Can Kin Selection Facilitate the Evolution of the Genetic Program of Senescence?
A. V. Markov
A. A. Borissiak Paleontological Institute, Russian Academy of Sciences, Profsoyuznaya ul. 123, 117997 Moscow, Russia; E-mail: markov_a@inbox.ru
Received February 13, 2012; Revision received February 22, 2012
The theory of adaptive senescence, or phenoptosis (“altruistic suicide” of the organism), implies that mutations enhancing mortality growth with age (“senescence genes”) can be favored by selection under some circumstances, although the nature of these circumstances and the frequency of their occurrence are not clear. Here I demonstrate by means of computer simulation that senescence genes can spread in the population’s gene pool via the mechanism of kin selection if two conditions are met. First, the population must have high viscosity (low intermixing), which provides positive correlation between spatial proximity of individuals and their relatedness, an important precondition for kin selection. Second, prior to acquisition of the senescence genes, there must be a sufficiently fast decline in the reproductive potential with age, while viability should decrease slower or remain constant. These conditions are probably met in some territorial and social species with severe competition for social rank and mating partners.
KEY WORDS: Hamilton’s rule, kin selection, phenoptosis, senescence, modelingDOI: 10.1134/S0006297912070061
The idea of senescence being a phenotypic expression of a special “genetic aging program” developed in the course of evolution as a beneficial adaptation [1] is rejected by the majority of gerontologists [2]. The lack of a thoroughly developed theory may be one of the reasons for this rejection. Attempts to provide a theory supporting this idea are hardly ever met with understanding of the scientific community, for they are based on the idea of “adaptation for the sake of species”, models of group or interpopulation selection [3], and this type of models are considered to work only within a very narrow range of conditions [4, 5]. That is why the issue of other possible theoretical rationale for the idea of adaptive aging and phenoptosis (adaptive “biochemical suicide of an organism”) [1, 6, 7] based on the generally accepted gene-centered evolutionary models seems to be rather urgent. In this work, the kin selection theory is presented as such a model potentially capable of providing a theoretical basis for the idea of “adaptive aging” [5, 8-10].
The idea of kin selection being able to influence the evolution of senescence is widely discussed in the scientific literature [11]. But we know of only three studies demonstrating the theoretical possibility of kin selection favoring “senescence genes” (mutations that accelerate age-dependent increase in death rate and have no other beneficial pleiotropic effects) [12-14]. According to this idea, the accelerated (due to senescence) death of an adult individual can theoretically increase its “inclusive fitness”, for the resources used by the dead individual can be used more effectively by its younger kin [11]. “Inheritance of resources” – a rather widespread situation when the resources released by the dead individual (a nest, territory, or social rank) are inherited by its offspring – should support this phenomenon [15].
In spite of the fact that the adaptive senescence hypothesis has not been generally accepted, theoretical models of evolution of life cycles (strategies) usually include the possibility of adaptive changes in life expectancy, including its reduction [16]. But in the case of these models, explanation of the causes of evolutionary development of adaptations is different from the one within the framework of the theory of adaptive senescence and phenoptosis. Models of life cycle evolution are usually based on the notion of trade-off between the amount of resources that an organism may use for reproduction and self-maintenance, in particular for reparation systems counteracting the deterioration of the body [17]. Increased costs of reproduction (reproductive effort) are likely to facilitate individual deterioration, this being why age-dependent decrease in viability and increase in death rate (senescence) may be seen as a byproduct of evolutional optimization of life cycle and reproductive effort growth [18-21]. For instance, it is not difficult to prove that in many cases (i.e. within a wide range of the model input parameters) selection will support a fertility-increasing mutation causing as a side effect the decrease of lifespan [22].
In addition, evolutionary theory of life cycles traditionally relies on the ideas developed by W. Hamilton in his fundamental paper on evolution of aging [23]. In particular, according to Hamilton, the later phenotypic effect of a harmful (viability-reducing) mutation is expressed in the course of ontogenesis, the weaker is the impact of purifying selection on such a mutation. Even non-aging individuals die for many different reasons (infections, predators, accidental traumas, natural disasters, etc.). Absence of aging does not mean absence of mortality – it is rather the absence of mortality increase with age. The number of individuals of a certain age will be decreasing with the increase in age even in case of non-aging individuals. The probability of reaching the age A in the case of non-aging organisms should decrease exponentially with the growth of A. The lower is the probability of reaching a certain age, the weaker is the impact of purifying selection on the mutations that start to be expressed at this age. Hence, accumulation of harmful mutations that decrease viability at the age rarely reached by individuals in the wild is unavoidable, and here lies the explanation of senescence being so universal. B. Charlesworth provides a review of these ideas that are the basis of the classic “evolutionary senescence theory” [24].
If we consider senescence to be a byproduct of evolutionary trade-off, then the idea of a special genetic senescence program seems to be superfluous. Natural deterioration caused by the fact that resources used for reparation are rather limited seems to be a good enough explanation of the senescence phenomenon [22]. It is fertility or reproductive effort growth that appears to be true adaptation in this case, while accelerated body deterioration is but a side effect of the phenomenon, the “price” that needs to be paid for the increase in reproduction efficiency. The concept of V. P. Skulachev, on the contrary, suggests aging per se to be of adaptive character. In other words, selection is suggested to favor the spread of alleles supporting the viability decrease with age (senescence) due to the very fact that they support aging, and not because of their positive impact on fertility increase coupled to aging as an unwelcome but unavoidable side-effect.
In this work we demonstrate by means of computer simulation that kin selection can support (at least theoretically) evolutionary development of senescence under certain conditions, i.e. support mutations accelerating viability decrease with age. It seems to be possible even in case of senescence being the only phenotypic expression of these mutations, and not a side effect of reproductive effort or fertility growth. Thus, a model of evolutionary development of senescence is suggested, a model which is based neither on group selection nor a teleological idea of “adaptations for the sake of species”, nor the concept of a compromise resource allocation between the bodily functions of reproduction and maintenance of viability, nor the idea of purifying selection weakening with age. The results indicate that at least theoretically a genetic senescence program can be formed under certain conditions. It is the reduction of life span that can become the reason for life-reducing mutations to be selected, even when they possess no other phenotypic expressions and provide no compensation for accelerated death. The results generally coincide with the conclusions of Travis [12], who was analyzing a similar type of model, which was nevertheless different in some essential details.
DESCRIPTION OF THE MODEL
As aging is a universal phenomenon, generalized and simple models should be used for the analysis of the basic principles of senescence evolution. These models should have minimal attachment to the realities of concrete species, life strategies, and habitat. Our model operates with two universal variables characterizing life strategy: fertility and mortality, each of them being dependent (or independent) on the age of an individual. Individual migratory activity, which determines the “population viscosity” or the degree of population mixing (which influences the relationship between the spatial proximity of individuals and their genetic relatedness), is an additional parameter. A population should be “viscous” enough for kin selection to operate in it [8, 25].
The life of a population of asexual organisms of no more than 900 individuals is simulated. A matrix of 30 × 30 with each cell being capable of supporting life of an adult individual is studied. Every adult individual at every given time (i.e. at every step of the model, which can be roughly equated to one year), is characterized by the following parameters: a –individual age; f – fertility (the number of offspring produced by an individual within a year); d – probability of death during the year; 1 – d value reflects the vitality (resistance to all factors that can lead to death); s – the value that determines the growth rate of d with age (due to “natural deterioration” or as a result of operation of “senescence genes”; the s value is inherited and does not change during individual life span; with s = 0, aging is absent, i.e. the probability of death does not increase with age); coordinates (x, y) of the cell occupied by an individual.
The following parameters common to all individuals are determined before starting the program: f0 – basic or maximum fertility, characteristic of all individuals in the first reproductive season (with a = 1); d0 – basic or minimum mortality characteristic for all individuals at a = 1; u – the value that determines the decline rate of fertility (f) with age. Fertility is expected to decline (or not decline when u = 0) due to “natural deterioration”; v – “migration cost” of juvenile individuals, i.e. probability of death of an individual juvenile in the process of moving to a neighboring cell of the matrix (“viscosity” of the population depends on v).
At the beginning of the program, the matrix is inhabited by an initial population of 900 young (a = 1) individuals. Each individual of the initial population is assigned an arbitrary value of s. We assigned half of the population low, and the other half high value of s (s1 and s2, respectively). The first group was assumed to have no “senescence genes”, and the low s value corresponded to vitality decline with age due to “natural deterioration”, while the second group was assumed to be the carriers of a “senescence gene”, i.e. an allele accelerating the decrease in viability with age. On the basis of observing the subsequent changes of the size of these two groups, one can conclude whether the carriers of “senescence gene” possess a selective advantage under these conditions (for a given set of input parameters) or vice versa, it is the individuals with no “senescence gene” that will be favored by selection. Several other parameters (besides s values), f0, d0, u and v, are set by the experimenter before the program starts. The following processes take place at every step of the model (during every year).
Reproduction. Every adult individual produces offspring (juvenile individuals) according to their current fertility. The number of offspring produced by an individual is determined by the formula f0 – [au] + 1 (square brackets denote the integer part of the bracketed value). Juvenile individuals initially belong to the same cell as their parental adult individual. Thus, all the individuals produce the maximum number of offspring f0 during their first reproductive season (at a = 1), and during the subsequent reproductive seasons the number of produced offspring either remains the same (if u = 0) or decreases gradually from season to season (if u > 0), and the rate of fertility decline is determined by u.
Death of adult individuals. The probability of an individual death is equal to d0 + 0.1as – 0.1. Thus, if there is no aging (s = 0), then mortality does not depend on age, it is always equal to d0, and the size of the cohort decreases exponentially. When s > 0 individuals age, i.e. the probability of their death increases with age. The rate of aging is determined by the value s, which is inherited (it is the same for offspring and their parent). This parameter is assumed to depend on both natural deterioration and the presence of senescence genes. The d0 value corresponds to the baseline of natural decrease (changes in this parameter can imitate, e.g. changes in the intensity of predator pressure). Mortality of all the individuals is minimal, i.e. equal to d0 during the first reproductive season; it is only starting from the second reproductive season that it may start growing if s > 0. This is consistent with the accepted understanding of the beginning of reproduction being that critical point after which (but not before) aging may start [19].
Offspring search for a place to settle. A juvenile individual will survive only in case it finds a free cell in the matrix of “living space”. The process is simulated as follows. One individual is randomly chosen out of all the juveniles produced during this season. If the cell of this juvenile is not occupied by an adult individual, then the juvenile occupies this cell and is considered to be an adult (a = 1) starting from this point. Then this cell is no longer available for other juveniles. In case of the cell being occupied, the juvenile migrates to one of the neighboring cells. The direction of this move is selected randomly keeping in mind that individuals cannot leave the matrix. In the course of migration, the juvenile faces risk that may lead to death with probability v. If it managed to survive and the cell where it moved happened to be free, this individual settles there. If this cell is also occupied, the individual migrates again while facing the risk v. This process continues until the individual dies or finds a free cell. After that, a new juvenile individual is randomly chosen and the process starts all over again. Juvenile settling continues until all the matrix cells are occupied (and then all the remaining homeless juveniles die) or until there are no more juvenile individuals. Thus, the value v determines the distance at which juveniles can migrate from their birth place. The level of population mixing as well as the level of correlation between the spatial proximity of individuals and their relatedness (an important condition of kin selection efficiency) depend on this value. Thus, in the case of this model all the juveniles initially have equal chances of survival; there are no more or less viable individuals among them. In fact, survival of each juvenile depends only on the availability of free cells in the vicinity of their birthplace. In the case of high values of v, the death of an individual parent or its closest neighbors greatly increases the chances of survival of the juvenile.
When the three above-listed processes are completed, there starts the next “year”: the age of all the adult individuals (a) is increased by one, and the cycle is repeated.
RESULTS AND DISCUSSION
Genocentric character of the model. It is necessary to clarify the biological meaning of the model and the processes occurring in it before turning to simulation results. The model is inherently genocentric. Group selection and “evolution for the sake of species” are not possible within its framework. Selection takes place only at individual level, a fact coinciding with the selection at gene level in case of clonal reproduction. Individuals differ only in terms of possessing (or not possessing) a senescence gene, i.e. an allele or a set of alleles that are phenotypically expressed only in acceleration of aging. Aging in this model is understood as decrease in viability (increase in mortality) with age. We arbitrarily assume that individuals with assigned low initial s values possess no senescence gene, whereas those with a high s value have this gene. If the frequency of the senescence gene in the model population grows with time (which also means the growth of the proportion of individuals – carriers of this gene), it means that for a given set of conditions (input parameters) the senescence gene has a selective benefit over a competing “null allele” that has no impact on the rate of aging. In other words, in such a situation accelerated aging turns out to be a beneficial adaptation. However, it is not the individual, which due to the presence of the senescence gene dies earlier and leaves fewer offspring (than it would if it had not had this gene), that receives this benefit. This adaptation increases efficiency of distribution of the gene that determines its appearance (senescence gene) in the gene pool of population, i.e. it is beneficial not for an individual, but for the gene [4, 5, 26]. The only mechanism by means of which a senescence gene can enhance the efficiency of spreading its copies in the population gene pool is “altruistic self-elimination” (phenoptosis) of this gene carrier due to accelerated aging. It is due to the latter phenomenon that an old individual with decreased fertility frees its living space earlier than it would be done by an individual with no senescence gene. At a sufficiently high probability of occupation of the freed space by another senescence gene carrier (and not unrelated individual with no senescence gene), such “altruism” may increase the efficiency of the spread of the senescence gene in the gene pool. Thus, this is a typical case of the development of an “altruistic” life strategy under the influence of kin selection [5, 8-10].
The model and Hamilton’s rule. The central idea of the kin selection theory is reflected in “Hamilton’s rule”, which can be formulated as follows: an allele promoting altruistic behavior will be favored by selection and will be spread in the population gene pool if RB > C, where R (relatedness) corresponds to the degree of relatedness between the “altruist” and the addressee of an altruistic act, B (benefit) represents reproductive benefit received by the addressee as a result of an altruistic act, and C (cost) is a reproductive damage experienced by the “altruist”.
Hamilton’s rule is not an empirical generalization – it is a strict logical consequence of the definitions of the variables R, B, and C (just as the theorems of Euclidean geometry are derived from its own axioms). It has to be clarified that the relatedness in the above definition of R value is important not per se, but only as an indicator of probability of the recipient having the same altruism allele as the contributor. It is this probability, rather than relatedness as such, that is crucial to the efficiency of kin selection [5].
The model description implies that Hamilton’s rule has to be strictly observed in it. This fact allows predictions to be made based on Hamilton’s rule, and then to test them using simulation. Phenoptosis (accelerated death of an adult individual) that frees the living space for a juvenile individual is an “altruistic act” in this case. We can adjust the C value in the model by changing the s2 parameter for individuals possessing the “senescence gene”: the higher is s2, the sooner the individual dies and the greater is reproductive damage experienced by this individual. The u parameter also effects the C value: the faster is fertility decline with age, the less is the reproductive damage experienced by the “altruist” who has committed the act of phenoptosis. We can also adjust the R value by changing the v parameter: increase in v causes increase in the probability of the living space vacated by the contributor being inhabited by another senescence gene carrier (e.g. the contributor’s offspring), and not by the carrier of the competing allele of senescence absence. It would seem that the B value could be considered constant in the model: some juvenile individual always gets a free cell, i.e. in fact the “right to live”. But the truth is that for a rigorous calculation of the values B, C, and R, one also needs to take into consideration characteristics of the population at the given time, e.g. if there are many free cells around the concrete adult individual, then its “altruistic self-elimination” would increase the probability of its offspring survival less radically (B will be less) than in case of all the adjacent cells being occupied.
Simulation results in the absence of “natural deterioration”. Let us first consider the simplest (and hardly possible in reality) situation with no deterioration either in the field of reproduction (u = 0) or in the field of maintenance of viability (in the case of individuals with no senescence gene s1 = 0). In such a case, individuals with no senescence gene do not age (mortality does not increase with age and is always equal to d0). Fertility also does not decrease – it remains equal to f0. Hence, the expected number of offspring that will be produced by a given individual within the remaining lifetime, and the expected number of years still in front of this individual, always remain constant. This means that the C value will be constant for any age. By committing the act of “altruistic self-elimination” at the age of 100 years, an individual will experience exactly the same reproductive damage as when the same act would be committed at the age of 2 years. It seems to be paradoxical, but it is an inevitable consequence of our not less paradoxical assumption of complete absence of deterioration.
The B value, which can be estimated as the expected number of offspring that will be produced by the juvenile individual that received the right to live due to the phenoptosis of the “altruist”, also will not depend on the age of the contributor. This expected number of offspring will be exactly the same for any adult individual of any age. Thus, in this case B = C. As R cannot be higher than 1, we conclude that the inequality RB > C cannot be fulfilled in the complete absence of deterioration. RB will be practically always smaller than C. The maximum we can reach when increasing R is reaching the equality RB = C with R = 1 (in the model R = 1 only if the juvenile individual has absolutely no chance to move to an adjacent cell, i.e. at v = 1, which is obviously unrealistic).
Thus, it can be predicted on the basis of Hamilton’s rule that in case of absence of deterioration the allele of absence of senescence will always have selective benefit over the senescence allele, and it is only at R = 1 that both of the competing alleles will have equal reproduction efficiency.
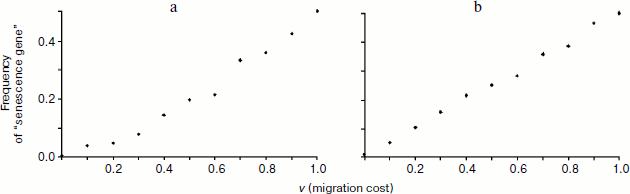
The results of simulation support this prediction. The graph in Fig. 1a shows the frequency of senescence gene (proportion of individuals aging with the rate of s2 = 1) after 20 years of life in the population, which initially contained equal numbers of non-aging (s1 = 0) and aging individuals. After 20 years the frequency of the senescence gene is always lower than the initial one (0.5) with the exception of the situation of complete absence of migration (v = 1). Thus, according to theoretical expectations, the allele of senescence absence has selective benefit, while the senescence allele is eliminated from the gene pool. Growth in the frequency of the senescence gene with increasing “viscosity” of the population (and, hence, increasing R value in the Hamilton formula) confirms that the higher is R, the less is the benefit of non-aging individuals over aging ones, or, in other words, the weaker is the impact of purifying selection on the harmful allele which accelerates aging.
Will the situation change if “natural deterioration” is spread over the function of self-preservation, but will not influence reproduction (u = 0, s1 > 0)? It may seem that in this case the chances of the senescence allele being favored by kin selection should be higher than in the previous case, for individual life expectancy becomes lower with age, and the “cost” of phenoptosis (C) also decreases. However, this argument ignores the fact that if probability of death increases with age because of deterioration, phenoptosis of such an individual will not only cause a smaller reduction in life expectancy (and hence the C value will be lower), but will also bring less benefit to the related individuals because its freed cell will be at their disposal for a reduced number of years. Consequently, when compared to the previous situation, both C and B values will be equally decreased. Since fertility does not depend on age, C and B values are determined only by the number of years “given away” by the dead individual and received by its young congener, and these numbers are equal. Hence, similarly to the previously discussed situation, one may expect selection to favor individuals possessing no senescence gene in this case as well. Simulation results support this conclusion (Fig. 1b).Fig. 1. Dependence of the frequency of “senescence gene” on the intensity of juvenile migration after 20 years of development of the model population (f0 = 10, d0 = 0.1, u = 0, s2 = 1). a) Complete lack of natural deterioration (s1 = 0); b) deterioration applies only to the sphere of self-preservation and does not affect fertility (s1 = 0.2).
Simulation results in case of fertility decreasing with age. If natural deterioration extends to the area of reproduction (u > 0), then fertility should decline with age. In this case the decrease in C with age is not necessarily accompanied by a corresponding decrease in B. An old individual with a low reproductive potential (low expected number of offspring that it is still capable of producing) does not sacrifice a lot due to premature death. The higher is the u value, the more rapid will be the decrease in C value with age. However, a young individual, who has won the vacant cell, gains more because its reproductive potential is still high. It is important to understand that the mere fertility decrease with age does not lead to a decrease in B. If B does decrease, then it would happen only because for the above-described reasons (if s1 > 0). Thus, if u > 0, we can predict on the basis of Hamilton’s rule that the advancement of the contributor’s age will cause the C value to decrease more rapidly than B (if s1 > 0), or only C and not B will decrease (if s1 = 0). In both cases, sooner or later the contributor can reach such an age when Hamilton’s inequality will be fulfilled. It seems obvious that the higher is R value, the sooner it will happen.
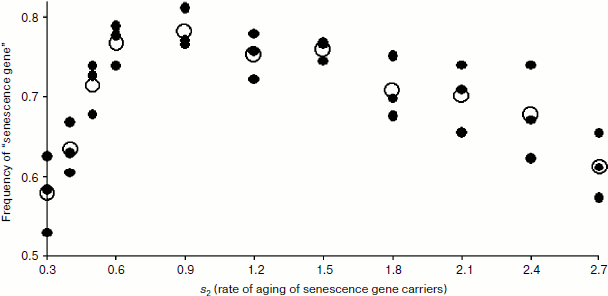
Model experiments have confirmed that for u > 0 it is indeed the carriers of the senescence allele that may have a selective benefit if the values of v and u are high enough (Fig. 2). The fact that the carriers of the senescence allele do possess a selective benefit can be derived from the fact that the frequency of the senescence allele after 50 years of life of the population is above 0.5 (see Fig. 2). This figure also shows that the selective benefit of the senescence allele depends on its “power”, that is, the rate of aging provided by this allele. For a set of input parameters, as reflected in Fig. 2, the optimal aging rate is close to s2 = 0.9. The senescence gene providing such an aging rate gives its carriers the greatest selective benefit over individuals lacking the senescence gene.
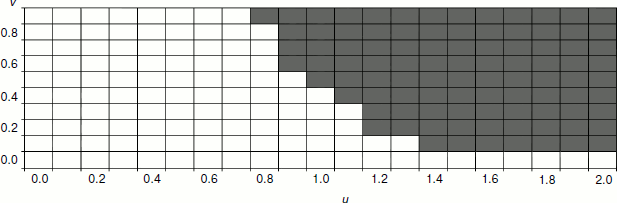
The dependence of the selective benefit of senescence allele carriers on the combination of parameters v and u is presented in Fig. 3. This figure shows that for kin selection to favor phenoptosis, the u value should be sufficiently high, i.e. there should be substantial “natural deterioration” of reproductive function.Fig. 2. Dependence of the frequency of a senescence gene on the power of this gene (s2) after 50 years of development of the model population (f0 = 10, d0 = 0.1, u = 1, s1 = 0.2, v = 0.8). The graph shows the nonlinear dependence of selective benefit of aging individuals on the aging rate with maximum benefit at values s2 0.6-1.2. Black circles indicate the results of three model runs at a given s2, and white circles show average values.
Interestingly enough, in the border area of parameters v and u (where gray and white areas in Fig. 3 come into contact), usually the frequency of the senescence gene first decreases (for the first 30-50 years), and only after that it begins to rise. The reason is that to ensure the realization of the benefits of accelerated aging in this border region, a subpopulation of slowly aging individuals should achieve middle-age, which leads to a decrease in their total fertility.
In our model, the efficiency of the distribution of “senescence genes” due to kin selection depends also on the basic (age-independent) mortality rate (d0). Decrease in the basic mortality rate causes increase in the selective advantage of the senescence allele carriers, while its increase has the opposite effect. For instance, at f0 = 10, d0 = 0.1, u = 1, s1 = 0.2, v = 0.8, the optimal aging rate s2 is about 0.9, and the proportion of aging individuals after 50 years of development of the model population reaches 0.77-0.81 (see Fig. 2). If the basic mortality rate is increased to d0 = 0.3, the optimal aging rate is reduced to 0.6, and the proportion of aging individuals after 50 years at this optimal rate does not exceed 0.59. If the basic mortality is reduced to d0 = 0.05, the optimal aging rate is increased to 1.0, and the proportion of aging individuals after 50 years reaches 0.87-0.92. Thus, aging turns out to be a more beneficial strategy at a low basic mortality rate than at a high one. Apparently, this phenomenon is due to the fact that an increase in basic mortality causes a decrease in B value: adult individuals often die because of reasons not related to age; that is why phenoptosis as a rule is less beneficial for young individuals (when compared to the absence of phenoptosis); the population is effectively “thinned” even without aging. In addition, slowly aging individuals (because of high basic mortality) simply have no time to live to the age when their fertility is significantly reduced, this fact reducing the selective benefit of rapidly aging individuals.Fig. 3. Dependence of selective benefit of senescence allele carriers on the combination of parameters v and u. The area where rapidly aging individuals (s2 = 1) have a selective benefit over slowly aging individuals (s1 = 0.2) is shaded gray. White cells correspond to the area where slowly aging individuals have selective benefit over rapidly aging ones. Other parameters: f0 = 10, d0 = 0.1.
Differences from the Travis model. A similar model was studied earlier by Travis [12]. We were not aware of the publication by Travis in the course of our work on this model, so similarities originate from the parallel line of thinking rather than borrowing. These are the most important differences of our model.
1) In the Travis model, adult individuals produce offspring one at a time, and fertility is understood as the probability of giving birth to the offspring at some concrete moment, while in our model offspring is produced in “broods” once a year, and fertility refers to the size of the brood.
2) In the Travis model, a young individual “jumps” from the place of its birth in a random direction; each individual is entitled to only one jump. If it gets into an empty slot, it survives, and if the cell it occupied, it dies. The population viscosity is regulated by the range of jumps. In our model an individual successively moves from cell to cell, at the risk of dying at each step. If the individual first gets into an occupied cell, it still has the chance to find a free cell at the next step. Population viscosity is determined by the probability of death at each step.
3) In the Travis model “aging” is defined as the age when an individual dies; in our model aging is understood as the increase in probability of death with age. At the same state of the “senescence gene” in the Travis model, individuals die at a specific age, while in our model the exact age of death is not known beforehand.
Our results are basically consistent with those of Travis, this fact indicating the sustainability of these results to changes in key assumptions and principles of modeling; it also improves the reliability of conclusions. Given that the fundamentally different analytical model also confirms the conclusion about the possibility of kin selection favoring the genetic program of aging under the conditions of high viscosity of the population and rapid decrease in fertility with age [14], validity of this conclusion becomes even higher (see also [11]).
Our results confirm the conclusions made earlier by Travis [12] on the basis of analysis of a model similar to ours in many respects, but different in a number of significant details. Similarly to the model of Travis, our model shows that for kin selection to favor mutations accelerating the reduction of viability with age (“senescence genes”), the following two conditions should be met.
1) The intensity of the movement (intermixing) of individuals in the population should be sufficiently low for the living space freed by a deceased individual to be occupied rather by its relatives and not by any random individuals of this population. This condition is called “population viscosity”, and it was Hamilton who spoke about it as an important prerequisite for the functioning of kin selection and evolution of altruistic behavior [8]. Hamilton rightly pointed out that individuals often cannot or do not want to move long distances from their birth places in many natural populations. Every living being is born in a certain place, and any migration is usually connected with risk. Even in the case of long migrations being a mandatory part of a life cycle, organisms often return to breed to their birth place, for it is generally safer than trying to explore new and unfamiliar places for nesting or spawning. It is due to this fact that there develops a positive correlation between the spatial proximity of individuals and the degree of their genetic relatedness. The presence of such a correlation is an important condition necessary for the efficiency of kin selection in general and for the evolution of altruistic phenoptosis in particular.
2) There should be a significant discrepancy between the rates of “natural deterioration” of different functional systems of an organism. More specifically, fertility should decline with age more rapidly than viability (this must have been happening originally, before the appearance of a special “genetic aging program”). It does not matter what the reason of fertility decline is – reduction in the number or quality of gametes, reduced sexual attractiveness, decreased efficiency of care for the offspring, or the loss of leadership in the group. Specific mechanisms of viability reduction also do not seem to be important, whether it is the reduction of running speed and subsequent increase in probability of being caught by a predator, or weakened immunity, or reduced efficiency of procuring food or something else. There is only one condition to be met for this mechanism to work: “natural deterioration” should cause reproductive potential of an individual to decrease more rapidly than its viability over time. In this case, kin selection can favor mutations correcting this imbalance, i.e. accelerating viability decrease with age in such a way as to conform to the natural decline in fertility rates. This mechanism will work also in the case of initial viability not being the object of deterioration, i.e. not decreasing with age (in the absence of aging). However, it will not work in case of individual reproductive potential not undergoing deterioration. If in the course of evolution the species managed to develop adaptations that ensure continuity of reproductive potential for an unlimited time, this mechanism will not favor “senescence genes”. Quite the opposite, it is the alleles that prolong life that will be strongly favored. The same phenomenon will be observed in all the cases where natural deterioration reduces viability faster than fertility. Thus, “natural deterioration”, even when affecting only one of the functions of the organism, a reproductive one, should be initially present for the mechanism discussed in this article to ensure the spread of senescence alleles in a population.
Ronce and Promislow [14] using another (analytical) model confirmed the conclusion made by Travis and supported by our work, that kin selection can favor “senescence genes” if population viscosity is sufficiently high and fertility decreases with age. However, these authors doubt that consolidation of senescence genes under the pressure of kin selection often occurs in nature. They emphasize that for kin selection to consolidate a “senescence allele”, individuals first need to experience sufficiently fast natural deterioration of reproductive function. In other words, aging should already exist before kin selection can speed it up. This observation is consistent with our results: indeed, fertility reduction with age is a prerequisite for senescence gene consolidation by kin selection to take place. However, mortality increase with age does not seem to be such an essential condition. If we assume “aging” to be the process of mortality increase with age (rather than fertility decrease), then the initial presence of aging is not required for our mechanism to work.
Ronce and Promislow [14] also note that if fertility rapidly decreases with age due to deterioration, then selection will favor not only accelerated aging, but also redistribution of reproduction potential by ages. In other words, instead of acquiring senescence genes, individuals can simply make sure that their fertility does not decrease that rapidly with age, and then no senescence genes will be favored by kin selection. However, we can assume that the probability of appearance of mutations accelerating aging is usually higher than that of mutations increasing fertility at old age. This issue requires further special studies.
It was also shown that not only the rate of aging can evolve depending on population viscosity, but also the migration activity (which directly determines viscosity) can evolve under the influence of aging rate. On the one hand, analysis of the models proves high viscosity to support evolution of accelerated aging, while low viscosity favors slow aging. On the other hand, slow aging may contribute to the evolution of increased migration activity, which leads to viscosity decrease [13]. Similar to the previous case, we can assume that the emergence of mutations that accelerate aging is as a rule a much more likely event than the appearance of mutations decreasing migration risks. It is particularly evident for social animals, whose departure from the original group is usually accompanied by a significant increase in mortality.
For our model, growth of the basic mortality rate (which may correspond, for example, to increased predator pressure) makes evolution of a genetic program of aging more difficult. This contradicts the well-known pattern, according to which the increase in predator pressure in wild populations leads, as a rule, to evolution of a shortened life cycle (including early maturation, the shift towards r-strategy, high fertility at young age, small size) [27]. Kin selection appears to be irrelevant to this pattern. It may be better understood within the classical model of a trade-off redistribution of resources between the functions of reproduction and survival accompanied by a decreased impact of purifying selection on the mutations that are expressed late in life.
The increase in mortality with age (aging) seems to develop for many different reasons in different living organisms. Consolidation of senescence genes caused by kin selection appears to be but one of the possible mechanisms of evolution of aging. It is not yet clear how often this mechanism operates in nature [11].
How realistic is the situation when individual reproductive potential is reduced much faster than the ability for self-preservation because of deterioration? It seems that this phenomenon may occur in animals, the reproductive success of which depends primarily on the outcome of kin competition for the right to reproduction, for example, in species with complex algorithms for choosing a mating partner, in polygamous species with intense competition for the right to mate, in social animals, whose reproductive success is highly dependent on their hierarchy status. In such situations, an animal while still being quite healthy and viable may have no chances to reproduce because younger individuals will beat it in competition for mating partners. In this case even a minimal loss of health and strength may result in drastic loss of chances for reproduction. In such situations, we can expect viability to be reduced in proportion to age, and fertility in proportion to the square of age. It is in this type of situations that kin selection will be efficient in favoring senescence genes, especially in territorial animals with high probability of “inheriting the resources” [15].
The results suggest that in nature there may be rather chaotic, hardly predictable distributions of forms having a genetic program of aging and those without such a program. Moreover, phenotypic manifestations and signs of aging, life expectancy, and other features of the life cycle are likely not to be fundamentally different in these forms. For some species with previously existent discrepancy between the rates of deterioration of different functional systems (accelerated fertility decline in comparison with viability), mutations which accelerate aging will be recorded in the genotype. At the same time, other species with no such previously existent imbalance will not have such mutations, and they will age only because of deterioration, the rate of which can be controlled through the mechanisms described by traditional models (such as accumulation of late-manifested harmful mutations and trade-off distribution of resources between the functions of reproduction and self-preservation).
Thus, the proposed model, along with the model of Travis [12] and in accordance with the conclusions of other authors [11, 13, 14], shows that the possibility of evolution of a genetic aging program stems from the theory of kin selection. It seems quite important that this possibility can be justified within the framework of classical population genetic approach, without invoking group selection and “evolution for the sake of species”. This model does not contradict the classical theories of evolution of life strategies. Apparently, the causes of aging are manifold, and the phenoptosis theory should be considered as complimentary rather than alternative to other models, including the classic Hamilton evolution theory of aging and the models of evolution of life strategies discussed in the literature.
REFERENCES
1.Skulachev, V. P. (2009) J. Russ. Chem. Soc.,
53, 125-140.
2.Khokhlov, A. N. (2009) J. Russ. Chem. Soc.,
53, 111-116.
3.Mitteldorf, J. (2006) Evol. Ecol. Res.,
8, 561-574.
4.Williams, G. C. (1966) Adaptation and Natural
Selection, Princeton University Press, Princeton.
5.Dawkins, R. (1999) The Extended Phenotype: The
Long Reach of the Gene, Oxford University Press, Oxford.
6.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
7.Longo, V. D., Mitteldorf, J., and Skulachev, V. P.
(2005) Nat. Rev. Genet., 6, 866-872.
8.Hamilton, W. D. (1964) J. Theor. Biol.,
7, 1-52.
9.Michod, R. E. (1982) Ann. Rev. Ecol. Syst.,
13, 23-55.
10.Foster, K. R., Wenseleers, T., and Ratnieks, F.
L. W. (2006) Trends Ecol. Evol., 21, 57-60.
11.Bourke, A. F. G. (2007) Annu. Rev. Ecol. Evol.
Syst., 38, 103-128.
12.Travis, J. M. J. (2004) J. Gerontol.,
59A, 301-305.
13.Dytham, C., and Travis, J. M. J. (2006)
Oikos, 113, 530-538.
14.Ronce, O., and Promislow, D. (2010) Proc. R.
Soc. B., 277, 3659-3667.
15.Ragsdale, J. E. (1999) Evol. Ecol. Res.,
1, 859-874.
16.Budilova, E. V., and Terekhin, A. T. (2010) J.
Gen. Biol., 71, 275-286.
17.Barnes, A. I., and Partridge, L. (2003) Anim.
Behav., 66, 199-204.
18.Stearns, S. C. (1977) Ann. Rev. Ecol.
Syst., 8, 145-171.
19.Stearns, S. C. (1992) The Evolution of Life
Histories, Oxford University Press, Oxford.
20.Partridge, L., and Sibly, R. (1991) Phil.
Trans. R. Soc. Lond. B., 332, 3-13.
21.Roff, D. A. (1992) The Evolution of Life
Histories: Theory and Analysis, Chapman and Hall, N. Y.
22.Roff, D. A. (1984) Can. J. Fish. Aquat.
Sci., 41, 989-1000.
23.Hamilton, W. D. (1966) J. Theor. Biol.,
12, 12-45.
24.Charlesworth, B. (2000) Genetics,
156, 927-931.
25.Wilson, D. S., Pollock, G. B., and Dugatkin, L.
A. (1992) Evol. Ecol., 6, 331-341.
26.Dawkins, R. (1989) The Selfish Gene,
Oxford University Press, Oxford, N. Y.
27.Reznick, D. N., Shaw, F. H., Rodd, F. H., and
Shaw, R. G. (1997) Science, 275, 1934-1937.