even though Darwin himself was perfectly aware of the limitations of his hypothesis (evolution follows only those directions that are favorable for an individual) in case of sexual selection and group adaptation. That is what he wrote in his second famous book “The Descent of Man”: “There can be no doubt that a tribe including many members who… are always ready to aid one other, and sacrifice themselves for the common good would be victorious over most other tribes; and this would be natural selection” [43].
Modern critics of Weismann usually quote Medawar, who suggested that aging while being useful for a population, could not have appeared as a mechanism produced by means of evolution. Medawar assumed that under natural conditions the great majority of animals die before they become old [44]. But today the falsity of this statement is obvious. Aging starts long before it can become a direct cause of the death of an individual [5, 44a, 44b]. At the same time, it can indirectly facilitate the future death. For instance, the age-dependent progressive weakening of the body undoubtedly increases the probability of death in case of it being attacked by predators, pathogens, etc. A. Loyson and colleagues [45] and independently R. Bonduryansky and C. Brussil [46] directly demonstrated that under natural conditions both long-lived mammals and short-lived insects suffer from aging. This result is not really surprising if we take into account the fact that, e.g. the reduction in skeletal muscle mass develops soon after the organism stops growing, and certain components of the immune system start to decay even earlier (in case of humans – between the 10th and 15th years of age).
Another objection to the programmed character of aging, often considered by Kirkwood, is based on the observation that the deviations of lifespan of individuals are too great for it to be determined by some kind of genetic program. The fallacy of this objection was directly shown in one of the articles of this issue [47]. The authors compared the deviations of values of the times of onset of menstruations, their termination, and death of the same women in the USA. In each of the three cases, the deviation values were normalized to the average value. The deviations for the onset and termination of menstruations were practically identical. In the case of mortality, deviations were only slightly higher3.
3 Such an effect is not surprising, since death is caused not only by aging but also by age-independent causes. There is another argument used by gerontologists-pessimists: “What is the reason for programmed aging if the organism is so complex that in any case it will sooner or later wear out and break?” [48]. However, it is not obvious that the systems selected over billions of years of biological evolution should spontaneously degrade during short individual lifespans. For instance, bacteria discovered in deposits of crystalline salt that were about 250 million years old were found to be quite viable [49]. A crystallin in the whale eye lens, once synthesized, can exist for at least 200 years, with the only change observed during this time being the spontaneous L-D isomerization of its amino acids (2% per decade) [50]. In addition, we should keep in mind that living systems try to control all the internal phenomena so as to avoid or at least to minimize any spontaneous processes. The question of life and death is of primary importance for an organism, and therefore it is extremely unlikely for this issue to be left uncontrolled by biological evolution. Moreover, the hypothesis of spontaneous aging as a cause of death contradicts the fact that even among animals, i.e. the kingdom of organisms of the most complex structure, there are examples of non-aging species: invertebrates (sea urchins, big crabs, pearl mussels), fish (pike, shark, northern rockfish species), amphibians (toad), reptiles (giant turtle, crocodile), birds (albatross, raven), and even mammals (the largest species of whales, naked mole-rat, likely a bat, and some others). Even a brief look at these examples indicates that it is not their greater simplicity, but something completely different that constitutes the difference between the aging and non-aging organisms. The non-aging northern rockfish is hardly less complex than its aging southern relative. The levels of complexity of such pairs as non-aging toad and aging frog, crocodile and lizard, bat and shrew, naked mole-rat and mouse do not differ from each other. The last pair seems to be especially interesting, since both animals, the aging and non-aging ones, are of similar small size and tolerate captivity relatively well.
The naked mole-rat (Heterocephalus glaber, Rodentia: Bathyergidae) is a species of small rodents that live in laboratory conditions for over 30 years. These animals do not suffer from cancer, cardiovascular diseases, immunodeficiencies, diabetes, or atherosclerosis. They live in large colonies composed of about 200-300 “subordinates”, a “queen”, and her several breeding partners. Only the queen and her “husbands” breed. The queen’s apartment is situated in the center of a vast underground labyrinth the size of a football field and, being well protected by their “subordinates”, they have no enemies. When in laboratory conditions, the naked mole-rats die very rarely and for unknown reasons. The level of their mortality does not depend on age [51]. Unlike social insects like bees or ants, the “queen” and the “husbands” of the naked mole-rat do not possess any initial morphological differences from the “subordinates”. In the case of the queen’s death, she can be replaced by any female that previously used to be her subordinate.
The striking and obvious difference between a naked mole-rat and a mouse is that the function of reproduction is monopolized by the “queen” and her “husbands”, who are well protected against external enemies and therefore no longer experience the pressure of natural selection. The absence of enemies is the common feature of all species of listed-above non-aging animals: sea urchins with poisonous spines, toads with skin glands forming the strongest poisons, large crabs, turtles, and pearl mussels with their strong shells, large predators of shark type and large birds with sharp beaks, giant whales that are too large to have enemies, and, finally, the naked mole-rat – a small creature that has created large societies with a few privileged individuals that have been entrusted with the function of reproduction4.
4 In the wild (equatorial Africa) the “subordinates” of the naked mole-rat live for only about 3 years. They die when fighting with snakes or hordes of naked mole-rats from neighboring communities. As they do not breed, their participation in the evolution of this species should be strongly limited.
SLOW AGING AS A WAY TO ACCELERATE EVOLUTION
It 1989, R. Dawkins [52] introduced the term “evolvability” (ability to evolve). According to Kirschner and Gerhart [53], such ability is inherent in all organisms that have appeared during biological evolution, though it varies in different species. In those species that took over new areas of habitat, natural selection was mainly directed towards an increase in evolvability.
In 1997-2003 we introduced a hypothesis that aging may be a way to accelerate evolution [2, 3, 8]. Aesop once said that a hare would always outrun a fox, because it is a matter of life or death for the hare, while for the fox it is but a matter of lunch. This would suggest that foxes are not involved in natural selection of hares. Such a statement is likely to be true with respect to strong young hares. But will it be still valid for older hares, who run slower and slower because of aging? Let us consider the following imaginable experiment. Two young hares, a clever one and the second not so clever, upon meeting a fox have almost equal chances to escape from the enemy, since they can run much faster than the fox. However, with age, this principle ceases to function: the clever hare will gain advantage over the stupid one, since they will run slower due to sarcopenia. Now the clever hare, as soon as it sees a fox, will immediately take to heels and will have much more chance to save itself than the stupid one that will be delayed at the start. This means that only the clever hare will continue producing leverets. As a result, the hare population will grow wiser [8].
It is important to note that the most actively reproducing young part of the population will not be involved in such an experiment, being a guarantor of the stability of what has already been achieved in the course of evolution. At the same time, the aging part of the population can afford to slightly change the genotype of the species by selecting certain new traits. If these traits prove to be useful, they will eventually be fixed in the offspring by means of natural selection. At the same time, in case of a new trait having some unfavorable effects, it will not pass through the sieve of selection, and the experiment per se will have no serious consequences for the species as the number of old individuals in a population is rather limited, they do not reproduce as actively as the young ones, and they are destined to die out [54].
THE MYSTERY OF SELECTING TRAITS COUNTERPRODUCTIVE FOR AN
ORGANISM
There is one objection used by “pessimists” that “optimists” still find difficult to address. This is the question of how the aging program might have been selected by natural selection while being counterproductive for an individual.
It should be clear from the material discussed above on acute phenoptosis that there are a number of counterproductive programs that have developed in the genomes of living organisms in the course of biological evolution in spite of their obviously negative impact on the individual. They include not only dying immediately after breeding, but also rapid aging of many annual plants and certain aminals. In plants, genes have been discovered that are required for killing the plants after seed maturation; also, one of the poisons used as the instrument of such murder – abscisic acid – has been identified. Aging of animals differs from these phenomena only in one respect – it is much slower.
One possibility to explain the selection of counterproductive programs is that they are carried out by bifunctional proteins that simultaneously fulfill two functions: one harmful to the organism and another one useful, the disappearance of which proves to be lethal. For example, cytochrome c is involved in apoptosis caused by ROS, which, in our opinion, play the key role in the process of animal aging (see below). Cytochrome c fulfills this counter-productive function (participation in the aging process) when leaving mitochondria for the cytosol where it interacts with the protein Apaf-1. It would seem that a mutation inactivating the cytochrome c gene could prevent aging, thereby providing advantages for the mutant animal in the struggle for survival. However, cytochrome c also has another, positive function – the transfer of electrons in the mitochondrial respiratory chain. The disappearance of cytochrome c would stop mitochondrial respiration, as if the animal had been poisoned by cyanide. Interestingly enough, the binding of cytochrome c with Apaf-1 and its partners in the respiratory chain (cytochrome c1 and cytochrome oxidase) is due to ionic interaction of the “corolla” of cationic groups of cytochrome c lysines with anionic groups of dicarboxylic acids of Apaf-1, cytochrome c1, and cytochrome oxidase [54]. Evolution might have selected cytochrome c to participate in aging because of the fact that this small (104 amino acids) single-domain protein had been already involved in a vitally important function of respiration, and a mutation violating its interaction with Apaf-1 would very likely lead to the disruption of respiratory function as well. Nevertheless, experiments carried out in our group have shown that the replacement of one of the lysines from the cationic corolla of cytochrome c (K72) by tryptophan produces a protein fully active in its respiratory function but forming an inactive complex with Apaf-1 [55]. Any replacement of K72 by amino acids other than W affected both functions or neither of them. The K72W mutant was quite viable in vivo (at least in experiments with embryonic fibroblasts [55, 56]). We are now studying aging of the K72W mutant mice.
WORKING HYPOTHESIS ON THE MECHANISM OF SLOW AGING OF
ANIMALS
There is much evidence that mitochondrial DNA damage participates specifically in the regulatory cascade that causes aging of different organisms from yeast to animals. This statement has been recently directly proved in the elegant experiments carried out in the laboratories of N. G. Larsson, T. A. Prolla, and H. P. Zassenhaus. They found that the expression of the mutant mitochondrial DNA-polymerase, which can still synthesize DNA but cannot carry out identification of mistakes in this synthesis and their correction (“proof reading”), leads to a substantial increase in the mutation frequency in mitochondrial DNA, especially in the area of cytochrome b. It also causes a substantial shortening of lifespan and development of many typical signs of aging [57-59]. Injection of cyclosporin A, the inhibitor of pores in the inner mitochondrial membrane, was found to prevent the effect of the mutation (these studies were conducted in Zassenhaus’ group; only DNA-polymerase from heart muscle was modified) [57].
About 50 years ago, D. Harman in the USA [60] and then N. M. Emmanuel in Russia [61] suggested aging to be the result of ROS-induced damage to biopolymers (especially to DNA). Over the years, much evidence supporting this postulate has been found, and it is mitochondrial DNA that seems to be the primary ROS target in the course of aging [8, 13, 54].
There is no doubt that aging leads to such a change in the balance of the systems of ROS generation and scavenging that the levels of both ROS and ROS-induced damage increase. This situation inevitably leads to gradual increase in the number of apoptotic cells. Not all dying cells are replaced by new ones, and as a result the overall number of cells in organs and tissues is reduced. The latter phenomenon leads to a decrease in vitality, the main sign of aging [62, 63]. It is not the fact that during aging each cell starts to work worse. It is far more important that the number of these cells decreases. Senile sarcopenia, i.e. the reduction of the number of cells in skeletal muscles, is a typical example of this phenomenon [54]5.
5 Skeletal muscles are known to begin aging much earlier than the majority of other tissues [8]. B. Szczesny and colleagues [63a] recently discovered a possible cause for this phenomenon. Two systems of reparation of damaged mitochondrial DNA were shown to change with age: they are stimulated in liver, kidneys, and heart muscle and are inhibited in skeletal muscles. Reparation of nuclear DNA in the cells of the same tissues does not change with age. Besides that, the levels of the key antioxidant enzymes, “manganese” and “copper-zinc” superoxide dismutases and catalase were shown to be much lower in skeletal muscles than in the three other tissues that age substantially slower. Accordingly, aging caused faster increase in carbonylation of proteins in skeletal muscle than in liver, kidney, and heart. It was found that H2O2 generation by mitochondria of different tissues is much lower in birds than in mammals of similar weight [54]. According to S. Austad and coworkers, similar differences can also be observed between flying and terrestrial mammals – a bat and a shrew [64].
Research conducted independently in the groups of R. Sohal [65], G. Barja [66, 67], and M. Brand [68] showed the lifespan of warm-blooded animals to shorten parallel to increase in ROS generation during reverse electron transfer in complex I of the heart mitochondrial respiratory chain. Such a correlation could not be observed in the case of ROS being generated during direct electron transfer in the same span of the respiratory chain [68]. The experiments of the Brand’s group were the most thorough: 12 different species of mammals (including mice, baboons, and cows) and birds (quails and pigeons) were studied. Eleven species fit perfectly well the pattern describing their lifespan as an inverse function of the ROS generation rate. Only one species was an exception to the rule. This was the naked mole-rat. This rodent of mouse size lives almost 10 times longer than a mouse while producing ROS much faster. This proved to be the case though when an exception confirms a rule, for the naked-mole rat was the only non-aging animal among the studied species. Apparently the mitochondrial ROS-linked aging program is switched off in the naked mole-rat.
So, we can suppose the realization of the aging program is triggered by the enhancement of the level of mitochondrial ROS, which is programmed in the genome.
There comes a question – why it is ROS that was selected during evolution as an aging instrument? There is no doubt that ROS still present a problem for current aerobic forms of life. Perhaps that is the reason for aging, while being a specialized evolutionary mechanism, to function so as to facilitate the improvement of the antioxidant system of an organism. If it is the case, ROS may be compared to the fox in the example discussed above of the improvement of hare breeds, but here the selection deals not with better intellect, but with better system of protection against ROS. This assumption might account for ROS participating in the realization of programs of self-destruction of mitochondria, cells, organs, and organisms [54].
There is no doubt the actuation of the ROS-mediated aging program starts relatively early, most likely when an organism stops growing, or even earlier, at the completion of puberty. The simplest version is that the “chronometer”, which counts time and gives the signal for the cessation of growth, not only switches off the growth program, but also triggers the aging program [41, 69, 70]. It is worth noting that such non-aging long-lived animals as pearl oyster, pike, big crabs, turtles, and whales never stop growing [41, 71].
The next questions are, what is that “chronometer” and where may it be found? If our organism counts our years similar to the way it counts hours, then the aging “chronometer” should be looked for in the suprachiasmatic nucleus of the hypothalamus, which is responsible for the maintenance of the circadian rhythm of mammals. In the case of birds, it is the pineal gland that fulfills the same function. This gland participates in the circadian rhythm of mammals as well, but here it is triggered by the suprachiasmatic nucleus.
Periodical biochemical reactions have been discovered in the cells of the suprachiasmatic nucleus. These cycles correlate with circadian rhythm. Neurons transmit corresponding signals to the pineal gland, which responds to this by fluctuations in production of melatonin, the hormone that is released by this gland and transported in the blood to all organs and tissues. Diurnal melatonin fluctuations in blood are the mediator of the circadian rhythm of the organism’s functioning. Melatonin happens to be an antioxidant, and, what seems to be even more important, an inducer of the synthesis of a group of the proteins of antioxidant cascades. It is interesting to note that the magnitude of circadian fluctuations of melatonin concentration in blood strongly decreases with age, and it is this decrease that seems to be the earliest aging sign, for in the case of humans it can be observed starting from 7 years of age [35]. V. N. Gladyshev and coworkers recently discovered severe disturbances in the melatonin mechanism in the naked mole-rat [72].
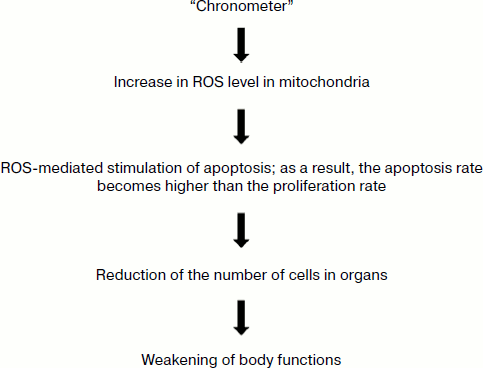
On the whole, the hypothetical aging program of mammals can be presented as follows (Fig. 3).
Fig. 3. Working hypothesis on the biochemical mechanism of programmed aging of organisms [12].
SCAVENGING OF MITOCHONDRIAL ROS AS A POSSIBLE WAY TO RETARD THE AGING PROGRAM
If the aging program can be described by the scheme shown in Fig. 3, an approach that could interrupt it seems to be quite obvious. To this end, it might be sufficient to scavenge ROS in mitochondria in such a way that prevents an increase of their effect with age. Natural antioxidants, such as vitamin E that accumulates in membranes, seem to be good candidates for this role. But the truth is that so far nobody has succeeded in radically slowing human or animal aging using an antioxidant. An attempt to cancel aging (as well as any other biological program) seems to face the resistance of the organism that tries to fulfill it by any means. For instance, the increase in vitamin E consumption was shown to induce a special form of cytochrome P450 in liver, this protein being responsible for the destruction of the excess vitamin E [54]. That is why it is synthetic antioxidants that seem to be better candidates for canceling the aging program. These antioxidants need to be effective in very small concentrations so as not to activate cytochrome P450; they also need to be specifically addressed to mitochondria, for their primary targets should be intramitochondrial ROS. Thus, the question is: how to provide addressed targeted delivery of antioxidants to mitochondria?
In the late 1960s, hydrophobic cations and anions having their charge strongly delocalized over relatively large organic molecules and easily penetrating the mitochondrial membranes were described in our group and the group of E. A. Liberman [73]. Later these compounds were given the name Skulachev ions (Sk+ and Sk−) by the prominent American biochemist D. Green [74]. Due to their low level of hydration (water dipoles cannot form a “water coat” around an ion if its charge is delocalized and screened by bulky hydrophobic substituents), Sk+ and Sk− can penetrate membranes. As a result, their distribution between the two compartments separated by a membrane follows the Nernst rule (10-fold gradient of ion concentration per 60 mV of membrane potential, Δψ). As any ion gradient depends logarithmically on ΔΨ value, a mitochondrion with Δψ = 180 mV (negative inside) is capable of maintaining a 1000-fold Sk+ gradient. The tetraphenylphosphonium cation and tetraphenylboron anion were studied as examples of penetrating ions. These two oppositely charged compounds differ by only one atom: phosphorus in the cation and boron in the anion. The flows of cations and anions were in different directions in mitochondria: cations are accumulated in these organelles, while anions are ejected from them [73, 75]. We suggested that Sk+-type ions can serve as “molecular electric locomotives”: if compound X needs to be delivered to mitochondria, it would be enough to link it to Sk+ [76]. Such a compound should be specifically delivered to the mitochondrial matrix, the only intracellular compartment surrounded by a membrane that is charged negatively in relation to the cytosol [75].
We applied such logic when explaining the role of the carnitine cation group in the process of transport of fatty acids residues into mitochondria [76]; later it was also used by M. Murphy and R. Smith for the construction of mitochondria-targeted antioxidants [77]. For their experiments, they mainly used the compound called MitoQ with ubiquinone serving as the antioxidant and decyltriphenylphosphonium cation as Sk+ [78-80].
We confirmed micromolar concentrations of MitoQ to be able to accumulate in mitochondria and to protect them from oxidative stress. But at the same time, M. Yu. Vyssokikh in our group showed even a small overdose of this compound causes the opposite effect: MitoQ was turned into a powerful prooxidant catalyzing H2O2 generation in mitochondria, the rate of this process being so high that it was comparable with the respiration rate of mitochondria in the absence of ADP [81, 82]. Similar observations were obtained in three other laboratories, as well as in Murphy’s group [83-85, 88a].
When it became obvious that MitoQ, due to its prooxidant effect, can hardly be the mitochondria-targeted antioxidant capable of decelerating the aging program, we turned to plastoquinone, an electron carrier that is found in the photosynthetic electron transport chains of plant chloroplasts and cyanobacteria. It is the better antioxidant properties of plastoquinone (in comparison to ubiquinone) that might have become the reason that ubiquinone of the mitochondrial respiratory chain is replaced by plastoquinone in the chloroplast photosynthetic chain of the same plant cell. Oxygen-producing chloroplasts constantly experience much stronger oxidative stress than oxygen-consuming mitochondria. Plastoquinone has methyl groups where ubiquinone has methoxyl groups, and a methyl group of ubiquinone is replaced by hydrogen in plastoquinone. These substitutions sharply increase the antioxidant activity of the quinone in model lipid systems (this was already shown by J. Kruk [86] and V. A. Roginsky [87] when we started our studies of plastoquinone derivatives). As was shown in our group by Vyssokikh, the MitoQ concentrations causing anti- and prooxidant effects differ by less than half (300 and 500 nM). For the plastoquinone derivative of decyltriphenylphosphonium, which was coined SkQ1, this difference increased to 32-fold (25 and 800 nM) [81, 82, 88]. Having obtained this result, we realized that SkQ1 is a remarkably efficient mitochondria-targeted antioxidant with little risk of prooxidant activity6.
6 Quite recently, Fink et al. in Iowa [88a] repeated such a comparison of MitoQ and SkQ and confirmed our data that two compounds at submicromolar concentrations are of similar prooxidant activity. Unfortunately, they did not study antioxidant activity of lower concentrations (for cell monolayers studied, below 5·10–8 M). An international investment project dedicated to the analysis of SkQ1 and related compounds was started in 2005. The A. N. Belozersky Institute of Physico-Chemical Biology (Moscow State University) became the parental organization of the project, which has embraced over 30 different research groups from Russia, Ukraine, Sweden, Germany, and USA. First of all, a reproducible method for synthesis and purification of the new compound and various derivatives was developed (G. A. Korshunova et al.) [81]. The results of further studies with SkQ1 and some of its analogs can be summarized as follows.
1) SkQ1 has low solubility both in water and hydrocarbons, but it is highly soluble in octanol (the distribution coefficient in the octanol/water system is about 104), indicating the high affinity of this compound to membrane structures (V. N. Tashlitsky et al.) [89, 90].
2) Experiments on planar bilayer phospholipid membrane (BLM) show that the SkQ1 cation penetrates the membrane (I. I. Severina, Yu. N. Antonenko, et al.) [81, 82].
3) SkQ1 is a very active antioxidant in BLM, liposomes, micelles, and mitochondria (M. Yu. Vyssokikh) [81, 82, 89, 91]. Peroxidation of mitochondrial lipids, first showing the destruction of cardiolipin, is strongly inhibited by nanomolar concentrations of SkQ1 (M. Yu. Vyssokikh) [81, 82, 89, 91].
4) Energization of mitochondria in vitro leads to SkQ1 accumulation in them (M. S. Muntyan) [81].
5) SkQ1 is reduced by the mitochondrial respiratory chain in the i center of complex III, this effect being blocked by antimycin A. The oxidation of reduced SkQ1 (SkQ1H2) in the o center is much slower than the reduction of SkQ1 in the i center. Therefore SkQH2 can serve as a rechargeable antioxidant that exists in mitochondria mostly in its active (reduced) form (M. Yu. Vyssokikh et al.) [81, 89].
6) At nanomolar concentrations, SkQ1 acts as an antioxidant due to its plastoquinone part (its plastoquinone-lacking analog, C12TPP, is inactive). Increasing SkQ1 concentration leads to the appearance of another antioxidant effect, which is reproduced by C12TPP and requires the presence of free fatty acids. It was shown that SkQ1 and C12TPP could serve as carriers of fatty acid anions, enhancing their uncoupling effect on mitochondria (F. F. Severin et al.) [91, 92].
7) In human cell cultures, SkQR1 (a fluorescent analog of SkQ1) specifically stains all of the mitochondria and only mitochondria (B. V. Chernyak et al.) [81, 82, 91].
8) In the same cell cultures, SkQ1 is a potent inhibitor of apoptosis caused by the addition of H2O2. It also prevents fragmentation of extended mitochondria into small spherical organelles, an effect accompanying apoptosis (B. V. Chernyak et al.) [81, 82, 89].
9) When added to food or water for the entire lifetime, SkQ1 increases lifespan of the mycelial fungus Podospora anserina, the crustacean Ceriodaphnia affinis, the insect Drosophila melanogaster, the fish Nothobranchius furzeri, and mammals: mice, mole-voles, and hamsters (V. N. Anisimov et al., B. Cannon, J. Nedergaard et al., E. A. Novikov, K. A. Rogovin et al.) [91, 93, 94]. The largest (two-fold) effect on the rodent lifespan is achieved when keeping animals in a non-sterile vivarium; it is accompanied by deceleration of such characteristic signs of age-dependent decay of the immune system as involution of thymus and spleen follicles and reduction of the ratio of lymphocytes to neutrophils in blood. This results in a sharp decrease in mortality of the mice from infections [82, 91, 93]. At the same time, the mortality of old mice from cancer remains virtually unchanged7. The result is that SkQ1 reduces mortality in a non-sterile vivarium at the early and middle stages of aging, but not at later stages, when cancer becomes the predominant cause of death. In a sterile vivarium, SkQ has minimal effect on the lifespan of mice since practically every mouse lives long enough for some cancer to develop (it is particularly true for females, who are prone to mammary gland cancer).
7 SkQ1 increases the lifespan of mice suffering from lymphoma and reverses certain types of cancer cells proliferating in cell culture into normal cells [82, 102], but it has no effect on mammary gland cancer in old female mice [82, 93, 94]. 10) Geroprotective effect of SkQ1 is accompanied by inhibition, termination, and in some cases reversal of the development of a large group of typical traits of aging, such as osteoporosis and lordokyphosis, canities, alopecia, loss of whiskers, slow wound healing, sarcopenia, extension of the left ventricle, disappearance of estrous cycles in females and male libido, as well as investigative reflex, etc. SkQ1 has particularly pronounced effect on the aging of the visual system of animals and humans. We have described inhibition of the development of retinopathies and their reversal by means of SkQ1; similar results were obtained for cataract, glaucoma, uveitis, conjunctivitis, and dry eye syndrome. In the latter case, we have successfully conducted clinical trials on humans, and the Russian Ministry of Health has granted permission to use SkQ1-eye drops for treating people suffering from this slowly progressing (but previously regarded as incurable) disease. Currently, the eye drops “Vizomitin” with 250 nM SkQ1 as the active ingredient are already on sale in Moscow pharmacies (M. V. Skulachev, A. A. Zamyatin, N. G. Kolosova, V. N. Anisimov, B. Cannon, J. Nedergaard, M. V. Yegorov et al.) [82, 91, 93-97].
11) A single injection of SkQ1 or SkQR1 prevents death after crisis and greatly facilitates recovery from ischemia of kidneys, rhabdomyolysis, and pyelonephritis. It reduces the areas of heart infarction and brain damage during stroke in experimental ischemia in rats (D. B. Zorov, E. Y. Plotnikov, N. K. Isaev et al.) [81, 82, 89, 98-101]. The reasons for these effects will be discussed in the last section of this article.
Summarizing the results presented above, we concluded that compounds of SkQ type are the most active of all known geroprotectors; they reduce mortality at the early and middle stages of aging and prevent the development of a large group of senile defects. In other words, SkQ can prolong the youth of experimental animals8.
8 When speaking of the extension of youth, we would like to stress that canceling of the aging program (viewed as the age-dependent weakening of the main life functions) does not mean individual immortality. If we look at the aging program as a biological mechanism that accelerates evolution, then the increase in the maximum lifespan is not likely to be an indispensable consequence of canceling of such a program. The input of the most long-lived individuals to the general productivity of a population will be rather small because of their paucity. Moreover, genome damage can accumulate with age not only because of the programmed increase in ROS level, but also for other reasons that last for a lifetime. This is especially dangerous for females: the DNA of their oocytes should remain intact throughout the entire individual lifetime. It is not surprising that females lose their ability to reproduce starting from a certain age. Menopause begins, which is particularly long in humans and whales, but exists even in the short-lived fruit fly Drosophila. It is no coincidence that the contribution of the aging program to the mortality rate is especially high at early and middle stages of aging, and its removal by SkQ significantly increases life expectancy. However, further aging brings forward other causes of death, which are, as a rule, independent of the aging program and therefore cannot be cancelled by SkQ. Cancer is one of the main death causes in this group. It may also be a phenoptotic program, but of a different function: in this case, it can be directed at discarding of individuals whose genomes have become overloaded with injuries [103-106].
COMPARISON OF THE EFFECTS OF SkQ AND FOOD RESTRICTION
The best evidence in favor of the hypothesis of programmed aging could be provided by a successful attempt at canceling it. There are some reasons to suggest that such an attempt gave a positive result as early as 1934-1943, when C. M. McCay, T. B. Robertson, and coworkers managed to extend the lifespans of rats and mice by restricting their diet [107-110]. This restriction was introduced at early life stages and initially resulted in retardation of growth. When the food restriction was canceled, the rats rapidly increased in size to reach the norm, but they lived longer by 70% (males) and 48% (females) than rats that were fed ad libitum over their whole life [107-109]. As to the reasons for deaths, a strong decrease in death rates from pulmonary diseases and certain types of tumors was observed. The researchers noted that long-lived animals looked mobile and young irrespective of their age [107-110]. Later the positive effect of dietary restriction on lifespan was demonstrated for a great variety of organisms – from yeast to rhesus monkeys and humans. With the appearance of Harman’s hypothesis on the role of ROS in aging [60], these effects came to be explained in terms of decreased amount of food oxidized by oxygen and, as a consequence, decreased production of ROS and reduction of ROS-induced damage to biopolymers. The inconsistency of this assumption was evident already in early works on dietary restriction when the same group of researchers found that the heat production per kilogram of body weight in the experimental rat cohort was higher than in the control [111]. Further research gave direct evidence against the viewpoint that had already become commonly accepted.
First, it was found that it is enough for Drosophila to live starving for only two days to become as long-lived as when the fly was subjected to dietary restriction over its whole life [112]. Second, it turned out that not only an excess of food, but also its smell attenuate the geroprotector effect provided by dietary restriction in Drosophila and the nematode C. elegans [113, 114]. These observations are unlikely to be specific for invertebrates. As far back as 1934, T. B. Robertson et al. discovered in their experiments on albino mice that a two-day-long “fasting” every week is quite sufficient for their lifespan to be extended by 50-60% [110]. C. J. Carr et al. observed, again on mice, that the reproductive ability lost by the end of the first year in case of unrestricted feeding was preserved until at least the 21st month of life when mice were limited in food over the first 11-15 months and then were fed ad libitum [115]. According to the data of Czech researchers E. Stuchlikova et al., rats, mice, and golden hamsters restricted in food by 50% for two years lived longer by 20% than the control. Dietary restriction only during the first year of life prolonged the lifespan by 40-60%, and during the second year by 30-40% [116].
Further research showed that the effect of dietary restriction is contributed by both the carbohydrate and protein components of food. The effect of proteins is associated with one, and only one, amino acid – methionine [117-120]. Methionine belongs to the group of essential amino acids, which are not synthesized in a mammal’s body, so food is the only source of this compound. It was found that a diet in which proteins were replaced by a mixture of amino acids containing no methionine not only favors a longer life, but also decreases mitochondrial ROS generation and oxidative damage to mitochondrial DNA [119, 120]. Interestingly, dietary restriction has no effect on the oxidation of nuclear DNA [121].
In our opinion, calorie restriction is perceived by an organism as a worrying signal of starvation. Even partial starvation is known to entail decreased fecundity. And this, in turn, jeopardizes the very existence of the population. To prevent, at least partially, such turn of events, it is sufficient to cancel the aging program, thereby prolonging the reproductive period of an individual, i.e. increasing the total number of its possible descendants [4, 122]. In other words, the influence of dietary restriction on lifespan is only indirectly related to ROS, being a regulatory effect. This is first of all biology, and not chemistry. That is why such evidently signaling effects as a short-term fasting (or, vice versa, smell of food), and not life-long food shortage (or excess) exert a powerful effect on the life cycle parameters. It is remarkable that a temporary dietary restriction (fasting) is better than a constant one. The signal can be given within a fairly short time, whereas, as a matter of fact, to live starving for a long time is not useful for an organism9. Gluttony is characteristic of certain people rather than of animals, the latter not tending to eat in excess when having enough food.
9 Religious fastings probably prolong life by short-term dietary restrictions. Believers are generally known to live longer than atheists [132]. The signaling nature of the effect of dietary restriction provides a good rationale for the experiments with methionine. Apparently, the organism determines the amount of available food and, first of all, essential amino acids required for protein synthesis, by monitoring the level of only one of them, specifically – methionine.
It seems quite significant that dietary restriction not only extends lifespan, but also prolongs youth, as already mentioned by the discoverer of this phenomenon McCay [107-109]. Age-related diseases step back and old animals become indistinguishable from young ones in behavior and even exterior appearance. The recently reported results of research of R. Weindruch and his group on primates are quite illustrative in this respect [123]. Twenty-year-long experiments on 76 macaques (adult animals from 7- to 14-year-old) showed that a long-term 30% dietary restriction gives the following effects: (i) a sharp decrease in age-related death rate (20% of over 30-year-old animals against 50% in the control group fed ad libitum), (ii) exclusion of diabetes from the list of causes of death, (iii) halving the death rate from cancer (in macaques this is primarily intestine adenocarcinoma), (iv) decrease in the death rate from cardiovascular diseases, (v) decrease in osteoporosis, (vi) arrest of the development of such age-related traits as sarcopenia, decline in brain grey matter, alopecia, canities, etc. By the age of 30 years, 80% of the surviving control macaques showed some traits of aging, whereas in the experimental group such traits were observed in only about 20% of the animals.
The experiment on monkeys is still not completed, and, therefore, we can say nothing about the effect of dietary restriction on the maximum lifespan of primates. At the same time, some evidence on this subject is available for rodents. This evidence shows that the median lifespan in mice, rats, and hamsters increases much more strongly than the maximum lifespan. The simplest explanation of the mechanism of this phenomenon is an arrest (or a strong retardation) of the aging program [54, 122]. Therewith, other ontogenetic programs, especially body growth, can also be retarded. These phenomena are observed for a rather strong and long-term fasting. However, a more moderate dietary restriction can prolong life while not causing inhibition of growth.
The effects of dietary restriction and SkQ antioxidants have much in common. In both cases, rectangularization of survival curves and decrease in early mortality rates are observed, and the median lifespan increases much more than maximum. Both of these factors operate not only, and not so much, for prolongation life as such, as to prolong a healthy young life. Both are very effective in organisms quite different in their systematic position (dietary restriction works well in yeast, insects, and mammals, and SkQ in mycelial fungi, crustaceans, insects, fish, and mammals). The effect of both factors is clearly pleiotropic in nature, i.e. they influence a variety of physiological functions. Cardiovascular diseases, osteoporosis, vision disorders, and certain types of cancer step back; graying and hair loss, as well as age-related depression (torpor) do not occur. Contradictory data concerning effects of dietary restriction on sarcopenia and immune responses have been reported. Some authors state that such exposure adversely affects both the muscle system and immunity10. On the other hand, R. Weindruch and coworkers [123] reported the lack of sarcopenia in monkeys and C. M. McCay and coworkers [107-109] described resistance to pulmonary diseases in rats subjected to dietary restriction (cf. sharp decrease in mortality from infectious diseases and inhibition in age-dependent involution of the thymus and follicular spleen compartments in rats exposed to SkQ [82, 126]). However, the effects of dietary restriction and SkQ on wound healing are opposite to each other: starvation inhibited, while the antioxidant SkQ1 stimulated this process [54, 127]. Dietary restriction decreased body temperature and inhibited growth of animals; these phenomena were not observed with SkQ [54, 122]. These data rule out such a trivial interpretation of our data as the assumption that SkQ1 decreases animal food intake, say, by decreasing appetite. Direct measurements on mice treated with SkQ1 revealed no decrease in food intake [54].
10 J. Fernandes and coworkers [124] noted that dietary restriction, especially at young age, inhibits interleukin secretion by macrophages as a response to bacterial polysaccharides, and this adversely affects resistance to sepsis and peritonitis. According to E. M. Gardner, partial starvation reduces resistance to influenza virus [125]. The fact that dietary restriction adversely affects some vitally important parameters is not surprising. It has already been mentioned that animals do not tend to overeat even if they are not restricted in food. Therefore, long-term dietary restriction entails some disorders in vital functions. It is also clear that such disorders are the more probable the longer is starvation. We have already stated that long-term and continuous dietary restriction is not at all necessary for the geroprotector effect. This explains the controversy in data on the effect of dietary restriction on lifespan and the organism’s state. In cases where dietary restriction was not too strong and not too long, positive effects were observed, whereas when gerontologists overdid the restriction, unfavorable side effects took place. Thus, it is commonly accepted that long-term dietary restriction decreases the frequency of estrous cycles (sometimes until they cease completely) [128], but in 1949 C. J. Carr and coworkers showed that temporary dietary restriction with its subsequent cancellation prolongs estrous cycles and favors their preservation until extreme old age [115]. Let us remind you that the same effect was observed with SkQ [82, 93, 94]. In principle, there is no need to starve through the whole life if starvation prolongs life by switching off the aging program. However, there is a probability that too weak or delayed dietary restriction will only partially retard the program, and the geroprotective effect will be weak.
One more circumstance should be taken into account when considering under-eating as a geroprotector for humans. Actually, if dietary restriction is a signal to warn about infertility and death from starvation, then the organism should respond not only by prolonging life to compensate for the decay of birth rates in lean years. Other responses also seem quite possible, and some of them are not as attractive as extension of a healthy life. For example, it was noted that a hungry mouse, once entering a squirrel wheel, does not want to leave it and travels from 6 to 8 km overnight (with normal feeding, this distance is always less than 1 km) [128]. Obviously, this effect cannot be explained by starvation-induced exhaustion and muscle weakness. More likely, we deal here with one more response to the starvation signal: extreme anxiety and attempted scanning of as large a space as possible in a search for food. Paraphrasing Strugatsky’s novel “Monday Begins on Sunday”, we deal here with a mouse “unsatisfied stomachically”. Had this effect been characteristic of SkQ, we would have observed enhanced food intake by SkQ-receiving animals, which is not the case. It seems that the use of SkQ is a “purer” way to switch off the aging program, not overburdened with undesirable side effects [54, 122].
FROM Homo sapiens TO Homo sapiens liberatus
There is at least one more aspect where dietary restriction is unlikely to replace SkQ. The material discussed above gives us grounds to suggest that SkQ is a quite promising agent for creation of new-generation medicines capable of influencing the aging processes – mitochondria-targeted antioxidants. It is important to note that SkQ acts not only as a geroprotector. Experiments on cardiac and renal infarctions, stroke, rhabdomyolysis, pyelonephritis, and a number of other experimental pathologies have been conducted on young animals. Surely, we might suggest that SkQ merely “cleans the dirtiest place in a cell” and thus exerts a certain nonspecific favorable effect on various aspects of its vital functions. However, such explanation is unlikely to be true. In this case, it remains unclear why during their evolution the plants that synthesize separately plastoquinone and the penetrating cation berberine have not formed their combination capable of removing ROS from the interior of plant mitochondria experiencing a higher “oxygen threat” than animal mitochondria, since the cytosol of plant cells is saturated with oxygen formed by chloroplasts.
An alternative possibility is that the program triggering aging as a slow phenoptosis is also used in other deadly programs which trigger rapid, or acute, phenoptosis. Here we come to a fundamental question of biology: how could genetic programs counterproductive for an individual come into existence? C. Darwin, A. R. Wallace, and A. Weismann suggested as early as the XIX century that the death of an individual may be altruistic, i.e. serving a family or a community [24, 43]. In 1964, W. D. Hamilton [129, 130] published a series of two papers entitled “The Genetic Evolution of Social Behavior”, where he calculated the quantitative aspect (the role of the level of relatedness) in this phenomenon. And in 1976, R. Dawkins wrote the book “The Selfish Gene”, in which he developed and popularized Hamilton’s concept concluding that “the main selection unit is a gene rather than an individual” [32]. In essence, the issue here is already not social well-being, but the “dictatorship” of the genome, the only self-reproducing biological structure whose preservation, development, and expansion have taken priority over the well-being of an individual or a group of individuals. In terms of this concept, an organism is nothing but a machine serving the genome’s interests [88, 131].
Some time ago, I formulated a principle called the “samurai” law of biology: “It is better to die than to be wrong” or, in a more extended form, “complex biological systems possess self-elimination programs that are activated when the system in question becomes dangerous for any other system of higher position in the biological hierarchy” [8]. Together with the “genome dictatorship” concept, this law implies that any critical state of an organism, when it no longer guarantees safety of its genome and, in case of convalescence, can generate offspring with a changed genome, should be a signal for the organism’s self-elimination, i.e. acute phenoptosis [88]. As Monsieur Bahays, a good-for-nothing physician in Moliere’s comedy “Love as a Healer” used to say: “It is better to die according to the rules than to recover against the rules”.
It seems quite possible that the mechanism of rapid phenoptosis, like that of slow phenoptosis (aging), is mediated by intramitochondrial ROS at early stages of the process. If this hypothesis is true, then the positive effect of SkQ not only on aging, but also on a variety of acute pathologies in both young and old organisms, can be explained in terms of quenching of these ROS.
Thus, it seems possible that SkQ can serve as a tool in the “rise of the machines”, an attempt of Homo sapiens to overcome the genome tyranny and to cancel those genome-dictated programs that are useful for genome evolution but unfavorable for an individual. Their cancellation would symbolize human conversion into Homo sapiens liberatus (liberatus means liberated in Latin), which might become the highest achievement of medicine of the XXI century.
REFERENCES
1.Skulachev, V. P., and Ozrina, R. D. (2011)
Biochemistry (Moscow), 76, 1-2.
2.Skulachev, V. P. (1997) Biochemistry
(Moscow), 62, 1191-1195.
3.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
4.Longo, V. D., Mitteldorf, J., and Skulachev, V. P.
(2005) Nat. Rev. Genet., 6, 866-872.
5.Libertini, G. (2012) Biochemistry (Moscow),
77, 707-715.
6.Koonin, E. V., and Aravind, L. (2002) Cell Death
Differ., 9, 394-404.
7.Lewis, K. (2000) Microbiol. Mol. Biol. Rev.,
64, 503-514.
8.Skulachev, V. P. (2003) in Model Systems in
Aging (Nystrom, T., and Osiewacz, H. D., eds.) Springer-Verlag,
Berlin-Heidelberg, pp. 191-238.
9.Severin, F. F., and Hyman, A. A. (2002) Curr.
Biol., 12, R233-R235.
10.Pozniakovsky, A. I., Knorre, D. A., Markova, O.
V., Hyman, A. A., Skulachev, V. P., and Severin, F. F. (2005) J.
Cell Biol., 168, 257-269.
11.Skulachev, V. P. (2002) Ann. N.Y. Acad.
Sci., 959, 214-237.
12.Severin, F. F., and Skulachev, V. P. (2009)
Advances Gerontol. (Russ.), 22, 37-48.
13.Skulachev, V. P. (2009) Rus. Chem. J.
(Russ.), LIII, 125-140.
14.Sukhanova, E. I., Rogov, A. G., Severin, F. F.,
and Zvyagilskaya, R. A. (2012) Biochemistry (Moscow), 77,
761-775.
15.Nooden, L. D., Guiamet, J. J., and John, I.
(1997) Physiol. Plant., 101, 746-753.
16.Leopold, A. C., Niedergangkamien, E., and Janick,
J. (1959) Plant Physiol., 34, 570-573.
17.Lindoo, S. J., and Nooden, L. D. (1977) Plant
Physiol., 59, 1136-1140.
18.Nooden, L. D., and Murray, B. J. (1982) Plant
Physiol., 69, 754-756.
19.Skulachev, V. P. (2011) Aging (Albany,
N.Y.), 3, 1120-1123.
20.Kirkwood, T. B. L., and Melov, S. (2011) Curr.
Biol., 21, R701-R707.
21.Cutler, S. R., Rodriguez, P. L., Finkelstein, R.
R., and Abrams, S. R. (2010) Ann. Rev. Plant Biol., 61,
651-679.
22.Cho, D., Shin, D. J., Jeon, B. W., and Kwak, J.
M. (2009) J. Plant Biol., 52, 102-113.
23.Melzer, S., Lens, F., Gennen, J.,Vanneste, S.,
Rohde, A., and Beeckman, T. (2008) Nature Genet., 40,
1489-1492.
24.Weismann, A. (1889) Essays upon Heredity and
Kindred Biological Problems, Clarendon Press, Oxford.
25.Carlquist, S. J. (1974) Island Biology,
Columbia University Press, New York.
26.Groover, A. T. (2005) Trends Plant Sci.,
10, 210-214.
27.Kim, S. C., Crawford, D. J., FranciscoOrtega, J.,
and SantosGuerra, A. (1996) PNAS, 93, 7743-7748.
28.Bohle, U. R., Hilger, H. H., and Martin, W. F.
(1996) PNAS, 93, 11740-11745.
29.Libbert, A. (1976) Physiology of Plants
[Russian translation], Mir, Moscow.
30.Wodinsky, J. (1977) Science, 198,
948-951.
31.Nesis, K. N. (1997) in Russian Science: to
Persevere and Be Reborn (Byalko, A. V., ed.) [in Russian],
Fizmatizdat, Moscow, pp. 358-365.
32.Dawkins, R. (1976) The Selfish Gene,
Oxford University Press, New York.
33.Bradley, A. J., McDonald, I. R., and Lee, A. K.
(1980) General Comparat. Endocrinol., 40, 188-200.
34.Skulachev, V. P. (2005) Vestnik RAN
(Russ.), 75, 831-843.
35.Austad, S. N. (2004) Aging Cell, 3,
249-251.
36.Maldonado, T. A., Jones, R. E., and Norris, D. O.
(2000) Brain Res., 858, 237-251.
37.Maldonado, T. A., Jones, R. E., and Norris, D. O.
(2002) J. Neurobiol., 53, 11-20.
38.Maldonado, T. A., Jones, R. E., and Norris, D. O.
(2002) J. Neurobiol., 53, 21-35.
39.Kipling, D., Davis, T., Ostler, E. L., and
Faragher, R. G. A. (2004) Science, 305, 1426-1431.
40.Terzibasi, E., Valenzano, D. R., and Cellerino,
A. (2007) Exp. Gerontol., 42, 81-89.
41.Comfort, A. (1979) The Biology of
Senescence, Elsevier, New York.
42.Schopenhauer, A. (1993) The World as Will and
Representation [Russian translation], MoskovskiiKlub, Moscow.
43.Darwin, C. (1871) The Descent of Man, and
Selection in Relation to Sex, D. Appleton and Company, New
York.
44.Medawar, P. B. (1952) An Unsolved Problem of
Biology, Published for the College by H. K. Lewis, London.
44a.Bowles, J. T. (1998) Med. Hypotheses, 51, 179-221.
44b.Bowles, J. T. (2000) Med. Hypotheses, 54, 236-339.
45.Loison, A., Festa-Bianchet, M., Gaillard, J. M.,
Jorgenson, J. T., and Jullien, J. M. (1999) Ecology, 80,
2539-2554.
46.Bonduriansky, R., and Brassil, C. E. (2002)
Nature, 420, 377-377.
47.Gavrilova, N. S., Gavrilov, L. A., Severin, F.
F., and Skulachev, V. P. (2012) Biochemistry (Moscow),
77, 754-760.
48.Khokhlov, A. N. (2009) Ros. Khim. Zh.,
LIII, 111-117.
49.Vreeland, R. H., Rosenzweig, W. D., and Powers,
D. W. (2000) Nature, 407, 897-900.
50.George, J. C., Bada, J., Zeh, J., Scott, L.,
Brown, S. E., O’Hara, T., and Suydam, R. (1999) Canad. J.
Zool., 77, 571-580.
51.Buffenstein, R. (2005) J. Gerontol. A. Biol.
Sci. Med. Sci., 60, 1369-1377.
52.Dawkins, R. (1989) in Artificial Life
Proceedings Reading (Langton, C., ed.) Addison Wesley,
Massachusetts, pp. 201-220.
53.Kirschner, M., and Gerhart, J. (1998)
PNAS, 95, 8420-8427.
54.Skulachev, V. P., Bogachev, A. V., and
Kasparinsky, F. O. (2010) Membrane Bioenergetics [in Russian],
Moscow University Publishers, Moscow.
55.Mufazalov, I. A., Penkov, D. N., Chernyak, B. V.,
Pletyushkina, O. Yu., Vyssokih, M. Yu., Kirpichnikov, M. P., Dolgikh,
D. A., Kruglov, A. A., Kuprash, D. V., Skulachev, V. P., and
Nedospasov, S. A. (2009) Mol. Biol. (Moscow), 43,
648-656.
56.Sharonov, G. V., Feofanov, A. V., Bocharova, O.
V., Astapova, M. V., Dedukhova, V. I., Chernyak, B. V., Dolgikh, D. A.,
Arseniev, A. S., Skulachev, V. P., and Kirpichnikov, M. P. (2005)
Apoptosis, 10, 797-808.
57.Mott, J. L., Zhang, D., Freeman, J. C.,
Mikolajczak, P., Chang, S. W., and Zassenhaus, H. P. (2004) Biochem.
Biophys. Res. Commun., 319, 1210-1215.
58.Trifunovic, A., Wredenberg, A., Falkenberg, M.,
Spelbrink, J. N., Rovio, A. T., Bruder, C. E., Bohlooly, Y. M., Gidlof,
S., Oldfors, A., Wibom, R., Tornell, J., Jacobs, H. T., and Larsson, N.
G. (2004) Nature, 429, 417-423.
59.Kujoth, G. C., Hiona, A., Pugh, T. D., Someya,
S., Panzer, K., Wohlgemuth, S. E., Hofer, T., Seo, A. Y., Sullivan, R.,
Jobling, W. A., Morrow, J. D., van Remmen, H., Sedivy, J. M., Yamasoba,
T., Tanokura, M., Weindruch, R., Leeuwenburgh, C., and Prolla, T. A.
(2005) Science, 309, 481-484.
60.Harman, D. (1956) J. Gerontol., 11,
298-300.
61.Emanuel, N. M. (1975) Izv. AN SSSR,
785-794.
62.Szilard, L. (1959) PNAS, 45,
30-45.
63.Skulachev, V. P., and Longo V. D. (2005) Ann.
N. Y. Acad. Sci., 1057, 145-164.
63a.Szczesny, B., Tann, A. W., and Mitra, S. (2010) Mech. Ageing
Develop., 131, 330-337.
64.Brunet-Rossinni, A. K., and Austad, S. N. (2004)
Biogerontology, 5, 211-222.
65.Ku, H. H., Brunk, U. T., and Sohal, R. S. (1993)
Free Radic. Biol. Med., 15, 621-627.
66.Barja, G. (1998) Ann. N. Y. Acad. Sci.,
854, 224-238.
67.Barja, G., and Herrero, A. (2000) FASEB
J., 14, 312-318.
68.Lambert, A. J., Boysen, H. M., Buckingham, J. A.,
Yang, T., Podlutsky, A., Austad, S. N., Kunz, T. H., Buffenstein, R.,
and Brand, M. D. (2007) Aging Cell, 6, 607-618.
69.Dilman, V. M. (1978) Mech. Ageing Dev.,
8, 153-173.
70.Dilman, V. M. (1982) Large Biological Clock
[in Russian], Znanie, Moscow.
71.Longo, V. D., and Finch, C. E. (2003)
Science, 299, 1342-1346.
72.Kim, E. B., Fang, X. D., Fushan, A. A., Huang, Z.
Y., Lobanov, A. V., Han, L. J., Marino, S. M., Sun, X. Q., Turanov, A.
A., Yang, P. C., Yim, S. H., Zhao, X., Kasaikina, M. V., Stoletzki, N.,
Peng, C. F., Polak, P., Xiong, Z. Q., Kiezun, A., Zhu, Y. B., Chen, Y.
X., Kryukov, G. V., Zhang, Q., Peshkin, L., Yang, L., Bronson, R. T.,
Buffenstein, R., Wang, B., Han, C. L., Li, Q. Y., Chen, L., Zhao, W.,
Sunyaev, S. R., Park, T. J., Zhang, G. J., Wang, J., and Gladyshev, V.
N. (2011) Nature, 479, 223-227.
73.Liberman, E. A., Topaly, V. P., Tsofina, L. M.,
Jasaitis, A. A., and Skulachev, V. P. (1969) Nature, 222,
1076-1078.
74.Green, D. E. (1974) Biochim. Biophys.
Acta, 346, 27-78.
75.Liberman, E. A., and Skulachev, V. P. (1970)
Biochim. Biophys. Acta, 216, 30-42.
76.Severin, S. Ye., Skulachev, V. P., and
Yaguzhinsky, L. S. (1970) Biokhimiya, 35, 1250-1257.
77.Burns, R. J., Smith, R. A., and Murphy, M. P.
(1995) Arch. Biochem. Biophys., 322, 60-68.
78.Smith, R. A., Porteous, C. M., Coulter, C. V.,
and Murphy, M. P. (1999) Eur. J. Biochem., 263,
709-716.
79.Kelso, G. F., Porteous, C. M., Coulter, C. V.,
Hughes, G., Porteous, W. K., Ledgerwood, E. C., Smith, R. A., and
Murphy, M. P. (2001) J. Biol. Chem., 276, 4588-4596.
80.Murphy, M. P., and Smith, R. A. (2007) Annu.
Rev. Pharmacol. Toxicol., 47, 629-656.
81.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L.
E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Y.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Y., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Y., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1273-1287.
82.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) Biochim. Biophys. Acta, 1787, 437-461.
83.James, A. M., Cocheme, H. M., Smith, R. A., and
Murphy, M. P. (2005) J. Biol. Chem., 280,
21295-21312.
84.O’Malley, Y., Fink, B. D., Ross, N. C.,
Prisinzano, T. E., and Sivitz, W. I. (2006) J. Biol. Chem.,
281, 39766-39775.
85.Doughan, A. K., and Dikalov, S. I. (2007)
Antioxid. Redox. Signal., 9, 1825-1836.
86.Kruk, J., Jemiola-Rzeminska, M., and Strzalka, K.
(1997) Chem. Phys. Lipids, 87, 73-80.
87.Roginsky, V., Barsukova, T., Loshadkin, D., and
Pliss, E. (2003) Chem. Phys. Lipids, 125, 49-58.
88.Skulachev, V. P. (2007) Biochemistry
(Moscow), 72, 1385-1396.
88a.Fink, B. D., Herlein, J. A., Yorek, M. A., Fenner, A. M., Kerns, R.
J., and Sivitz, W. I. (2012) J. Pharm. Exp. Therap., DOI:
10.1124/jpet.112.195586.
89.Skulachev, V. P., Antonenko, Y. N., Cherepanov,
D. A., Chernyak, B. V., Izyumov, D. S., Khailova, L. S., Klishin, S.
S., Korshunova, G. A., Lyamzaev, K. G., Pletjushkina, O. Y., Roginsky,
V. A., Rokitskaya, T. I., Severin, F. F., Severina, I. I., Simonyan, R.
A., Skulachev, M. V., Sumbatyan, N. V., Sukhanova, E. I., Tashlitsky,
V. N., Trendeleva, T. A., Vyssokikh, M. Y., and Zvyagilskaya, R. A.
(2010) Biochim. Biophys. Acta, 1797, 878-889.
90.Roginsky, V. A., Tashlitsky, V. N., and
Skulachev, V. P. (2009) Aging (Albany, NY), 1,
481-489.
91.Skulachev, M. V., Antonenko, Y. N., Anisimov, V.
N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov, M.
V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov, E.
Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F. F.,
Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M. Y.,
Zamyatnin, A. A., Zorov, D. B., and Skulachev, V. P. (2011) Curr.
Drug Targets, 12, 800-826.
92.Severin, F. F., Severina, I. I., Antonenko, Y.
N., Rokitskaya, T. I., Cherepanov, D. A., Mokhova, E. N., Vyssokikh, M.
Y., Pustovidko, A. V., Markova, O. V., Yaguzhinsky, L. S., Korshunova,
G. A., Sumbatyan, N. V., Skulachev, M. V., and Skulachev, V. P. (2010)
PNAS, 107, 663-668.
93.Anisimov, V. N., Egorov, M. V., Krasilshchikova,
M. S., Lyamzaev, K. G., Manskikh, V. N., Moshkin, M. P., Novikov, E.
A., Popovich, I. G., Rogovin, K. A., Shabalina, I. G., Shekarova, O.
N., Skulachev, M. V., Titova, T. V., Vygodin, V. A., Vyssokikh, M. Y.,
Yurova, M. N., Zabezhinsky, M. A., and Skulachev, V. P. (2011) Aging
(Albany NY), 3, 1110-1119.
94.Anisimov, V. N., Bakeeva, L. E., Egormin, P. A.,
Filenko, O. F., Isakova, E. F., Manskikh, V. N., Mikhelson, V. M.,
Panteleeva, A. A., Pasyukova, E. G., Pilipenko, D. I., Piskunova, T.
S., Popovich, I. G., Roshchina, N. V., Rybina, O. Y., Saprunova, V. B.,
Samoylova, T. A., Semenchenko, A. V., Skulachev, M. V., Spivak, I. M.,
Tsybul’ko, E. A., Tyndyk, M. L., Vyssokikh, M. Y., Yurova, M. N.,
Zabezhinsky, M. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1329-1342.
95.Stefanova, N. A., Fursova, A., and Kolosova, N.
G. (2010) J. Alzheimer’s Dis., 21, 479-491.
96.Skulachev, V. P. (2012) J. Alzheimer’s
Dis., 28, 283-289.
97.Neroev, V. V., Archipova, M. M., Bakeeva, L. E.,
Fursova, A., Grigorian, E. N., Grishanova, A. Y., Iomdina, E. N.,
Ivashchenko, Zh. N., Katargina, L. A., Khoroshilova-Maslova, I. P.,
Kilina, O. V., Kolosova, N. G., Kopenkin, E. P., Korshunov, S. S.,
Kovaleva, N. A., Novikova, Y. P., Philippov, P. P., Pilipenko, D. I.,
Robustova, O. V., Saprunova, V. B., Senin, I. I., Skulachev, M. V.,
Sotnikova, L. F., Stefanova, N. A., Tikhomirova, N. K., Tsapenko, I.
V., Shchipanova, A. I., Zinovkin, R. A., and Skulachev, V. P. (2008)
Biochemistry (Moscow), 73, 1317-1328.
98.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. B., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashook, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasilyeva, A. K., Victorov, I. V., Zorov, D. B., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1288-1299.
99.Plotnikov, E. Y., Silachev, D. N., Chupyrkina, A.
A., Danshina, M. I., Jankauskas, S. S., Morosanova, M. A., Stelmashook,
E. V., Vasileva, A. K., Goryacheva, E. S., Pirogov, Y. A., Isaev, N.
K., and Zorov, D. B. (2010) Biochemistry (Moscow), 75,
145-150.
100.Plotnikov, E. Y., Chupyrkina, A. A.,
Jankauskas, S. S., Pevzner, I. B., Silachev, D. N., Skulachev, V. P.,
and Zorov, D. B. (2011) Biochim. Biophys. Acta, 1812,
77-86.
101.Zorov, D. B., Plotnikov, E. Y., Yankauskas, S.
S., Isaev, N. K., Silachev, D. N., Zorova, L. D., Pevzner, I. B.,
Pul’kova, N. V., Zorov, S. D., and Morosanova, M. A. (2012)
Biochemistry (Moscow), 77, 742-753.
102.Agapova, L. S., Chernyak, B. V., Domnina, L.
V., Dugina, V. B., Efimenko, A. Y., Fetisova, E. K., Ivanova, O. Y.,
Kalinina, N. I., Khromova, N. V., Kopnin, B. P., Kopnin, P. B.,
Korotetskaya, M. V., Lichinitser, M. R., Lukashev, A. L., Pletjushkina,
O. Y., Popova, E. N., Skulachev, M. V., Shagieva, G. S., Stepanova, E.
V., Titova, E. V., Tkachuk, V. A., Vasiliev, J. M., and Skulachev, V.
P. (2008) Biochemistry (Moscow), 73, 1300-1316.
103.Sommer, S. S. (1994) Hum. Mutat.,
3, 166-169.
104.Manskikh, V. N. (2004) Essays on
Evolutionary Oncology [in Russian], SibGMU, Tomsk.
105.Lihtenstein, A. V. (2005) Biochemistry
(Moscow), 70, 1055-1064.
106.Manskikh, V. N. (2009) Ros. Khim. Zh.,
LIII, 57-63.
107.MacCay, C. M., and Crowell, M. F. (1934)
Sci. Mon., 39, 405-414.
108.MacCay, C. M., Crowell, M. F., and Maynard, L.
A. (1935) J. Nutr., 10, 63-79.
109.MacCay, C. M., Maynard, L. A., and Barnes, L.
L. (1943) Arch. Biochem., 2, 469-479.
110.Robertson, T. B., Marston, R., and Walters, J.
W. (1934) Aust. J. Exp. Biol. Med. Sci., 12, 33.
111.Will, L. C., and MacCay, C. M. (1943) Arch.
Biochem., 2, 481-484.
112.Mair, W., Goymer, P., Pletcher, S. D., and
Partridge, L. (2003) Science, 301, 1731-1733.
113.Libert, S., and Pletcher, S. D. (2007)
Cell, 131, 1231-1234.
114.Libert, S., Zwiener, J., Chu, X., Vanvoorhies,
W., Roman, G., and Pletcher, S. D. (2007) Science, 315,
1133-1137.
115.Carr, C. J., King, J. T., and Visscher, B.
(1949) Proc. Fedn. Am. Soc. Exp. Biol., 8, 22.
116.Stuchlikova, E., Juricova-Horakova, M., and
Deyl, Z. (1975) Exp. Gerontol., 10, 141-144.
117.Richie, J. P., Jr., Leutzinger, Y.,
Parthasarathy, S., Malloy, V., Orentreich, N., and Zimmerman, J. A.
(1994) FASEB J., 8, 1302-1307.
118.Miller, R. A., Buehner, G., Chang, Y., Harper,
J. M., Sigler, R., and Smith-Wheelock, M. (2005) Aging Cell,
4, 119-125.
119.Sanz, A., Caro, P., Ayala, V., Portero-Otin,
M., Pamplona, R., and Barja, G. (2006) FASEB J., 20,
1064-1073.
120.Caro, P., Gomez, J., Sanchez, I., Garcia, R.,
Lopez-Torres, M., Naudi, A., Portero-Otin, M., Pamplona, R., and Barja,
G. (2009) Biogerontology, 10, 579-592.
121.Edman, U., Garcia, A. M., Busuttil, R. A.,
Sorensen, D., Lundell, M., Kapahi, P., and Vijg, J. (2009) Aging
Cell, 8, 331-338.
122.Skulachev, V. P. (2011) Aging (Albany,
NY), 3, 1045-1050.
123.Colman, R. J., Anderson, R. M., Johnson, S. C.,
Kastman, E. K., Kosmatka, K. J., Beasley, T. M., Allison, D. B.,
Cruzen, C., Simmons, H. A., Kemnitz, J. W., and Weindruch, R. (2009)
Science, 325, 201-204.
124.Sun, D., Muthukumar, A. R., Lawrence, R. A.,
and Fernandes, G. (2001) Clin. Diagn. Lab. Immunol., 8,
1003-1011.
125.Gardner, E. M. (2005) J. Gerontol. A. Biol.
Sci. Med. Sci., 60, 688-694.
126.Obukhova, L. A., Skulachev, V. P., and
Kolosova, N. G. (2009) Aging (Albany NY), 1, 389-401.
127.Demianenko, I. A., Vasilieva, T. V., Domnina,
L. V., Dugina, V. B., Yegorov, M. V., Ivanova, O. Yu., Ilinskaya, O.
P., Pletiushkina, O. Yu., Popova, E. N., Saharov, I. Yu., Fedorov, A.
V., and Chernyak, B. V. (2010) Biochemistry (Moscow), 75,
274-280.
128.Hopkin, K. (2003) Sci. Aging Knowledge
Environ., 2003, NS4.
129.Hamilton, W. D. (1964) J. Theor. Biol.,
7, 1-16.
130.Hamilton, W. D. (1964) J. Theor. Biol.,
7, 17-52.
131.Dawkins, R. (2010) Extended Phenotype. Long
Arm of the Gene [Russian translation], Astrel, Moscow.
132.Eng, P. M., Rimm, E. B., Fitzmaurice, G., and
Kawachi, I. (2002) Am. J. Epidemiol., 155, 700-709.
NOTE IN PROOF to paper by V. P. Skulachev, Biochemistry (Moscow),
2012, 77, 689-706.
In 2012, F. Lens and S. Melzer published a review of studies on Arabidopsis thaliana mutants. In this review, two essential facts were mentioned, namely that (i) the lifespan of soc 1 and ful mutants is already measured (it is 18 months) and (ii) these mutants still form some flowers and seeds, but much later (and in relatively smaller quantities) than the wild type (F. Lens and S. Melzer, “Stem anatomy supports Arabidopsis thaliana as a model for insular woodiness”, New Phytologist, 2012, 193, 12-17).