REVIEW: Phenoptosis in Yeasts
E. I. Sukhanova1, A. G. Rogov1, F. F. Severin2, and R. A. Zvyagilskaya1*
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; fax: (495) 954-2732; E-mail: renata_z@inbi.ras.ru2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: severin@belozersky.msu.ru
* To whom correspondence should be addressed.
Received March 11, 2012
The current view on phenoptosis and apoptosis as genetic programs aimed at eliminating potentially dangerous organisms and cells, respectively, is given. Special emphasis is placed on apoptosis (phenoptosis) in yeasts: intracellular defects and a plethora of external stimuli inducing apoptosis in yeasts; distinctive morphological and biochemical hallmarks accompanying apoptosis in yeasts; pro- and antiapoptotic factors involved in yeast apoptosis signaling; consecutive stages of apoptosis from external stimulus to the cell death; a prominent role of mitochondria and other organelles in yeast apoptosis; possible pathways for release of apoptotic factors from the intermembrane mitochondrial space into the cytosol are described. Using some concrete examples, the obvious physiological importance and expediency of altruistic death of yeast cells is shown. Poorly known aspects of yeast apoptosis and prospects for yeast apoptosis study are defined.
KEY WORDS: yeasts, phenoptosis, apoptosis, mitochondria, endoplasmic reticulum, vacuoleDOI: 10.1134/S0006297912070097
Abbreviations: AIF, apoptosis inducing factor; AMID, AIF-homologous mitochondrion-associated inducer of death; CsA, cyclosporin A; ER, endoplasmic reticulum; HtrA, located in the intermembrane space high temperature required protease A; IAP, protein, inhibitor of apoptosis; mPTP (mitochondrial permeability transition pore), nonspecific permeability of the inner mitochondrial membrane; OST, oligosaccharyl transferase; Pi, inorganic phosphate; ROS, reactive oxygen species; SOD, superoxide dismutase; YMUC, yeast mitochondrial unspecific channel; Ysp1p and Ysp2p, mitochondrial yeast suicide proteins.
The hypothesis of programmed organism death as a mechanism developed by
natural selection for elimination of old, worn down individuals to make
life space and resources available for young generations was put
forward by August Weismann in the 1880s [1, 2]. This process was dubbed phenoptosis
by V. P. Skulachev [3, 4],
by analogy with apoptosis (from Greek words apo and
ptosis, meaning “falling off”) – an
active, regulated at genetic level mechanism of programmed cell death
aimed at precise eliminating of self-destructive, unused, damaged,
virus-infected, weakened, cell cycle-completed, and potentially
dangerous cells; mitoptosis – self-elimination of
mitochondria [5-8], and
organoptosis – the well-known degradation of some
organs emerging temporally in juvenile organisms and disappearing later
in development, probably, by virtue of apoptosis of cells composing
these organs.
According to Skulachev [3, 4], evolutionary events gave rise to a set of mechanisms designated as fast phenoptosis (sepsis, carcinogenesis, myocardial infarction, etc.) and slow phenoptosis (aging of the whole organism). The major role in slow phenoptosis belongs presumably to the mechanism of cell apoptosis often triggered by mitoptosis. The evolutionary mechanism for such a suicide program might be based on kin selection (when organisms die for the good of their offspring bearing useful new traits) or group selection (the death for the good of individuals unrelated by ties of kinship). Theoretically, aging can stabilize population size and prevent overpopulation; it can increase genetic diversity, shorten the effective generation cycle, and accelerate adaptation.
Since phenoptosis of multicellular organisms is based on apoptosis of cells composing the metazoan body, apoptosis in mammals deserves closer attention. According to the recommendation of the Nomenclature Committee on Cell Death [9], apoptosis is distinguished from other forms of cell death (autophagy, anoikis, entosis, mitotic catastrophe, necroptosis, NETosis, and others) by characteristic morphological and biochemical markers including retention of the intact cytoplasmic membrane (maintenance of plasma membrane integrity) up to late stages of the process. At earlier stages there are: disturbance of the cytoplasmic membrane symmetry (externalization of phosphatidylserine to the outer leaflet of the plasma membrane); shrinking of the cell volume; intensive vacuolation and vesiculation. At late stages there are: blebbing of the cytoplasmic membrane; in some cases release of cytochrome c from the intermembrane space of mitochondria into the cytosol and activation of cysteine proteases (caspases); generation of reactive oxygen species (ROS); breakage of nuclear DNA in internucleosomal sites; chromatin condensation on the periphery of the nucleus with further nuclear fragmentation (50-180 kb); and cell fragmentation leading to formation of apoptotic bodies.
A typical roughly simplified scenario of induction and regulation of apoptosis in mammalian cells is given in Fig. 1 in [9]. Several key points should be noted. Both external factors and intrinsic defects of intracellular processes can act as triggers for the onset of apoptosis in animal cells. A number of pro- and antiapoptotic factors have been found (the number of such factors is increasing every year), and their interactions are arranged into a complicated hierarchical structure. Caspase activation is not a strict requirement for many cases of apoptosis. It is obvious that mitochondria play a central role of in the choice between life and death of a cell. This particular role of mitochondria relies on the fact that mitochondria are not only the major intracellular generators of reactive oxygen species (ROS) (frequently potent triggers of apoptosis), but also the place (normally in the intermembrane space) of some proapoptotic factors. Rupture of the outer mitochondrial membrane is accompanied by the release of proapoptotic factors, which in turn induces and enhances a reaction cascade ultimately leading to cell death. So, AIF (apoptosis-inducing factor), a phylogenetically old, bifunctional FAD-containing protein with significant homology to plant ascorbate reductases and bacterial NADH oxidases [10], normally confined to the mitochondrial intermembrane space, after rupture of the outer mitochondrial membrane translocates to the cytosol and then relocates to the nucleus. Ectopic (extra-mitochondrial) AIF induces nuclear chromatin condensation as well as large scale (approximately 50 kb) DNA fragmentation [10, 11]. Similarly, endonuclease G, also normally confined to the mitochondrial intermembrane space, translocates to the cytosol and then relocates to the nucleus, mediating large-scale DNA fragmentation independently of caspases. The OMI protease, released into the cytosol from the intermembrane space, belongs to HtrA protease family; it activates apoptosis by means of its own protease activity [12] as it disturbs the normal dynamics of the cytoskeleton. These events are accompanied by inhibition of the respiratory chain, by decline of membrane potential, deenergization of mitochondria and cells, and excessive production of ROS. The simplified scenario of apoptosis does not consider the implication of caspases.
In case of caspase-dependent apoptosis, the released cytochrome c (the injury of mitochondrial outer membrane is obligatory but insufficient for cytochrome c release, since oxidation of cardiolipin existing in a complex with cytochrome c is also required) binds to the cytoplasmic adaptor protein Apaf-1, dATP or ATP, and the initiating procaspase-9 (cysteine protease) with the formation of the apoptosome, a high molecular weight heptamer activating effector procaspases 3 and 7 [13]. The initiated proteolytic cascade involves also other procaspases (2, 6, 8, and 10), which results in cell destruction. The Smac/DIABLO (second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI) protein released from the mitochondrial intermembrane space enhances this cascade by means of binding to the inhibitor of apoptosis (IAP) proteins. This scheme is still simplified: the apoptogenic signal transduction recruits also endoplasmic reticulum (ER) (see below); stress conditions promote activation of procaspase-12, with the total number of known procaspases being 14 already.
Until recently, there has been a general belief that apoptosis is uniquely metazoan, as in genomes of unicellular organisms genes encoding apoptotic factors analogous to those of metazoan have not been found and because there was reasonable skepticism about the physiological usefulness and evolutionary advantages of apoptosis in unicellular organisms, including yeasts.
The discovery of apoptosis-like death of yeast cells comes from experiments where pro- and antiapoptotic factors of animals were heterologously expressed in the yeast Saccharomyces cerevisiae and the physiology of the yeast cells was studied ([14], for review see [15]). In these experiments, yeast cells either died, showing a set of characteristic physiological markers of apoptosis, or survived, depending on the expression of pro- or antiapoptotic factors, respectively. Later, apoptosis-like process induced by heterologously expressed Bax (a proapoptotic factor of mammals) was described not only for S. cerevisiae [14-17], but also for Schizosaccharomyces pombe [18, 19], Pichia pastoris [20], Kluyveromyces lactis [21], and Candida albicans [22]. Collectively, these data are a solid indication of similarity of several elements in the mechanisms underlying apoptosis in yeast cells and in animals. In 1997, a temperature-sensitive S. cerevisiae strain carrying a mutation in the CDC48 gene was described (cdc48p plays an important role in many cellular processes, including protein degradation, membrane fusion, chaperone activity). Under non-permissive temperatures, the mutant cells died, displaying markers of apoptosis commonly observed in higher eukaryotes, such as exposure of phosphatidylserine on the outer leaflet of the plasma membrane, chromatin condensation, and cell fragmentation [23]. In 2002, in S. cerevisiae the first proapoptotic protein involved in apoptosis in yeast was identified, i.e. a metacaspase (cysteine protease) termed yeast metacaspase-1 (Yca1p) [24], a functional analog of caspases of animal cells.
It is now clear that microorganisms, including yeasts, in nature preferably live in multicellular communities (e.g. biofilms, colonies) and undergo a sort of differentiation coupled to apoptosis [25-29].
Saccharomyces cerevisiae cells secrete aromatic alcohols (tryptophol and phenylethanol) that stimulate morphogenesis. The production of these autosignaling alcohols is regulated by nitrogen: high ammonia restricts it by repressing the expression of their biosynthetic pathway, whereas nitrogen-poor conditions activate it. Formation of specific aromatic alcohols mediating social interactions between Saccharomyces cells has been described. The same autoregulatory molecules do not evoke the morphological switch in Candida albicans, suggesting that these molecular signals are species-specific [30]. An ammonia signal, emitted by aging colonies, triggers metabolic changes that localize yeast death only in the colony center. The remaining population can exploit the released nutrients and survives. The establishment of these zones required a functional SOK2 gene product, which functions in the ammonia signaling pathway. In colonies defective in Sok2p transcription factor that are unable to produce ammonia, death is spread throughout the whole population, thus decreasing the lifetime of the colony. The physical removal of the dead zone from growing colonies caused a reduction in growth at the colony periphery, strongly suggesting that apoptosis within the central dead zone benefits the population and thereby acts to preserve the whole colony [26, 28, 29, 31, 32]. Therefore, for unicellular organisms, including yeasts, apoptosis or phenoptosis is an active, highly genetically regulated mechanism of programmed cell death aimed at eliminating self-destructive, damaged, weakened cells that are potentially dangerous for the cell population.
Strictly speaking, phenoptosis is an appropriate term to describe the death of unicellular organisms, including yeast, when it refers to individual cells and uniform cell suspensions. The term apoptosis should be reserved for the death of individual cells in biofilms and cell colonies, by analogy to cell death in multicellular communities of higher organisms. However, in the analysis below we apply the term “apoptosis” because it was used by the authors of the cited papers.
COMPONENTS IMPLICATED IN YEAST APOPTOSIS
Numerous cases of apoptosis-like death of yeast cells (investigations have been made primarily on S. cerevisiae) have been described. Apoptosis in yeast cells implicates not only metacaspase(s) Yca1p [24, 33], but also another caspase-like cysteine protease (conserved in mammals) Esp1 [34]. This protease of yeast was recently reported to cleave yeast Mcd1 (ortholog of human cohesin Rad21/Mcd1) involved in the attachment of sister kinetochores for chromatid cohesion [34]. Among other components involved in apoptosis signaling in yeasts are the serine protease OMI (Nma111p – nuclear mediator of apoptosis) belonging to the family of HtrA (high temperature requirement A) proteins. Nma111p is able to promote apoptotic cell death by, at least in part, degradation of Bir1p, the only identified inhibitor of apoptosis in S. cerevisiae (see [35, 36]); AIF (an apoptosis-inducing factor) and AMID (AIF-homologous mitochondrion-associated inducer of death) [37, 38]; Dnm1p [39], a homolog of animal Drp1p (dynamin-related protein) responsible for fragmentation of mitochondria in humans; cytochrome c [40, 41]; the normally confined to the intermembrane space endonuclease Nuc1p [42, 43], the yeast homolog of endonuclease G; an endo-/exonuclease Tat-D (the enzyme activities are metal-dependent with Mg2+ as the optimal metal ion) [44]; mitochondrial proteins (Ysp1 and Ysp2 – yeast suicide proteins), specifically required for the mitochondrial thread–grain transition, de-energization, and cell death (in pheromone and amiodarone-induced apoptotic changes) [45, 46]; Rho5 GTPase (under apoptosis induced by ROS) [47]; Mcd1 (the yeast homolog of human Rad21) in hydrogen peroxide-induced apoptosis in yeast [48]; proapoptotic factor Ybh3p (yeast cells lacking Ybh3p display prolonged chronological and replicative lifespans and resistance to apoptosis induction) [49], and the antiapoptotic factor Bir1p (baculovirus IAP repeat), a substrate of Nma111p [34, 50, 51]. Bre1p (a product of the BRE1 gene), a ligase that is required for the ubiquitylation of histone H2B at lysine 123 in S. cerevisiae, also manifests antiapoptotic activity [52]. Enhanced levels of Bre1p protect from hydrogen peroxide-induced cell death, whereas deletion of BRE1 enhances cell death. Moreover, cells lacking Bre1p show reduced lifespan during chronological aging. Very recently [53], in S. cerevisiae a homolog of Bax inhibitor-1 (BI-1), an antiapoptotic protein that resides in the ER and is involved in the unfolded protein response (UPR) that is triggered by ER stress (see below) has been revealed. Deoxyuridine triphosphatase (dUTPase), a product of the mitochondrial variant of the human DUT gene (DUT-M), can prevent apoptosis in yeast in response to internal (Bax expression) and to exogenous (H2O2 and cadmium) stresses [54.] Of interest, cell death was not prevented under culture conditions modeling chronological aging, suggesting that DUT-M only protects dividing cells. Lithocholic acid modulates housekeeping longevity assurance pathways by suppressing mitochondria-controlled apoptosis, enhancing stability of nuclear and mitochondrial DNA, attenuating mitochondrial fragmentation, and enhancing resistance to oxidative and thermal stresses [55].
In the fission yeast S. pombe novel cell-death regulators, including metacaspase Pca1, BH3-domain protein Rad9 (a component of the Rad9–Hus1–Rad1 complex, a sensor of DNA damage and replication), and diacylglycerol-binding proteins Pck1 and Bzz1 [56], have been identified. Also, three conserved MAP kinase signaling pathways have been revealed in this yeast [57]. In yeasts, some of the abovementioned apoptotic factors are located in the mitochondrial intermembrane space, like in animal mitochondria.
INDUCERS OF YEAST APOPTOSIS
Numerous cases of apoptosis-like death of yeast cells caused by various environmental stimuli and intercellular defects have been described (see [50, 58-62]).
Defects of DNA, mutations. Apoptosis was found to occur in mutant S. cerevisiae yeast displaying defective initiation of DNA replication [63, 64]. The apoptosis-like phenotype of the initiation mutants includes the production of ROS and activation of the budding-yeast metacaspase Yca1p.
Cell death with predominant apoptotic features was mediated in S. cerevisiae by deletion of the histone chaperone Asf1p/Cia1p [65]. Deletion of the histone chaperone inhibits the normal assembly/disassembly of nucleosomes in the yeast and thereby initiates the active cell death system. Many changes were identified, such as fragmentation of the nuclei, condensation and fragmentation of chromatin, reduction of the mitochondrial membrane potential, dysfunction of the mitochondrial proton pump, and a discernible release of cytochrome c to the cytoplasm that resembles these features of apoptosis in multicellular organisms.
Pds5p is a cohesin-related protein. It is required for maintenance of sister chromatid cohesion in mitosis and meiosis. Cohesin is a multisubunit complex thought to embrace DNA as a ring-shaped structure that mediates sister chromatin cohesion and ensures accurate chromosome segregation [66, 67]. A reduction in the maintenance of sister chromatid cohesion as a result of a mutated cohesin-like gene, PDS5, led to apoptosis induction during early meiosis in S. cerevisiae. The pds5-1-caused cell death has characteristics of apoptosis and necrosis, including externalization of phosphatidylserine at the cytoplasmic membrane, accumulation of DNA breaks, chromatin condensation and fragmentation, nuclei fragmentation, and then degeneration of the membrane and cell [66].
Mutation in CDC48 (cdc48(S565G)), a gene essential in the ER-associated protein degradation (ERAD) pathway, led to the discovery of apoptosis as a mechanism of cell death in S. cerevisiae. Mitochondrial dysfunction in the cdc48(S565G) strain was accompanied by structural damage of mitochondria and ROS accumulation concomitantly, and caspase-like enzymatic activity emerged, suggesting a role for caspases in the cell death process [23].
Defects of RNA structure. Apoptosis was also found in cells exhibiting increased mRNA stability as a result of disruption of the efficiency of pre-mRNA splicing and mRNA decapping. Mitochondrial function and YCA1, which encodes a budding yeast metacaspase, is necessary for apoptosis triggered by stabilization of mRNAs. Deletion of YCA1 in yeast cells mutated in the LSM4 gene prevents mitochondrial fragmentation and rapid cell death during chronological aging of the culture, diminishes reactive oxygen species accumulation and DNA breakage, and increases resistance to H2O2 and acetic acid [68].
Defects of actin dynamics. Mutations (or addition of drugs) that reduce actin dynamics lead to the formation of F-actin aggregates, resulting in constitutive activation of a caspase-independent signaling mechanism, ROS production, and apoptosis [69]. As a reduction in the dynamic state of filamentous actin (F-actin) and the consequent formation of F-actin aggregates is commonly observed in aging yeast, an exciting possibility is that this apoptosis pathway provides a means to eliminate aged cells from a population. In actin-induced yeast apoptosis, ROS accumulation is thought to be the major killer, this being demonstrated by the fact that the addition of antioxidants, or the reduction of ROS at their source, can circumvent cell death [70, 71].
Defects of N-glycosylation. N-Glycosylation in the ER is an essential protein modification and highly conserved in evolution from yeast to man. Defects of N-glycosylation in humans lead to congenital disorders. The key step of this pathway is the transfer of the evolutionarily conserved core-oligosaccharide GlcNAc2Man9Glc3 linked to dolichyl pyrophosphate to selected asparagine residues of the nascent polypeptide chain, catalyzed by the heterooligomeric membrane complex oligosaccharyl transferase (OST). Biochemical and molecular genetic studies in S. cerevisiae have identified nine proteins, which are assembled into a complex consisting of Ost1p, Stt3p, Wbp1p, Ost3p, Ost6p, Swp1p, Ost2p, Ost5p, and Ost4p. One of the subunits, Ost2p, is 40% identical to Dad1 (defender against apoptotic death), a highly conserved protein in higher eukaryotes including plants. Ost mutants, such as ost2 and wbp1-1, display morphological and biochemical features of apoptosis upon induction of the glycosylation defect [72]. Nuclear condensation, DNA fragmentation, as well as externalization of phosphatidylserine have been observed. As deletion of metacaspase YCA1 does not seem to abrogate glycosylation-induced apoptosis, this suggests a different proteolytic process involved in this death pathway.
Heterologous expression of proteins. Expression of DR4 (a tumor necrosis factor-related apoptosis-inducing ligand receptor) in S. cerevisiae causes growth inhibition. Moreover, treatment of DR4-expressing yeast with a DNA damaging agent sustained cell growth inhibition accompanied with massive apoptotic cell death. Further analysis revealed that cell death in the presence of DNA damage and DR4 expression was not dependent on the yeast caspase YCA1 [73].
Human PDCD5 protein is a novel programmed cell death-promoting molecule. However, the function of Ymr074cp, a S. cerevisiae homolog of PDCD5p, is still unknown. YMR074c overexpression promotes H2O2-induced apoptosis in yeast, not only in a metacaspase Yca1-dependent manner but also in an Yca1-independent manner [74].
Huntington’s disease is caused by specific mutations in huntingtin protein. Expansion of a polyglutamine (polyQ) repeat of huntingtin leads to protein aggregation in neurons followed by cell death with apoptotic markers [46, 75]. The physiological consequences of expanded polyQ domain expression in yeast are similar to those in neurons. In particular, expression of expanded polyQ in yeast causes apoptotic changes in mitochondria, caspase activation, nuclear DNA fragmentation, disorder of the cell cycle, and death [46].
Bax, a proapoptotic protein of animals, induces apoptosis in several yeast species (see above) with characteristic morphological and biochemical markers including chromatin condensation on the periphery of the nucleus with further nucleus fragmentation, externalization of phosphatidylserine to the outer leaflet of the plasma membrane, ROS generation, and in some cases release of cytochrome c from mitochondria into the cytosol [15, 76]. Recently [77], it has been shown that protein kinase Cα (PKCα) increases the translocation and insertion of Bax c-myc (an active form of Bax) into the outer membrane of yeast mitochondria.
Schizosaccharomyces pombe cells expressing a lumenal version of calnexin (an ER transmembrane chaperone playing key roles in translocation, protein folding, and quality control of newly synthesized polypeptides) exhibited a 2-fold increase in the levels of apoptosis provoked by inositol starvation [78].
Oxidative stress. Oxygen radicals are important components of apoptosis. The ROS produced during mitochondrial respiration of yeast cells are the main mDNA damaging agents [79]. Yeast cells with nonfunctional mitochondrial DNA (petite) are known to be extremely stress-resistant [80].
Hydrogen peroxide (H2O2) induces oxidation of specific proteins in yeast cells, and the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (Tdh) is a major target. Using a 2D-gel system to study protein carbonylation, it was shown that both Tdh2p and Tdh3p isozymes are oxidized during exposure to H2O2. In addition, two other proteins were carbonylated and inactivated – Cu,Zn-superoxide dismutase (SOD) and phosphoglycerate mutase. The oxidative inactivation of SOD decreases the antioxidant capacity of yeast cells and probably contributes to H2O2-induced cell death [81-84] with classical parameters of apoptosis such as chromatin condensation, DNA fragmentation, and morphology changes of the cells [83]. Yeast cells were most sensitive to peroxide at the exponential growth phase. Minocycline (an antibiotic of the tetracycline family that displays cytoprotective potency) increased availability of peroxide-treated exponentially growing cells (young cells) [85].
Apoptosis can be induced in S. cerevisiae by depletion of glutathione [83]. Cycloheximide (a protein synthesis inhibitor) and hypoxia prevent apoptotic death, revealing active participation of the cell. A shift in the cellular GSH-to-GSSG redox balance in favor of the oxidized species, GSSG, constitutes an important signal that can decide the fate of a cell.
Allicin, a volatile prooxidant substance of garlic juice, displaying antimicrobial activity primarily due to impairment of thiol-disulfide exchange reactions with -SH groups in proteins and glutathione (the oxidative inactivation of essential thiol-containing enzymes), can activate apoptosis in S. cerevisiae cells. Caspase activation occurred after allicin treatment, and yeasts expressing a human antiapoptotic Bcl-XL construct were rendered more resistant to allicin [85].
Apoptosis in S. cerevisiae cells induced by UV irradiation caused chromatin condensation in the nucleus, cell shrinking, and DNA damage [86].
Hyperosmotic stress. Hyperosmotic stress caused by high glucose or sorbitol concentrations in the culture medium induced in S. cerevisiae a caspase-dependent cell death accompanied by morphological and biochemical indicators of apoptosis, namely chromatin condensation, mitochondrial swelling and reduction of cristae number, production of ROS, and DNA strand breaks with maintenance of plasma membrane integrity. The mutant strains cyc1Deltacyc7Delta and cyc3Delta, both lacking mature cytochrome c, displayed a decrease in caspase activation associated with increased cell survival when exposed to hyperosmotic stress. Yeast cells lacking mitochondrial DNA are hypersensitive to salt stress-induced apoptosis [87].
Excess of mating pheromones. Haploid S. cerevisiae yeast cells exist in two forms, a and α, each belonging to one of the two mating types (Mat a and Mat α). Sexual conjugation of haploid yeast cells of opposing mating type is mediated by the secretion and detection of mating pheromones (a-factor and α-factor). α-Cells secrete α-factor, while a-cells secrete a-factor. These short peptide pheromones are detected by heterotrimeric G-protein coupled receptors (Ste2p and Ste3p). This leads to polarization of the cell towards the pheromone source, remodeling of the cell wall and causing the formation of a mating projection [88]. Treatment of a-cells by the α-factor is routinely used for cell synchronization in the G1-stage of the cell cycle [62]. In modern practice, it is known that high concentrations of pheromones are toxic for cells. In 2002 [89], it was demonstrated for the first time that the exposure of haploid S. cerevisiae to high concentrations of the appropriate mating pheromone resulted in increased cytosolic Ca2+ concentrations, hyperpolarization of mitochondria, and enhanced ROS production, which, in turn, induced fragmentation of mitochondria and apoptosis-like cell death [45, 62, 89, 90]. This was the first evidence of apoptosis in a yeast cell induced by a natural factor.
Killer toxins. Moderate doses of the virally encoded killer toxins induce an apoptotic yeast cell response. The best known are three different killer toxins (K1, K2, and K28) produced and secreted by S. cerevisiae (for review, see [91]). Members of each group can kill non-killer yeasts as well as killer yeasts belonging to the other types. K1, K2, and K28 killer toxins are genetically encoded by medium-size double-stranded RNA (dsRNA) viruses grouped into three types, M1, M2, and M28, of 1.6, 1.5, and 1.8 kb, respectively. Saccharomyces cerevisiae viral toxins kill sensitive yeast cells in a receptor-mediated process by interacting with receptors located in the yeast cell wall and cytoplasmic membrane. After reaching the plasma membrane, ionophoric virus toxins, such as K1 (19-kDa α/β heterodimer) and most likely K2, disrupt the cytoplasmic membrane function by forming cation-selective ion channels. K28 toxin (21.5-kDa protein), after interacting with a sensitive cell, is taken up by endocytosis and travels the secretion pathway in reverse until it reaches the cytosol. This toxin blocks DNA synthesis and arrests cells in the early S phase of the cell cycle [91].
Zygocin, a monomeric 10-kDa protein toxin secreted by the osmotolerant spoilage yeast Zygosaccharomyces bailii, is encoded by a double-stranded (ds) RNA killer virus and kills a broad spectrum of human and phytopathogenic yeasts and filamentous fungi by disrupting cytoplasmic membrane function [92, 93].
The spectrum of action and the activity of killer toxins are influenced by temperature, salinity, pH of media, and the prevailing growth phase (high activity in the log phase of growth, decaying in the stationary phase). Low and moderate concentrations of K1, K28, zygocin, and killer toxin from Pichia acaciae [94] induced S. cerevisiae apoptotic cell death that was mediated through yeast caspase Yca1p and the generation of ROS [95-97]. High concentrations of killer toxins induced non-apoptotic necrotic cell death [63]. Treatment of S. cerevisiae cells by T-2 toxin induced externalization of phosphatidylserine, oxidative damage to mitochondria, nucleolytic damage to DNA, probably mitochondrial, and, ultimately, apoptotic cell death [98.]
Yeast cells harboring killer viruses were also shown to induce apoptosis when incubated with uninfected or susceptible yeast cells [96].
Antifungal and antimicrobial preparations, peptides. Plagiochin E (an antifungal active macrocyclic bis (bibenzyl) isolated from liverwort Marchantia polymorpha L.) induced apoptosis in C. albicans through a metacaspase-dependent apoptotic pathway [99]. Cells treated with plagiochin showed typical markers of apoptosis: chromatin condensation, nuclear fragmentation, and phosphatidylserine exposure. Besides, plagiochin promoted the release of cytochrome c and activated the metacaspase, which resulted in apoptosis of the yeast. The addition of L-cysteine prevented plagiochin-induced nuclear fragmentation, phosphatidylserine exposure, and metacaspase activation, indicating that ROS were an important mediator of plagiochin E-induced apoptosis. The expression of CDC28, CLB2, and CLB4 was downregulated by plagiochin. Gene CDC28 encodes Cdc28, a cyclin-dependent protein kinase, the central coordinator of the major events of the yeast cell division cycle. Genes CLB2 and CLB4 encode cyclin B-like proteins.
Amphotericin B (a polyene macrolide antibiotic; antifungal drug) induced an apoptotic process in biofilms of three Candida species (C. albicans, C. krusei and C. parapsilosis), probably activated by histone acetylation. Externalization of phosphatidylserine, chromatin condensation, accumulation of ROS, DNA degradation, and caspase activation were demonstrated [100].
Culturing the yeast C. utilis in the presence of low doses of doxorubicin (an anthracycline antibiotic) (25 µg/ml) caused morphological alteration of the plasma membrane. In the presence of higher doxorubicin doses (50 µg/ml), in addition to profound alterations in the plasma membrane, changes in mitochondrial shape and cristae organization were observed. Morphologically, doxorubicin doses of up to 200 µg/ml produced apoptosis, whereas higher doxorubicin doses produced necrosis [101].
Baicalein (one of the skullcap flavones; a compound that was originally extracted from Scutellaria baicaleinsis root, exhibits antioxidant, antibacterial, antiviral, and antifungal activities) induced apoptosis in the yeast C. albicans. Chromatin condensation and fragmentation, increasing intracellular levels of ROS, and upregulation of some redox-related genes (CAP1, which encodes the transcription factor Cap1p); SOD2, the manganese SOD gene; and TRR1, the thioredoxin reductase gene) were observed. Finally, mitochondrial de-energization resulted in loss of mitochondrial membrane potential accompanied by the release of cytochrome c [102]. The antifungal activity of baicalein in C. krusei isolates occurred through perturbation of mitochondrial homeostasis without causing elevation of the intracellular ROS level and does not involve apoptosis [103].
Acriflavine (an antiseptic fungicide agent against parasitic infections) caused both apoptosis and necrosis in C. utilis. Acriflavine induced uncoupling of oxidative phosphorylation, a collapse in the electrochemical proton gradient, decrease in ATP synthesis, cell number, and cytochrome c content, chromatin condensation, and subsequent cytolysis (necrosis) [104].
Metergoline (an antifungal agent) elicited cell death in C. krusei (a notorious yeast species) through elevation of the intracellular ROS level and perturbation of mitochondrial homeostasis, followed by damage to the nucleus in a concentration-dependent fashion [105].
The fungicidal piperazine-1-carboxamidine derivative BAR0329 induced caspase-dependent apoptosis in S. cerevisiae, in which the mitochondrial fission machinery consisting of Fis1 (an apoptotic factor), Drp1 (yeast Dnm1, dynamin-related GTPase), and Mdv1 is involved [106]. During mitochondrial division in yeast, Drp1 is recruited to the outer mitochondrial membrane by a conserved integral membrane protein, Fis1, and adaptor proteins Mdv1 and Caf4. Both Dnm1/Drp1 and Mdv1 are essential for mitochondrial fission in yeast [107].
The 26-kDa protein osmotin produced by cultured tobacco (Nicotiana tabacum var Wisconsin 38) cells adapted to grow under osmotic stress [108] belongs to the family of plant protection proteins with antifungal activity in vitro and in vivo [109]. Osmotin induces apoptosis in S. cerevisiae via a caspase-independent signaling mechanism. Osmotin, being a homolog of mammalian adiponectin, is believed to control apoptosis in yeast through a homolog of mammalian adiponectin receptor [109]. Osmotin-induced manifestations of apoptosis were progressive, dependent both on duration of treatment and osmotin concentration. After 2-h osmotin treatment, a decrease in the number of viable cells was revealed, as well as the appearance of cells with abnormal morphology, enhanced ROS accumulation, fragmentation of DNA and nucleus, membrane shrinkage and blebbing, and increased vacuolation. Cells exhibiting increased cytoplasmic shrinkage, formation of cytosol fragments resembling apoptotic bodies, or unusual mitochondrial morphology were most common after 3-h treatments. Higher osmotin doses produced necrosis. Accumulation of intracellular ROS in S. cerevisiae induced the synthesis of proteins with antioxidant properties, such as peroxidases, catalases, and SODs. Deletion of even one of these antioxidant proteins, namely the thioredoxin peroxidase TSA1, resulted in increased loss of viability and a greater proportion of the cell population presenting apoptotic markers, such as ROS accumulation, than in wild-type cells. Yeast cells were protected against osmotin toxicity in the presence of N-acetyl-L-cysteine, an antioxidant known to increase cellular pools of free radical scavengers [70, 71]. These results suggest that ROS are effectors of osmotin-mediated apoptosis.
Bostrycin is an anthracenedione with phytotoxic and antibacterial activity that belongs to a large family of quinines isolated from secondary metabolites of a mangrove endophytic fungus collected from the South China Sea. Bostrycin inhibits S. cerevisiae cell proliferation by blocking the cell cycle at G1 phase and ultimately leads to cell death [110]. Bostrycin-induced lethal cytotoxicity is accompanied by increased levels of intracellular ROS and hallmarks of apoptosis such as chromatin condensation, DNA fragmentation, and externalization of phosphatidylserine. Bostrycin induces apoptosis in yeast cells through a caspase-independent pathway.
Psacotheasin (a 34-residue antimicrobial peptide isolated from the yellow-spotted long-horned beetle Psacothea hilaris) possesses an antifungal property in opportunistic C. albicans via apoptosis. Cells treated with psacotheasin showed diagnostic markers in yeast apoptosis: phosphatidylserine externalization from the inner to the outer membrane surface, mitochondrial membrane depolarization, increase in metacaspase activity, and DNA fragmentation and condensation [111].
Pleurocidin isolated from skin mucous secretions of the winter flounder Pleuronectes americanus and possessing a high potency and broad-spectrum antimicrobial activity induced apoptosis in C. albicans cells [112]. Exposure to pleurocidin caused at earlier stages activation of yeast metacaspases, phosphatidylserine externalization, elevated ROS accumulation, DNA fragmentation, chromatin condensation, depolarization of the mitochondrial membrane, and release of proapoptotic factors [112].
Papiliocin, a 37-residue peptide isolated from the swallowtail butterfly Papilio xuthus, induced apoptosis in C. albicans. Cells treated with papiliocin showed a series of cellular changes normally seen in cells undergoing apoptosis: ROS accumulation, plasma membrane translocation of phosphatidylserine, dissipation of the mitochondrial membrane potential, and the presence of active metacaspases, and DNA condensation and fragmentation [113].
Melittin (the principal toxic component in the venom of the European honey bee Apis mellifera, a small linear amphiphilic 26-residue peptide) forms voltage-gated ion channels in planar lipid bilayers and induces apoptosis in C. albicans [114]. Yeast cells exposed to melittin showed increased ROS production, phosphatidylserine externalization, and DNA and nuclear fragmentation.
Metal ions. At moderately toxic levels, Cu2+ and Mn2+ induced extensive apoptosis in S. cerevisiae cells [115]. At even higher concentrations necrosis took over. Yeast metacaspase Yca1p is not involved in Cu-induced apoptosis, although it plays an important role in the Mn-induced process. Exposure of S. cerevisiae cells to 1 mM Pb2+ resulted in severe oxidative stress, which can be the trigger of programmed cell death by apoptosis [116]. The addition of ascorbic acid (a ROS scavenger) strongly reduced the oxidative stress. Very low doses of Cd2+ cause ER stress in S. cerevisiae, DNA damage, oxidative stress, and apoptosis [117]. Cd2+-induced ER stress and Cd2+ toxicity are a direct consequence of Cd2+ accumulation in the ER. Cd2+ activated the calcium channel Cch1/Mid1, which also contributed to Cd2+ entry into the cells.
Metabolites and other factors. Dwindling nutrients trigger the altruistic death of older yeast cells [118]. Apoptosis may also play a role in aging as prominent markers were observed in chronologically aged cells [58, 119]. Glucose administration to chronologically aged yeast cells resulted in a diminished efficacy of cells to enter quiescence, finally causing superoxide-mediated replicative stress and apoptosis [120].
Treatment of S. cerevisiae cells with high (21-23%) ethanol concentrations for 3-h triggers apoptosis with DNA cleavage, chromatin condensation, and Fis1-mediated mitochondrial fragmentation [121].
Inositol is a precursor for numerous molecules including inositol-containing phospholipids, inositol esters, and phosphorylated versions of inositol playing central roles in membrane integrity and cell signaling. Inositol starvation in S. pombe causes cell death with apoptotic features. This apoptotic death is dependent on the metacaspase Pca1p [78].
Farnesol, a precursor in the isoprenoid/sterol pathway, is involved in the inhibition of germination and biofilm formation by C. albicans. Farnesol promotes apoptosis in C. albicans through caspase activation with ROS accumulation and mitochondrial degradation [122].
The exposure of S. cerevisiae cells to 13-L-hydroperoxylinoleic acid (LOOH) caused their death with ROS accumulation, the degree of which was dependent on the growth phase of the cells. Antioxidants such as melatonin and vitamin E inhibited the LOOH-triggered cell death, while an inhibitor of glutathione synthetase enhanced the cell death by LOOH [123].
Low concentrations of formic acid induced apoptosis-like cell death in S. cerevisiae with several morphological and biochemical changes that are typical of apoptosis, including chromatin condensation, DNA fragmentation, externalization of phosphatidylserine, ROS production, loss of mitochondrial membrane potential, and mitochondrion destruction. This process may not be dependent on the activation of the yeast metacaspase Yca1p [124].
Acetic acid was identified as the principle cell-extrinsic mediator of cell death during chronological aging [40, 80, 125-127]. Cell death was dependent on metacaspase activation and was accompanied by typical hallmarks of apoptosis including ROS accumulation and DNA fragmentation, as well as by cell cycle arrest at the S-phase (instead of G1), possibly by cytosol acidification and by an increase in the level of protonated superoxide, the most reactive form of ROS [128]. The antioxidant N-acetylcysteine prevented acetic acid-induced programmed cell death by scavenging H2O2 and by inhibiting both cytochrome c release and caspase-like activation.
Valproic acid (2-propyl pentanoic acid, widely used in treating epilepsy and bipolar disorder) induced apoptosis in S. cerevisiae that was not dependent on metacaspase Yca1p [129]. Valproic acid inhibited the growth of yeast in a dose-dependent manner: when cells were exposed to 25 mM valproic acid, the wild-type died showing apoptotic markers, and when cells were exposed to 50 mM valproic acid, the wild-type died showing morphological features similar to those of the autophagic death, which was not dependent on Yca1p. Schizosaccharomyces pombe treated with valproic acid died with apoptotic markers (DNA fragmentation, loss of mitochondrial membrane potential, chromatin condensation) independently of metacaspase [130]. Sensitivity to valproic acid was strongly dependent on growth phase. Cells in a later growth phase were much more sensitive to valproic acid than those in an earlier one. Altering the pH of the medium also caused remarkable changes in sensitivity. Cells in an acidic medium were more sensitive to valproic acid.
Aspirin (acetylsalicylic acid) induced apoptosis in MnSOD-deficient S. cerevisiae cells when cultivated on the non-fermentable carbon source ethanol [131]. Aspirin had an inhibitory effect on cellular respiration and caused the release of most of the mitochondrial cytochrome c and a dramatic drop in the mitochondrial membrane potential.
Membrane-permeable C2-ceramide induced apoptotic and necrotic cell death of S. cerevisiae cells, causing generation and over-generation of ROS upon deletion of the mitochondrial genome [132]. Thus, mitochondrial function is strictly required for C2-ceramide-induced yeast lethality.
Propolis (a natural product of plant resins that is used by bees to seal holes in their honeycombs and protect the hive entrance) was able to induce an apoptotic cell death response in S. cerevisiae cells [133]. Cytochrome c but not endonuclease G (Nuc1p) was involved in the propolis-mediated cell death in S. cerevisiae. The metacaspase YCA1 gene was important for propolis-mediated cell death.
A solution of 10-µM tributyltin chloride, a widely occurring environmental pollutant toxic to a variety of eukaryotic and prokaryotic organisms, induced apoptosis-like cell death in S. cerevisiae via a Yca1p-dependent pathway, possibly downstream of ROS production, with nuclear fragmentation and chromatin condensation [134].
Amiodarone (a pharmaceutical substance used for treating a number of diseases) was shown to cause apoptosis in S. cerevisiae [135]. Amiodarone caused the appearance of small and separated, slightly swollen mitochondria. Chromatin displacement to the periphery of the nucleus, nuclear sectioning, and nuclear envelope disturbances were observed in the cells under these conditions.
Gefitinib, a selective inhibitor of the epidermal growth factor receptor tyrosine kinase, induced apoptotic cell death in the S. cerevisiae through a mitochondrial-dependent pathway in a time- and dose-dependent manner [136]. When cells were exposed to 15 µM gefitinib, typical apoptotic markers including phosphatidylserine exposure, DNA fragmentation, ROS production, and decrease in mitochondrial membrane potential were observed.
PHYSIOLOGICAL IMPORTANCE AND USEFULNESS OF APOPTOSIS FOR YEAST
CELLS
Intensive current studies have helped to clarify the physiological importance and usefulness of apoptosis for yeast cells. It is now clear that microorganisms, including yeasts, in nature preferably live in multicellular communities (e.g. biofilms, colonies) [25, 31, 137]. Yeast colonies can be considered as a multicellular organism that undergoes a sort of differentiation coupled to apoptosis. According to [138], apoptosis of yeast cells usually occurs only in the colony center. The physical removal of the dead zone from growing colonies caused a reduction in growth at the colony periphery, this strongly suggesting the altruistic (from Latin alter – another, alternative; antonym – selfish) nature of the cell death in these social communities (preservation of the whole colony at the expense of death of a part of the population) and advantages of cell suicide.
In some cases, advantages are obvious when apoptosis is induced by pheromones or aging. Elimination of infertile (i.e. incapable of mating) and damaged cells can offer advantages for diploid cells better (as compared to haploid cells) adapted to changing environmental conditions [45]. Removal of old and damaged cells under starvation enhances the chance for the rest of the population to survive and sporulate, therefore increasing probability for clones to survive [45]. Moreover, the increased production of reactive oxygen species increases the likelihood for generation of genetic variants that can adapt to continuously changing conditions. Therefore, altruistic cell death via the activation of highly conservative, self-destructive, enzymatic machinery confers an advantage on the population, enhancing its genetic diversity both via sexual reproduction and somatic mutations. At the same time, apoptosis, while limiting longevity, would favor genetic conservatism.
ORDER OF THE MAIN STEPS IN YEAST APOPTOSIS
A scheme of the mitochondria-mediated death cascade in yeast S. cerevisiae is proposed [62] (figure). Treatment of yeast cells (Mat α) with α-factor or amiodarone leads to an intracellular increase in Ca2+ concentration. In turn the strong boost in cytosolic [Ca2+] acts on mitochondrial respiration, possibly by stimulating the activity of respiratory enzymes and by increasing energy coupling, which is followed by hyperpolarization of the mitochondrial membrane potential (ΔΨM). Elevation of ΔΨM promotes ROS production mainly in complex III of the respiratory chain, which then initiates the mitochondrial thread–grain transition (mitochondrial fragmentation) and de-energization. This process was shown to be dependent on a mitochondrial protein named yeast suicide protein 1, or Ysp1p. It is well conceivable that several distinct inducers of yeast apoptosis acting via the mitochondrial pathway trigger a similar chain of events [45, 50].
Order of the main steps in mitochondria-mediated yeast apoptosis
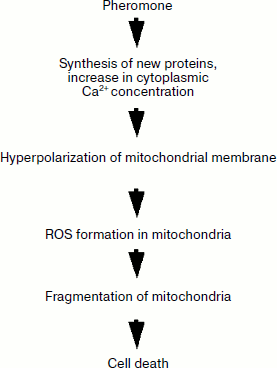
ROLE OF INTRACELLULAR ORGANELLES IN YEAST APOPTOSIS
A crucial role of mitochondria in apoptosis signaling has been proved for animals [9]. The early studies of apoptosis in yeasts [40, 76, 125] were performed with the facultative anaerobe S. cerevisiae grown in the presence of high glucose concentration, i.e. under conditions of glucose repression, when the oxidative cell metabolism is replaced with glycolytic metabolism under which fully functional mitochondria are replaced by so-called pro-mitochondria exhibiting reduced mitochondrial functions. These data seemed to indicate that the mitochondrial energetic function is not always important for apoptogenic signal transduction. However, evidence is rapidly accumulating indicating that mitochondria play the crucial role in the transduction of apoptogenic signal in yeasts. The first evidence that mitochondria are implicated in the programmed cell death process was presented upon induction of apoptosis in S. cerevisiae by acetic acid [40]. The apoptosis was accompanied by the release of cytochrome c from mitochondria into the cytosol, by suppression of mitochondrial respiration, by depletion of cytochrome oxidase subunit II and cytochromes a + a3 [40], as well as by implication of mitochondrial proteins Fis1p, Dnm1p, Mdv1p, Aif1p, Nuc1p Aac, and Por1p in this process [37, 39, 42, 139]. Amiodarone [45], hydrogen peroxide [139], and other drugs caused the release of the mitochondrial cytochrome c. In accordance with these observations, rho0 mutants lacking functional mitochondrial DNA and mitochondria were found to be more resistant to apoptosis inducers [40, 89, 124]. Apoptosis induced in Zygosaccharomyces boidinii by acetic acid was accompanied by mitochondrial ultrastructural changes, namely decrease in number of cristae, formation of myelinic bodies, and swelling [125]. The importance of functional mitochondria and respiration in determining yeast chronological lifespan and apoptosis was confirmed [140].
The fragmentation of mitochondria was documented for treatments with various apoptosis inducers [39, 45, 68, 95, 121] and was proven for the heterologous expression of Bax [141]; it is supposed to be a common trait of apoptosis in yeast [139]. It should be noted that mitochondrial fragmentation per se is attributed not only to apoptosis but also to some other processes, e.g. autophagy. It is thought that such fragmentation facilitates the removal of damaged mitochondria.
The most recent studies focused also on other organelles that are capable of participating in the transduction of the apoptotic signal in animal and yeast cells, such as ER and the vacuole.
The ER is devoted to several crucial cellular processes including lipid synthesis, regulation of calcium homeostasis, and biosynthesis of proteins destined for either intracellular organelles or the cell surface. The ER is the main intracellular Ca2+ store. Overloading the cell with Ca2+ or changes in intracellular Ca2+ stores lead to cytotoxic stress and the induction of apoptosis. Mitochondria and ER are physically close and physiologically connected, which can be spectacularly demonstrated by their mutual influence during transduction of the Ca2+ signal in animal cells. After the inositol trisphosphate-mediated Ca2+ release from ER, the ER–mitochondria complexes become the sites where microdomains with a high (50-100 µM) Ca2+ concentration segregate. These events activate the mitochondrial uniporter having a comparatively low Ca2+ affinity. The uptake of Ca2+ by mitochondria modulates activities of Ca2+-dependent enzymes in the Krebs cycle and induces the Ca2+-dependent permeabilization of mitochondria, which can play a significant role in the induction of an irreversible apoptosis stage in response to various stimuli [9].
It is known that proapoptotic proteins Bax and Bak in animal cells are not confined to mitochondria only but localize also to ER, and that overexpression of these proteins promotes Ca2+ release from ER with a concomitant Ca2+ uptake by mitochondria during apoptosis. Conversely, the deficiency of Bax and Bak proteins results in a dramatic retardation of Ca2+ uptake by mitochondria and confers extreme cell resistance to Ca2+-dependent inducers of apoptosis.
The ER is also the site where secretory proteins are primarily assembled and folded. ER stress results from an imbalance between ER client protein load and folding capacity. Exposure of animal cells to long-chain saturated fatty acids induces apoptosis due to so-called ER stress. Ultimately, failure to handle ER stress can result in apoptosis, a process that is believed to contribute to fatty acid-induced cell death. The budding yeast S. cerevisiae, which is not routinely exposed to high-fat substrates, also undergoes ER stress under lipotoxic conditions. In both cell types, which may seem largely unrelated in terms of function and physiology, the impacts of non-esterified fatty acids (NEFA) on cell viability appear to be very similar. In both types of organisms, the deleterious effects of NEFA vary significantly depending on the length and the unsaturation level of their fatty acid chain. Overall, it appears that NEFA with long saturated chains (such as palmitate (C16:0) and stearate (C18:0)) are highly detrimental to cells, whereas shorter saturated (C14:0 and below) and long-chain fatty acids bearing at least one unsaturation (unsaturated fatty acids (UFAs), such as palmitoleate (C16:1), oleate (C18:1), linoleate (C18:2), and linolenate (C18:3)) are relatively harmless. Moreover, unsaturated fatty acids can efficiently counteract cell death induced by saturated fatty acids [142].
It was recently shown that yeast vacuoles, which share many similarities to plant vacuoles and mammalian lysosomes, are also involved in the regulation of yeast apoptosis and that the vacuolar protease Pep4p, an ortholog of the human CatD (cathepsin D), is released from the vacuole into the cytosol in response to acetic acid and can trigger a mitochondrial apoptotic cascade and necrosis [143].
RELEASE OF APOPTOTIC FACTORS FROM MITOCHONDRIA
For animal mitochondria, two major mechanisms for release of mitochondrial apoptotic factors from the intermembrane space have been suggested. One involves activation, conformational rearrangement, and insertion in the mitochondrial outer membrane of proapoptotic Bax, a member of the Bcl-2 family proteins. Insertion of Bax into the mitochondrial outer membrane is a very complicated process [144, 145] modulated by the mitochondrial complex of translocases of the outer membrane [146], and it requires cardiolipin [147]. The other mechanism relies on increased mitochondrial conductance due to opening of some pores in the inner mitochondrial membranes: a nonspecific, Ca2+/Pi-dependent, cyclosporin A (CsA)-sensitive pore (known as the mitochondrial permeability transition pore, mPTP), a megachannel with diameter of 2.6-2.9 nm enabling free passage of molecules of <1.5 kDa [148-150]; a non-protein pore formed by Ca2+ in the presence of saturated fatty acids (palmitate and stearate) and distinguished from the classical pore (mPTP) by its insensitivity to CsA and Pi, a nonselective permeability to divalent cations, and an ability to be spontaneously closed [151, 152]; a CsA-sensitive pore induced by high extramitochondrial phosphate concentrations at acidic pH values [153, 154]; a CsA-sensitive pore induced by anaerobiosis [155, 156]. Pore opening is accompanied by dissipation of the membrane potential, loss of the energy-transducing function of mitochondria, disruption of ionic homeostasis, high-amplitude swelling of mitochondria, and hence, rupture of the outer membrane and release of proapoptotic factors located in the intermembrane space [148].
Yeast cells are lacking proteins of the Bcl-2 family (the yeast genome does not contain genes encoding these proteins). We still know practically nothing about the role of cristae structural remodeling in release of apoptotic factors from yeast mitochondria. Information on an mPTP-like pore in yeast mitochondria has been until recently scarce, fragmentary, and contradictory (for a review see [157, 158]).
Very recently [157-160] we showed that mitochondria from Yarrowia lipolytica and Dipodascus (Endomyces) magnusii yeasts, which are aerobes possessing the fully competent respiratory chain with all three points of energy conservation and well-structured mitochondria, do not undergo permeabilization when subjected to almost all conditions known to induce permeability transition in animal mitochondria. All our attempts to induce the Ca2+/Pi-dependent pore in tightly-coupled, de-energized, or adenine nucleotide-depleted yeast mitochondria from Y. lipolytica and D. magnusii have failed, even when the incubation medium was supplemented with agents (phenylarsine oxide, for example) known to promote mPTP opening in mammalian mitochondria [157-160]. The general conclusion is that yeast mitochondria are devoid of Ca2+-dependent permeabilization; this feature makes them reminiscent of protozoan mitochondria [161].
In S. cerevisiae the existence of the YMUC (yeast mitochondrial unspecific channel) has been reported [162-168]. The YMUC exhibited a similar cutoff size to that of the mammalian mPTP, opened in response to ATP addition and closed upon ATP depletion, and was active in situ [166]. All our attempts to detect a similar channel in mitochondria of D. magnusii and Y. lipolytica have been unsuccessful [158]. Just the opposite, we succeeded, to our knowledge for the first time, in revealing in mitochondria from Y. lipolytica and D. magnusii the existence of an ATP-dependent K+-channel, closed, as in animal mitochondria, by ATP and opened upon ATP depletion [161, 169]. Moreover, according to our preliminary results, under oxidative stress the selective ATP-dependent K+-channel could be transformed into a nonspecific pore permeable for apoptotic factors normally located in the intermembrane space.
CONCLUSION AND PROSPECTS
Thus, there are currently known numerous factors triggering yeast apoptosis, some pro- and antiapoptotic factors, and the majority of morphological and biochemical alterations accompanying yeast apoptosis. A crucial role of mitochondria and other organelles in yeast apoptosis signaling is proved. A scheme of the mitochondrial death cascade in yeast S. cerevisiae is proposed. In some cases the physiological importance and usefulness of apoptosis for yeast cells are obvious. Much progress has been achieved toward understanding of the way for releasing of apoptotic factors from mitochondria into the cytosol, inducing the reversible stage of apoptosis. Moreover, it has been recognized that yeasts, relatively simple unicellular organisms, vigorously growing on simple and inexpensive media, having small and well-characterized genomes, relatively easily changing their physiological and genetic status, are extremely promising models for functional analysis not only of known pro- and antiapoptotic factors, but also for revealing novel ones [51, 170-173]. The yeast S. cerevisiae was employed to study the apoptosis mechanisms underlying life-threatening human disorders such as Alzheimer’s disease (e.g. [174]) and Huntington’s disease [46, 176]. As a result, there has been a boom in research on yeast apoptosis in the last 3-4 years: six specialized symposia have been held, tens of reviews have appeared [50, 56, 58, 62, 89, 95, 176, 177], and a special issue of Biochimica et Biophysica Acta was devoted to this topic.
However, some questions remain open because the emerging picture is still incomplete and warrants further studies. Admittedly, the current trend consists in widening rather than deepening of research on apoptosis induction. The crucial factors leading to apoptosis after its induction by any particular agent have been fully clarified only in few cases. Elucidation of the pathways for apoptogenic signal transduction remains also problematic in many cases. The first evidence for the crosstalk between the Ca2+/calmodulin/calcineurin system and yeast apoptosis was deduced from study of apoptosis induced in the calcineurin-deficient yeast mutant (cnbΔ) by salt stress [178]. It was established that calcineurin (PP2B phosphatase) regulates Na+ homeostasis, dephosphorylating a transcription factor Tcn1p/Crz1p. As a result, Tcn1p/Crz1p is translocated into the nucleus and activates transcription of many genes, including ENA1, which encodes a P-type ATPase primarily responsible for efflux of the cation across the plasma membrane. Osmotin (a tobacco PR-5 protein that has antifungal activity and is implicated in host-plant defense) induced apoptosis in S. cerevisiae [70], and this induction correlated with intracellular accumulation of ROS due to activation of the RAS2/cAMP-dependent protein kinase, which suppressed transcription of YRE (Yap-response element) and STRE (stress response element) and therefore suppressed synthesis of enzymes of antioxidant defense. However, such studies remain scarce. The role of cytochrome c release from mitochondria into the cytosol is still unclear (no apoptosomes have been found in yeasts). It is still not known which pathways are engaged in activation of metacaspase and what is the role of other proteases in the transduction of apoptogenic signal.
REFERENCES
1.Weismann, A. (1882) Uber die Dauer des
Lebens, Verlag von Gustav Fisher, Jena, Germany.
2.Weismann, A. (1884) Leben and Tod, Verlag
von Gustav Fisher, Jena, Germany.
3.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
4.Skulachev, V. P. (2002)'' Ann. N. Y. Acad Sci.,
959, 214-237.
5.Skulachev, V. P. (2006) Apoptosis,
11, 473-485.
6.Tinari, A., Garofalo, T., Sorice, M., Esposti,
M. D., and Malorni, W. (2007), 3, 282-284.
7.Lyamzaev, K. G., Nepryakhina, O. K., Saprunova, V.
B., Bakeeva, L. E., Pletjushkina, O. Y., Chernyak, B. V., and
Skulachev, V. P. (2008) Biochim. Biophys. Acta, 1777,
817-825.
8.Mijaljica, D., Prescott, M., and Devenish, R.
J. (2010) Methods Mol. Biol., 648, 93-106.
9.Galluzzi, L., Vitale, I., Abrams, J. M.,
Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., Dawson,
T. M., Dawson, V. L., El-Deiry, W. S., Fulda, S., Gottlieb, E.,
Green, D. R., Hengartner, M. O., Kepp, O., Knight, R. A., Kumar, S.,
Lipton, S. A., Lu, X., Madeo, F., Malorni, W., Mehlen, P., Nunez, G.,
Peter, M. E., Piacentini, M., Rubinsztein, D. C., Shi, Y., Simon,
H. U., Vandenabeele, P., White, E., Yuan, J., Zhivotovsky, B., Melino,
G., and Kroemer, G. (2012) Cell Death Differ., 19,
107-120.
10.Daugas, E., Nochy, D., Ravagnan, L.,
Loeffler, M., Susin, S. A., Zamzami, N., and Kroemer, G. (2000)
FEBS Lett., 476, 118-123.
11.Punj, V., and Chakrabarty, A. M. (2003)
Cell Microbiol., 5, 225-231.
12.Hedge, V. L., and Williams, G. T. (2002)
Apoptosis, 7, 123-132.
13.Zou, H., Li, Y., Liu, X., and Wang, X. (1999) J.
Biol. Chem., 274, 11549-11556.
14.Hanada, M., Aime-Sempe, C., Sato, T., and Reed,
J. C. (1995) J. Biol. Chem., 270, 11962-11969.
15.Priault, M., Camougrand, N., Kinnally, K.
W., Vallette, F. M., and Manon, S. (2003) FEMS Yeast Res.,
4, 15-27.
16.Greenhalf, W., Stephan, C., and Chaudhuri, B.
(1996) FEBS Lett., 380, 169-175.
17.Manon, S. (2004) Antioxid. Redox. Signal.,
6, 259-267.
18.Ink, B., Zornig, M., Baum, B., Hajibagheri, N.,
James, C., and Chittenden, T. (1997) Mol. Cell. Biol.,
17, 2468-2474.
19.Jurgensmeier, J. M., Krajewski, S., Armstrong, R.
C., Wilson, G. M., Oltersdorf, T., and Fritz, L. C. (1997) Mol.
Biol. Cell, 8, 325-339.
20.Martinet, W., van den Plas, D., Raes, H.,
Reekmans, R., and Contreras, R. (1999) Biotechnol. Lett.,
21, 821-829.
21.Poliakova, D., Sokolikova, B., Kolarov, J., and
Sabova, L. (2002) Microbiology, 148, 2789-2795.
22.De Smet, K., Eberhardt, I., Reekmans, R., and
Contreras, R. (2004) Yeast, 21, 1325-1334.
23.Madeo, F., Frohlich, E., and Frohlich, K. U.
(1997) J. Cell Biol., 139, 729-734.
24.Madeo, F., Herker, E., Maldener, C., Wissing, S.,
Lachelt, S., Herlan, M., Fehr, M., Lauber, K., Sigrist, S.
J., Wesselborg, S., and Frohlich, K. U. (2002) Mol. Cell,
9, 911-917.
25.Webb, J. S., Givskov, M., and Kjelleberg, S.
(2003) Curr. Opin. Microbiol., 6, 578-585.
26.Vachova, L., and Palkova, Z. (2005) J. Cell.
Biol., 169, 711-717.
27.Palkova, Z., Vachova, L., Gaskova, D., and
Kucerova, H. (2009) Mol. Membr. Biol., 26,
228-235.
28.Vachova, L., Chernyavskiy, O., Strachotova, D.,
Bianchini, P., Burdikova, Z., Fercikova, I., Kubinova, L., and Palkova,
Z. (2009) Environ. Microbiol., 11, 1866-1877.
29.Vachova, L., Kucerova, H., Devaux, F.,
Ulehlova, M., and Palkova, Z. (2009) Environ. Microbiol.,
11, 494-504.
30.Chen, H., and Fink, G. R. (2006) Genes Dev.,
20, 1150-1161.
31.Palkova, Z., and Vachova, L. (2006) FEMS
Microbiol. Rev., 30, 806-824.
32.Cap, M., Vachova, L., and Palkova, Z. (2010)
Commun. Integr. Biol., 3, 198-200.
33.Lee, R. E., Brunette, S., Puente, L. G., and
Megeney, L. A. (2010) Proc. Natl. Acad. Sci. USA, 107,
13348-13353.
34.Cheng, W. C., Leach, K. M., and Hardwick,
J. M. (2008) Biochim. Biophys. Acta, 1783,
1272-1279.
35.Belanger, K. D., Walter, D., Henderson, T. A.,
Yelton, A. L., O’Brien, T. G., Belanger, K. G., Geier, S. J., and
Fahrenkrog, B. (2009) J. Cell Sci., 122, 3931-3941.
36.Fahrenkrog, B. (2011) Biochem. Soc. Trans.,
39, 1499-1501.
37.Wissing, S., Ludovico, P., Herker, E., Buttner,
S., Engelhardt, S. M., Decker, T., Link, A., Proksch, A., Rodrigues,
F., Corte-Real, M., Frohlich, K. U., Manns, J., Cande, C., Sigrist, S.
J., Kroemer, G., and Madeo, F. (2004) J. Cell. Biol.,
166, 969-974.
38.Li, W., Sun, L., Liang, Q., Wang, J., Mo, W., and
Zhou, B. (2006) Mol. Biol. Cell, 17,
1802-1811.
39.Fannjiang, Y., Cheng, W. C., Lee, S. J., Qi, B.,
Pevsner, J., McCaffer, J. M., Hill, R. B., Basanez, G., and Hardwick,
J. M. (2004) Genes Dev., 18, 2785-2797.
40.Ludovico, P., Rodrigues, F., Almeida, A., Silva,
M. T., Barrientos, A., and Corte-Real, M. (2002) Mol. Biol.
Cell, 13, 2598-2606.
41.Silva, R. D., Sotoca, R., Johansson, B.,
Ludovico, P., Sansonetty, F., Silva, M. T., Peinado, J. M., and
Corte-Real, M. (2005) Mol. Microbiol., 58, 824-834.
42.Buttner, S., Eisenberg, T., Carmona-Gutierrez,
D., Ruli, D., Knauer, H., Ruckenstuhl, C., Sigrist, C., Wissing, S.,
Kollroser, M., Frohlich, K. U., Sigrist, S., and Madeo, F. (2007)
Mol. Cell., 25, 233-246.
43.Cymerman, I. A., Chung, I., Beckmann, B. M.,
Bujnicki, J. M., and Meiss, G. (2008) Nucleic Acids Res.,
36, 1369-1379.
44.Qiu, J., Yoon, J. H., and Shen, B. (2005) J.
Biol. Chem., 280, 15370-15379.
45.Pozniakovsky, A. I., Knorre, D. A., Markova, O.
V., Hyman, A. A., Skulachev, V. P., and Severin, F. F. (2005) J.
Cell Biol., 168, 257-269.
46.Sokolov, S., Pozniakovsky, A., Bocharova, N.,
Knorre, D., and Severin, F. (2006) Biochim. Biophys. Acta, 1757,
660-666.
47.Singh, K., Kang, P. J., and Park, H. O. (2008)
Proc. Natl. Acad. Sci. USA, 105, 1522-1527.
48.Yang, H., Ren, Q., and Zhang, Z. (2008) Mol.
Biol. Cell., 19, 2127-2134.
49.Buttner, S., Ruli, D., Vogtle, F. N., Galluzzi,
L., Moitzi, B., Eisenberg, T., Kepp, O., Habernig, L,
Carmona-Gutierrez, D., Rockenfeller, P., Laun, P.,
Breitenbach, M., Khoury, C., Frohlich, K. U., Rechberger, G.,
Meisinger, C., Kroemer, G., and Madeo, F. (2011) EMBO J., 30,
2779-2792.
50.Eisenberg, T., Buttner, S., Kroemer, G., and
Madeo, F. (2007) Apoptosis, 12, 1011-1023.
51.Greenwood, M. T., and Ludovico, P. (2010) Cell
Death Differ., 17, 737-745.
52.Walter, D., Matter, A., and Fahrenkrog, B. M. E.
(2010) J. Cell Sci., 123, 1931-1939.
33.Cebulski, J., Malouin, J., Pinches, N.,
Cascio, V., and Austriaco, N. (2011) PLoS One,
6, e20882.
54.Williams, D., Norman, G., Khoury, C., Metcalfe,
N., Briard, J., Laporte, A., Sheibani, S., Portt, L., Mandato, C. A.,
and Greenwood, M. T. (2011) Biochim. Biophys. Acta, 1813,
315-321.
55.Goldberg, A. A., Richard, V. R., Kyryakov, P.,
Bourque, S. D., Beach, A., Burstein, M. T., Glebov, A., Koupaki, O.,
Boukh-Viner, T., Gregg, C., Juneau, M., English, A. M.,
Thomas, D. Y., and Titorenko, V. I. (2010) Aging(Albany
N.Y.), 2, 393-414.
56.Low, C. P., Shui, G., Liew, L. P., Buttner, S.,
Madeo, F., Dawes, I. W., Wenk, M. R., and Yang, H. (2008) J. Cell.
Sci., 121, 2671-2684.
57.Perez, P., and Cansado, J. (2010) Curr.
Protein Pept. Sci., 11, 680-692.
58.Almeida, B., Buttner, S., Ohlmeier, S., Silva,
A., Mesquita, A., Sampaio-Marques, B., Osorio, N. S., Kollau, A.,
Mayer, B., Leao, C., Laranjinha, J., Rodrigues, F., Madeo, F., and
Ludovico, P. (2007) J. Cell. Sci., 120,
3279-3288.
59.Almeida, B., Silva, A., Mesquita, A.,
Sampaio-Marques, B., Rodrigues, F., and Ludovico, P. (2008) Biochim.
Biophys. Acta, 1783, 1436-1448.
60.Madeo, F., and Frohlich, K. U. (2008) Biochim.
Biophys. Acta, 1783, 1271.
61.Schmitt, M. J., and Reiter, J. (2008) Biochim.
Biophys. Acta, 1783, 1413-1417.
62.Severin, F. F., Meer, M. V., Smirnova, E. A.,
Knorre, D. A., and Skulachev, V. P. (2008) Biochim. Biophys.
Acta, 1783, 1350-1353.
63.Weinberger, M., Ramachandran, L., Feng, L.,
Sharma, K., Sun, X., Marchetti, M., Huberman, J. A., and Burhans, W. C.
(2005) J. Cell. Sci., 118, 3543-3553.
64.Weinberger, M., Feng, L., Paul, A., Smith,
D. L., Jr., Hontz, R. D., Smith, J. S., Vujcic, M., Singh, K. K.,
Huberman, J. A., and Burhans, W. C. (2007) PLoS One, 2,
e748.
65.Yamaki, M., Umehara, T., Chimura, T., and
Horikoshi, M. (2001) Genes Cells, 6, 1043-1054.
66.Ren, Q., Yang, H., Rosinski, M., Conrad, M. N.,
Dresser, M. E., Guacci, V., and Zhang, Z. (2005) Mutat. Res.,
570, 163-173.
67.Caron, P., Aymard, F., Iacovoni, J. S., Briois,
S., Canitrot, Y., Bugler, B., Massip, L., Losada, A., and Legube, G.
(2012) PLoS Genet., 8, e1002460.
68.Mazzoni, C., Herker, E., Palermo, V., Jungwirth,
H., Eisenberg, T., Madeo, F., and Falcone, C. (2005) EMBO
Rep., 6, 1076-1081.
69.Gourlay, C. W., and Ayscough, K. R. (2006)
Mol. Cell. Biol., 26, 6487-6501.
70.Narasimhan, M. L., Damsz, B., Coca, M. A., Ibeas,
J. I., Yun, D. J., Pardo, J. M., Hasegawa, P. M., and Bressan, R.
A. (2001) Mol. Cell., 8, 921-930.
71.Gourlay, C. W., and Ayscough, K. R. (2005) J.
Cell. Sci., 118, 2119-2132.
72.Hauptmann, P., Riel, C., Kunz-Schughart, L. A.,
Frohlich, K. U., Madeo, F., and Lehle, L. (2006) Mol.
Microbiol., 59, 765-778.
73.Kang, M. S., Lee, S. K., Park, C. S., Kang, J.
H., Bae, S. H., and Yu, S. L. (2008) Biochem. Biophys. Res.
Commun.'', 376, 305-309.
74.Hong, J., Zhang, J., Liu, Z., Qin, S., Wu, J.,
and Shi, Y. (2009) Biochemistry, 48, 6824-6834.
75.Walling, H. W., Baldassare, J. J., and Westfall,
T. C. (1998) J. Neurosci. Res., 54, 301-308.
76.Madeo, F., Frohlich, E., Ligr, M., Grey, M.,
Sigrist, S. J., Wolf, D. H., and Frohlich, K. U. (1999) J. Cell.
Biol., 145, 757-767.
77.Silva, R. D., Manon, S., Goncalves, J., Saraiva,
L., and Corte-Real, M. (2011) Exp. Cell Res., 317,
781-790.
78.Guerin, R., Beauregard, P. B., Leroux, A., and
Rokeach, L. A. (2009) PLoS One, 4, e6244.
79.Arcangioli, B., and Ben Hassine, S. (2009)
Cell Cycle, 8, 2326-2331.
80.Kochmak, S. A., Knorre, D. A., Sokolov, S. S.,
and Severin, F. F. (2011) Biochemistry (Moscow), 76,
167-171.
81.Godon, C., Lagniel, G., Lee, J., Buhler,
J. M., Kieffer, S., Perrot, M., Boucherie, H., Toledano, M.
B., and Labarre, J. (1998) J. Biol. Chem., 273,
22480-22489.
82.Costa, V. M., Amorim, M. A., Quintanilha,
A., and Moradas-Ferreira, P. (2002) Free Radic. Biol. Med.,
33, 1507-1515.
83.Magherini, F., Tani, C., Gamberi, T., Caselli,
A., Bianchi, L., Bini, L., and Modesti, A. (2007) Proteomics,
7, 1434-1445.
84.Sollner, S., Durchschlag, M., Frohlich, K.
U., and Macheroux, P. (2009) FEMS Yeast Res., 9,
885-891.
85.Gruhlke, M. C., Portz, D., Stitz, M., Anwar,
A., Schneider, T., Jacob, C., Schlaich, N. L., and Slusarenko, A. J.
(2010) Free Radic. Biol. Med., 49, 1916-1924.
86.Del Carratore, R., Della Croce, C., Simili, M.,
Taccini, E., Scavuzzo, M., and Sbrana, S. (2002) Mutat. Res.,
513, 183-191.
87.Gao, Q., Ren, Q., Liou, L. C., Bao, X., and
Zhang, Z. (2011) FEBS Lett., 585, 2507-2512.
88.Gourlay, C. W., Du, W., and Ayscough, K. R.
(2006) Mol. Microbiol., 62, 1515-1521.
89.Severin, F. F., and Hyman, A. A. (2002) Curr.
Biol., 12, R233-R235.
90.Knorre, D. A., Smirnova, E. A., and Severin, F.
F. (2005) Biochemistry (Moscow), 70, 264-266.
91.Rodriguez-Cousino, N., Maqueda, M., Ambrona, J.,
Zamora, E., Esteban, R., and Ramirez, M. (2011) Appl. Environ.
Microbiol., 77, 1822-1832.
92.Weiler, F., Rehfeldt, K., Bautz, F., and Schmitt,
M. J. (2002) Mol. Microbiol., 46, 1095-1105.
93.Weiler, F., and Schmitt, M. J. (2003) FEMS
Yeast Res., 3, 69-76.
94.Klassen, R., and Meinhardt, F. (2005) Cell.
Microbiol., 7, 393-401.
95.Reiter, J., Herker, E., Madeo, F., and Schmitt,
M. J. (2005) J. Cell Biol., 168, 353-358.
96.Ivanovska, I., and Hardwick, J. M. (2005)
J. Cell Biol., 170, 391-399.
97.Baeza, M. E., Sanhueza, M. A., and Cifuentes, V.
H. (2008) Biol. Res., 41, 173-182.
98.Josse, L., Li, X., Coker, R. D., Gourlay, C. W.,
and Evans, I. H. (2011) FEMS Yeast Res., 11, 133-150.
99.Wu, X. Z., Chang, W. Q., Cheng, A. X., Sun,
L. M., and Lou, H. X. (2010) Biochim. Biophys. Acta,
1800, 439-447.
100.Al-Dhaheri, R. S., and Douglas, L. J. (2010)
J. Med. Microbiol., 59, 149-157.
101.Keyhani, E., and Keyhani, J. (2004) Ann. N.
Y. Acad. Sci., 1030, 369-376.
102.Dai, B. D., Cao, Y. Y., Huang, S., Xu, Y. G.,
Gao, P. H., Wang, Y., and Jiang, Y. Y. (2009) J. Microbiol.
Biotechnol., 19, 803-809.
103.Kang, K., Fong, W. P., and Tsang, P. W. (2010)
Mycopathology, 170, 391-396.
104.Keyhani, E., Khavari-Nejad, S., Keyhani, J.,
and Attar, F. (2009) Ann. N. Y. Acad. Sci., 1171,
284-291.
105.Kang, K., Wong, K. S., Fong, W. P., and Tsang,
P. W. (2011) Fungal Biol., 115, 302-309.
106.Bink, A., Govaert, G., Francois, I. E.,
Pellens, K., Meerpoel, L., Borgers, M., van Minnebruggen, G.,
Vroome, V., Cammue, B. P., and Thevissen, K. (2010)'' FEMS
Yeast Res., 10, 812-818.
107.Mears, J. A., Lackner, L. L., Fang, S.,
Ingerman, E., Nunnari, J., and Hinshaw, J. E. (2011) Nat. Struct. Mol.
Biol., 18, 20-26.
108.Singh, N. K., Bracker, C. A., Hasegawa,
P. M., Handa, A. K., Buckel, S., Hermodson, M. A., Pfankoch, E.,
Regnier, F. E., and Bressan, R. A. (1987) Plant. Physiol., 85,
529-536.
109.Narasimhan, M. L., Coca, M. A., Jin, J.,
Yamauchi, T., Ito, Y., Kadowaki, T., Kim, K. K., Pardo, J. M., Damsz,
B., Hasegawa, P. M., Yun, D. J., and Bressan, R. A. (2005) Mol.
Cell, 17, 171-180.
110.Xu, C., Wang, J., Gao, Y., Lin, H., Du, L.,
Yang, S., Long, S., She, Z., Cai, X., Zhou, S., and Lu, Y. (2010)
FEMS Yeast Res., 10, 297-308.
111.Hwang, B., Hwang, J. S., Lee, J., and Lee, D.
G. (2011) Biochem. Biophys. Res. Commun., 405,
267-271.
112.Cho, J., and Lee, D. G. (2011) Biochimie,
93, 1873-1879.
113.Hwang, B., Hwang, J. S., Lee, J., Kim, J. K.,
Kim, S. R., Kim, Y., and Lee, D. G. (2011) Biochem. Biophys. Res.
Commun., 408, 89-93.
114.Park, C., and Lee, D. G. (2010) Biochem.
Biophys. Res. Commun., 394, 170-172.
115.Liang, Q., and Zhou, B. (2007) Mol. Biol.
Cell, 18, 4741-4749.
116.Bussche, J. V., and Soares, E. V.
(2011) Appl. Microbiol. Biotechnol., 90, 679-687.
117.Gardarin, A., Chedin, S., Lagniel, G., Aude, J.
C., Godat, E., Catty, P., and Labarre, J. (2010) Mol.
Microbiol., 76, 1034-1048.
118.Buttner, S., Eisenberg, T., Herker, E.,
Carmona-Gutierrez, D., Kroemer, G., and Madeo, F. (2006) J. Cell Biol.,
175, 521-525.
119.Lewinska, A., Macierzynska, E., Grzelak, A.,
and Bartosz, G. (2011) Biogerontology, 12, 309-320.
120.Ruckenstuhl, C., Carmona-Gutierrez, D., and
Madeo, F. (2010) Aging (Albany N.Y.), 2,
643-649.
121.Kitagaki, H., Araki, Y., Funato, K., and
Shimoi, H. (2007) FEBS Lett., 581, 2935-2942.
122.Shirtliff, M. E., Krom, B. P., Meijering, R.
A., Peters, B. M., Zhu, J., Scheper, M. A., Harris, M. L., and
Jabra-Rizk, M. A. (2009) Antimicrob. Agents. Chemother.,
53, 2392-2401.
123.Aoshima, H., Kadoya, K., Taniguchi, H., Satoh,
T., and Hatanaka, H. (1999) Biosci. Biotechnol. Biochem.,
63, 1025-1031.
124.Du, L., Su, Y., Sun, D., Zhu, W., Wang, J.,
Zhuang, X., Zhou, S., and Lu, Y. (2008) FEMS Yeast Res.,
8, 531-539.
125.Ludovico, P., Sansonetty, M. T., and
Corte-Real, M. (2003) FEMS Yeast Res., 3, 91-96.
126.Giannattasio, S., Guaragnella, N.,
Corte-Real, M., Passarella, S., and Marra, E. (2005) Gene,
354, 93-98.
127.Burhans, W. C., and Weinberger, M. (2009)
Cell Cycle, 8, 2300-2302.
128.Guaragnella, N., Passarella, S., Marra, E., and
Giannattasio, S. (2010) FEBS Lett., 584, 3655-3660.
129.Mitsui, K., Nakagawa, D., Nakamura, M.,
Okamoto, T., and Tsurugi, K. (2005) FEBS Lett., 579,
723-727.
130.Mutoh, N., Kitajima, S., and Ichihara, S.
(2011) Biosci. Biotechnol. Biochem., 75, 1113-1118.
131.Sapienza, K., Bannister, W., and Balzan, R.
(2008) Microbiology, 154, 2740-2747.
132.Carmona-Gutierrez, D., Reisenbichler, A.,
Heimbucher, P., Bauer, M. A., Braun, R. J., Ruckenstuhl, C., Buttner,
S., Eisenberg, T., Rockenfeller, P., Frohlich, K. U., Kroemer, G., and
Madeo, F. (2011) Cell Cycle, 10, 3973-3978.
133.De Castro, P. A., Savoldi, M., Bonatto,
D., Barros, M. H., Goldman, M. H., Berretta, A. A., and Goldman, G. H.
(2011) Eukaryot. Cell, 10, 398-411.
134.Chahomchuen, T., Akiyama, K., Sekito, T.,
Sugimoto, N., Okabe, M., Nishimoto, S., Sugahara, T., and
Kakinuma, Y. (2009)'' J. Toxicol. Sci., 34, 541-545.
135.Ozhovan, S. M., Knorre, D. A., Severin, F. F.,
and Bakeeva, L. E. (2009) Tsitologiya, 51, 911-916.
136.Wu, J., Min, R., Wu, M., and Chen, W.
(2011) Mol. Med. Rept., 4, 357-362.
137.Stoodley, P., Sauer, K., Davies, D. G., and
Costerton, J. W. (2002) Annu. Rev. Microbiol., 56,
187-209.
138.Vopalenska, I., Hulkova, M., Janderova,
B., and Palkova, Z. (2005) Res. Microbiol., 156,
921-931.
139.Pereira, C., Silva, R. D., Saraiva, L.,
Johansson, B., Sousa, M. J., and Corte-Real, M. (2008) Biochim.
Biophys. Acta, 1783, 1286-1302.
140.Aerts, A. M., Zabrocki, P., Govaert, G.,
Mathys, J., Carmona-Gutierrez, D., Madeo, F., Winderickx, J., Cammue,
B. P., and Thevissen, K. (2009) FEBS Lett., 583,
113-117.
141.Kissova, I., Plamondon, L. T., Brisson, L.,
Priault, M., Renouf, V., Schaeffer, J., Camougrand, N., and Manon, S.
(2006) J. Biol. Chem., 281, 36187-36197.
142.Pineau, L., and Ferreira, T. (2010) FEMS
Yeast Res., 10, 1035-1045.
143.Sousa, M. J., Azevedo, F., Pedras, A., Marques,
C., Coutinho, O. P., Preto, A., Geros, H., Chaves, S. R., and
Corte-Real, M. (2011) Biochem. Soc. Trans., 39,
1533-1537.
144.Sheridan, C., Delivani, P., Cullen, S. P., and
Martin, S. J. (2008) Mol. Cell, 31, 570-585.
145.Yamaguchi, R., Lartigue, L., Perkins, G.,
Scott, R. T., Dixit, A., Kushnareva, Y., Kuwana, T., Ellisman, V. Y.,
and Newmeyer, D. D. (2008) Mol. Cell, 31, 557-569.
146.Colin, J., Garibal, J., Mignotte, B., and
Guenal, I. (2009) Biochem. Biophys. Res. Commun., 379,
931-943.
147.Lucken-Ardjomande, S., Montessuit, S., and
Martinou, J. C. (2008) Cell Death Differ., 15,
923-937.
148.Bernardi, P., Krauskopf, A., Basso, E.,
Petronilli, V., Blachly-Dyson, E., Di Lisa, F., and Forte, M. A. (2006)
FEBS J., 273, 2077-2099.
149.Leung, A. W., and Halestrap, A. P. (2008)
Biochim. Biophys. Acta, 1777, 946-952.
150.Halestrap, A. P. (2009) J. Mol.
Cardiol., 46, 821-831.
151.Sultan, A., and Sokolove, P. (2001) Arch.
Biochem. Biophys., 386, 52-61.
152.Mironova, G. D., Gateau-Roesch, O., Levrat, C.,
Gritsenko, E., Pavlov, E., Lazareva, A. V., Limarenko, E., Rey, P.,
Louisot, P., and Saris, N.-E. L. (2001) J. Bioenerg. Biomembr.,
33, 319-331.
153.Kristian, T., Bernardi, P., and Siesjo, B. K.
(2001) J. Neurotrauma, 18, 1059-1074.
154.Knorre, D. A., Dedukhova, V. I., Vyssokikh, M.
Y., and Mokhova, E. N. (2003) Biosci. Rep., 23,
67-75.
155.Chavez, E., Moreno-Sanchez, R., Zazueta, C.,
Rodriguez, J. S., Bravo, C., and Reyes-Vivas, H. (1997) J. Bioenerg.
Biomembr., 29, 571-577.
156.Kuzminova, A. E., Zhuravlyova, A. V.,
Vyssokikh, M. Yu., Zorova, L. D., Krasnikov, B. F., and Zorov, D. B.
(1998) FEBS Lett., 434, 313-316.
157.Kovaleva, M. V., Sukhanova, E. I., Trendeleva,
T. A., Zyl’kova, M. V., Ural’skaya, L. A., Popova, K. M.,
Saris, N. E., and Zvyagilskaya, R. A. (2009) J. Bioenerg.
Biomembr., 41, 239-249.
158.Kovaleva, M. V., Sukhanova, E. I., Trendeleva,
T. A., Popova, K. M., Zylkova, M. V., Uralskaya, L. A., and
Zvyagilsksya, R. A. (2010) Biochemistry (Moscow), 75,
297-303.
159.Trendeleva, T., Sukhanova, E.,
Ural’skaya, L., Saris, N.-E., and Zvyagilskaya, R. (2011) J.
Bienerg. Biomembr., 43, 623-631.
160.Trendeleva, T., Sukhanova, E.,
Ural’skaya, L., Saris, N.-E., and Zvyagilskaya, R. (2011) J.
Bienerg. Biomembr., 43, 633-644.
161.Holman, J. D., and Hand, S. C. (2009) J.
Exp. Mar. Bio. Ecol., 376, 85-93.
162.Prieto, S., Bouillaud, F., Ricquier, D., and
Rial, E. (1992) Eur. J. Biochem., 208, 487-491.
163.Prieto, S., Bouillaud, F., and Rial, E. (1995)
Biochem. J., 307, 657-661.
164.Prieto, S., Bouillaud, F., and Rial, E. (1996)
Arch. Biochem. Biophys., 334, 43-49.
165.Roucou, X., Manon, S., and Guerin, M.
(1997) Biochem. Mol. Biol. Int., 43, 53-61.
166.Manon, S., and Guerin, M. (1998) Biochem.
Mol. Biol. Int., 44, 565-575.
167.Manon, S., Roucou, X., Guerin, M., Rigoulet,
M., and Guerin, B. (1998) J. Bioenerg. Biomembr., 30,
419-429.
168.Gutierrez-Aguilar, M., Perez-Vazquez, V.,
Bunoust, O., Manon, S., Rigoulet, M., and Uribe, S. (2007) Biochim.
Biophys. Acta, 1767, 1245-1251.
169.Trendeleva, T., Sukhanova, E. I., Kovaleva, M.
V., Uralskaya, L. A., and Zvyagilskaya, R. A. (2011) Papers Int.
Conf. “Receptors and Intercellular Signaling”
(Zinchenko, V. P., Kolesnikova, S. S., and Berezhnova, A. V., eds.)
Pushchino, Vol. 2, pp. 732-737.
170.Madeo, F., Herker, E., Wissing, S., Jungwirth,
H., Eisenberg, T., and Fröhlich, K. U. (2004) Curr. Opin.
Microbiol., 7, 655-660.
171.Ludovico, P., Madeo, F., and Silva, M.
(2005) IUBMB Life, 57, 129-135.
172.Lisa-Santamaria, P., Neiman, A. M.,
Cuesta-Marban, A., Mollinedo, F., Revuelta, J. L., and Jimenez, A.
(2009) Biochim. Biophys. Acta, 1793, 561-571.
173.Silva, R. D., Manon, S., Goncalves, J.,
Saraiva, L., and Corte-Real, M. (2011) Curr. Pharm. Des.,
17, 246-255.
174.Flower, T. R., Chesnokova, L. S., Froelich, C.
A., Dixon, C., and Witt, S. N. (2005) J. Mol. Biol., 351,
1081-1100.
175.Bocharova, N., Chave-Cox, R., Sokolov, S.,
Knorre, D., and Severin, F. (2009) Biochemistry (Moscow),
74, 231-234.
176.Owsianowski, E., Walter, D., and Fahrenkrog, B.
(2008) Biochim. Biophys. Acta, 1783, 1303-1310.
177.Perrone, G. G., Tan, S. X., and Dawes, I. W.
(2008) Biochim. Biophys. Acta, 1783, 1354-1368.
178.Huh, G. H., Damsz, B., Matsumoto, T. K., Reddy,
M. P., Rus, A. M., Ibeas, J. I., Narasimhan, M. L., Bressan, R. A., and
Hasegawa, P. M. (2002) Plant J., 29, 649-659.