MINI-REVIEW: Longevity and Mitochondrial Membrane Potential
D. A. Knorre and F. F. Severin*
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: severin@genebee.msu.ru; severin@belozersky.msu.ru* To whom correspondence should be addressed.
Received March 14, 2012
In Saccharomyces cerevisiae yeast cells a decrease in the mitochondrial membrane potential caused by protonophores or by a loss of mitochondrial DNA leads to an increase in longevity (replicative life span). The loss of mitochondrial DNA also activates retrograde signaling that results in certain changes in transcription. Recently, Miceli and coauthors ((2011) Front. Genet., 2, 102) showed that retrograde response is triggered by a drop in the membrane potential. Independently, it has been shown that retrograde response activates autophagic mitochondrial degradation (mitophagy). Together, it suggests that activation of selective mitophagy increases lifespan by protecting cells from accumulation of damaged mitochondria in cells. Low concentrations of protonophores can be beneficial by increasing the accuracy of the mitophagosomal degradation of mitochondria with deleterious mutations in their DNA.
KEY WORDS: yeast, mitochondria, mitophagy, retrograde signaling, agingDOI: 10.1134/S0006297912070127
Induction of a mild decrease in mitochondrial membrane potential can be beneficial for cells and organisms. For example, low doses of the protonophore dinitrophenol increase the lifespan of yeast cells, rats [1, 2], and mice [3]. A possible reason for this is that high membrane potential can trigger generation of reactive oxygen species (ROS) by the respiratory chain. The latter was shown first on isolated mitochondria [4] and later confirmed on intact Saccharomyces cerevisiae yeast cells [5].
A recent paper of Miceli et al. [6] suggests another explanation for the beneficial effects. Saccharomyces cerevisiae cells can survive without mitochondrial DNA (rho0 form, as opposed to Rho+). The rho0 cells grow slower than Rho+, but they are more resistant to a variety of harsh treatments and display increased replicative lifespan (the number of daughter cells produced by one mother). This is due to activation of retrograde response: dysfunctional mitochondria signal to the nucleus triggering certain transcriptional changes (see [7] for review). The paper of Miceli et al. [6] shows that activation of the retrograde pathway in rho0 cells is triggered by a drop in the membrane potential. To generate membrane potential, rho0 cells use combined work of F1 subunit of ATP-synthase and ADP/ATP antiporter instead of the respiratory chain. As a result, the level of the membrane potential in rho0 is much lower than in Rho+. The authors introduced a mutation in such cells that allowed partial reversal of the membrane potential decrease. The mutation (atp1-111) of ATP synthase partially suppresses petite phenotype and increases mitochondrial membrane potential. This increase inhibits both the retrograde pathway activation and the lifespan increase.
What is the connection between activation of retrograde response and lifespan increase? Recently, it has been found that retrograde signaling is linked to the process of mitochondrial degradation – mitophagy [8]. It is possible that activation of selective mitophagy protects yeast cells from propagation of mitochondria with low membrane potential (which may be a result of deleterious mutations in the mitochondrial genome) and in this way acts to increase lifespan. If this is the case, addition of low doses of protonophores is likely to strengthen such a selection (figure). Together, it suggests that even moderate lowering of mitochondrial membrane potential can result in retrograde signaling activation and lifespan increase.
Mammalian cells also have a mechanism of mitochondria-to-nucleus signaling. The mechanism is activated by mitochondrial malfunction [9]. Calcium release induced by mitochondrial depolarization (permeability transition pore opening) is a possible trigger of this signaling pathway. Ichas et al. [10] suggested that due to this process mitochondria can be regarded as an excitable organelle. At the same time, animal cells lack clear orthologs of yeast retrograde pathway proteins that are conserved in fungi. Mammalian mitophagy is directly related to mitochondria membrane potential: a drop in the potential slows the transport of PINK1 protein into mitochondria, causing re-localization of parkin (PINK1-associated protein) to the mitochondrial membrane [11]. Parkin is a ubiquitin-ligase, thus such a re-localization causes ubiquitination of mitochondrial proteins, leading to autophagic degradation of the mitochondria (mitophagy). In turn, selective mitophagy could counterpart the process of age-dependent accumulation of adverse mutations in mitochondrial DNA of somatic cells (figure).
Schematic representation of possible relationships between mitochondrial membrane potential, selective mitophagy, and aging
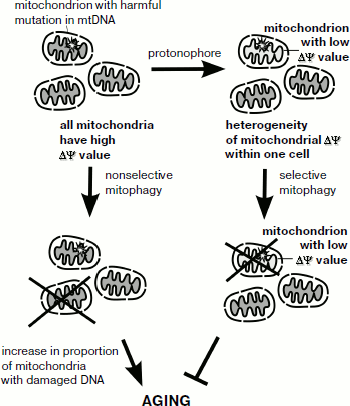
Therefore, similar to yeast, mild changes in mitochondrial membrane potential have long-term consequences for mammalian cells. Regardless of the sensing mechanism, it becomes clear that by decreasing the membrane potential one can not only prevent mitochondrial ROS production, but also induce potentially beneficial changes in transcription (retrograde response). Here it is important to mention that lowering the level of the potential below a critical value is damaging for mitochondria. Therefore, the aforementioned findings widen the area for medical application of mild (membrane potential-dependent) protonophores [12]. Finally, within the scope of this journal issue, it is tempting to speculate that a minor rise in the mitochondrial membrane potential can serve as an intermediate of a phenoptotic program [13].
This work was supported by the Russian Foundation for Basic Research grant 12-04-01412.
REFERENCES
1.Barros, M. H., Bandy, B., Tahara, E. B., and
Kowaltowski, A. J. (2004) J. Biol. Chem., 279,
49883-49888.
2.Tainter, M. L. (1938) J. Pharmacol. Exp.
Ter., 63, 51-57.
3.Caldeira da Silva, C. C., Cerqueira, F. M.,
Barbosa, L. F., Medeiros, M. H., and Kowaltowski, A. J. (2008) Aging
Cell, 7, 552-560.
4.Korshunov, S. S., Skulachev, V. P., and Starkov, A.
A. (1997) FEBS Lett., 416, 15-18.
5.Pozniakovsky, A. I., Knorre, D. A., Markova, O. V.,
Hyman, A. A., Skulachev, V. P., and Severin, F. F. (2005) J. Cell
Biol., 168, 257-269.
6.Miceli, M. V., Jiang, J. C., Tiwari, A.,
Rodriguez-Quinones, J. F., and Jazwinski, S. M. (2011) Front
Genet., 2, 102.
7.Liu, Z., and Butow, R. A. (2006) Annu. Rev.
Genet., 40, 159-185.
8.Journo, D., Mor, A., and Abeliovich, H. (2009)
J. Biol. Chem., 284, 35885-35895.
9.Biswas, G., Guha, M., and Avadhani, N. G. (2005)
Gene, 354, 132-139.
10.Ichas, F., Jouaville, L. S., and Mazat, J. P.
(1997) Cell, 89, 1145-1153.
11.Jin, S. M., Lazarou, M., Wang, C., Kane, L. A.,
Narendra, D. P., and Youle, R. J. (2010) J. Cell. Biol.,
191, 933-942.
12.Severin, F. F., Severina, I. I., Antonenko, Y.
N., Rokitskaya, T. I., Cherepanov, D. A., Mokhova, E. N., Vyssokikh, M.
Y., Pustovidko, A. V., Markova, O. V., Yaguzhinsky, L. S., Korshunova,
G. A., Sumbatyan, N. V., Skulachev, M. V., and Skulachev, V. P. (2010)
Proc. Natl. Acad. Sci. USA, 107, 663-668.
13.Skulachev, V. P. (2012) Biochemistry
(Moscow), 77, 689-706.