Oxidative Phosphorylation and Respiratory Control Phenomenon in Paracoccus denitrificans Plasma Membrane
T. V. Zharova and A. D. Vinogradov*
Department of Biochemistry, Faculty of Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (495) 939-1376; E-mail: adv@biochem.bio.msu.su* To whom correspondence should be addressed.
Received April 18, 2012; Revision received June 1, 2012
Changes in respiratory activity, transmembrane electric potential, and ATP synthesis as induced by additions of limited amounts of ADP and Pi to tightly coupled inverted (inside-out) Paracoccus denitrificans plasma membrane vesicles were traced. The pattern of the changes was qualitatively the same as those observed for coupled mitochondria during the classical State 4–State 3–State 4 transition. Bacterial vesicles devoid of energy-dependent permeability barriers for the substrates of oxidation and phosphorylation were used as a simple experimental model to investigate two possible mechanisms of respiratory control: (i) in State 4 phosphoryl transfer potential (ATP/ADP × Pi) is equilibrated with proton-motive force by reversibly operating F1·Fo-ATPase (thermodynamic control); (ii) in State 4 apparent “equilibrium” is reached by unidirectional operation of proton motive force-activated F1·Fo-ATP synthase. The data support the kinetic mechanism of the respiratory control phenomenon.
KEY WORDS: FoF1-H+-ATP synthase, oxidative phosphorylation, bacterial plasma membrane, Paracoccus denitrificansDOI: 10.1134/S0006297912090064
Abbreviations: ΔµH+, transmembrane electrochemical gradient of protons; F1 and Fo, hydrophilic and hydrophobic parts of ATP synthase, respectively; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; p, proton motive force (p = Δψ − ΔpH) where Δψ is a transmembrane electric potential and ΔpH is pH difference between inner (matrix) and outer space of mitochondria.
An adult human being consumes about 10 mmol of oxygen per minute in the
resting state (basal metabolic rate), and this value increases about
10-fold upon moderate physical exercise [1]. Since
most of the oxygen consumption is due to the respiratory activity of
mitochondria, a mechanism must exist which increases or decreases their
respiration depending on the energy demand. Such a mechanism, called
respiratory control, was originally observed in 1939 by Belitzer [2, 3] on minced muscle tissues.
Thirteen years later, Lardy and Wellman, using Warburg’s
manometric technique, described 5-15-fold stimulation of rat liver
mitochondrial oxygen consumption by ADP in the presence of inorganic
phosphate and a hexokinase trap [4]. The concept of
respiratory control was further developed qualitatively and
quantitatively in classical papers by Chance and Williams, where
reversible stimulation of respiration (measured by oxygen-sensitive
electrode) [5] and redox-state transitions of the
respiratory chain components [6] in response to the
addition of limited amounts of ADP (and Pi) to mitochondria
were analyzed. Since then measurement of respiratory control (ratio
between the rates of respiration in the presence (State 3) and absence
(State 4) of ADP) have become routine characteristics of isolated
mitochondria. The addition of a limited amount of ADP (apparent
“Km” is in µmolar range) to
tightly coupled mitochondria incubated aerobically in the presence of
an excess of the oxidizable substrate and Pi (State 4)
results in 2-20-fold (depending on particular preparation of
mitochondria), transitory increase of oxygen consumption rate, which
declines upon the conversion of ADP to ATP. This phenomenon provides a
simple explanation for the strong dependence of physiological
respiration on energy demand [1] and suggests that
ADP is a key regulatory factor of oxidative metabolism.
In light of current spectacular achievements in bioenergetics [7], the respiratory control phenomenon is now conventionally described in well-defined terms. Free energy of substrate oxidation is accumulated as proton motive force (p = Δψ − ΔpH) across the inner mitochondrial membrane, built up by operation of three proton-pumping respiratory chain complexes: p is used by F1·Fo-ATPase/synthase to accumulate free energy in the phosphoryl group transfer potential of ATP. When no ADP and Pi are available, F1·Fo does not turn over, and the oxidative capacity of the respiratory chain is limited by the leakage of protons across the coupling membrane. In the presence of ADP and Pi, the proton-translocating activity of F1·Fo results in increase in proton flow, thus allowing an increase in the proton-pumping activity of the respiratory chain.
Tightly coupled mitochondria, which show high respiratory control, provide naturally the preparation of choice for studies on regulation of respiration by ADP and/or Pi. It should be emphasized, however, that several enzymatic activities in addition to those of the respiratory chain components and F1·Fo, are involved in the overall process of State 3 or State 4 respiration of intact mitochondria. These are: (i) translocation of the respiratory substrate into the mitochondrial matrix; (ii) operation of matrix-located dehydrogenases; (iii) Pi/OH– exchange [8]; and (iv) adenine nucleotide translocation (ADP/ATP exchanger) [9]. A number of studies aimed toward defining the important parameters in the net ATP synthesis as related to the respiratory activity have been carried out, and two models have been extensively discussed. According to the first, the rate of respiration is a function of ADP availability [5, 10] or external ATP/ADP ratio [11] (kinetic control of respiration realized by the adenine nucleotide translocase). The other model postulates that extramitochondrial ATP/ADP × Pi ratio is the parameter that controls the rate of oxygen consumption (“thermodynamic” control of respiration) [12, 13] (see [14] for comprehensive discussion). Because the intramitochondrially (matrix) located F1·Fo-ATPase/synthase is an immediate device directly interacting with p, it seems of obvious importance to know how this extremely complex molecular machine operates at different ATP, ADP, and Pi concentrations and variable p in terms of the kinetic and thermodynamic parameters of ATP synthesis or hydrolysis.
Inside-out submitochondrial particles devoid of permeability barriers, a system where enzymes (i)-(iv) do not operate, although being capable of ATP synthesis, do not show ADP-induced reversible State 4–State 3–State 4 transition. Inverted (inside out) plasma membrane vesicles derived from Paracoccus denitrificans grown on succinate and nitrate are capable of ATP synthesis [15] and show the classical respiratory control phenomenon [16]. Some properties of P. denitrificans ATPase and ATP synthase activities have been described in previous reports from this laboratory [17-20]. In this paper we describe and analyze the phenomenon of respiratory control in this simple system where the respiratory chain and F1·Fo-ATP synthase machinery are the only players in the overall oxidative phosphorylation reaction. The results show complex kinetic control of ATP synthesis catalyzed by F1·Fo.
MATERIALS AND METHODS
Chemicals. ADP, Hepes, malonate, succinate, MgCl2, EDTA, sucrose, FCCP, gramicidin, and pyruvate kinase were from Sigma-Aldrich (USA), venturicidin was from A.G. Scientific Inc. (USA), and other chemicals were of the highest purity commercially available.
Rat heart mitochondria were prepared essentially as described [21].
Preparation of bacterial vesicles. Paracoccus denitrificans cells (strain Pd 1222) were grown anaerobically in the presence of succinate and nitrate. Tightly coupled plasma membrane vesicles were prepared essentially as described by John and Whatley with some modifications [16]. The final preparations were suspended in 0.25 M sucrose, 10 mM Tris-acetate (pH 7.3), 1 mM MgCl2, and 0.1 mM malonate (protein concentration ~20 mg/ml) and stored in liquid nitrogen. Protein content was determined by the biuret procedure. The respiratory control ratio, measured as the ratio of NADH oxidase activity in the presence and absence of uncoupler (gramicidin (0.05 μg/ml) and ammonium acetate (15 mM)) for different preparations, varied from 4.5 to 6.0. The content of inside-out vesicles in the preparations determined as the ratio of NADH oxidase activity in the presence and absence of alamethicin [22] was 80-90%.
Synthesis of ATP. ATP synthesis was measured by continuous registration of hydrogen ion concentration by a glass electrode according to the equation:
ADP3(Mg2+) + Pi2 + H+ ↔ ATP4(Mg2+) + H2O. (1)
When succinate is used as the respiratory substrate, its oxidation by oxygen does not result in any pH change because the pKa values of succinate and fumarate carboxylic group are almost the same, and thus the observed pH change is due to reaction (1) only.
The stoichiometry [H+]/[ATP] at pH 8.0 in the presence of Mg2+ (25-fold excess over nucleotides) of 1.0 [23] was checked by measurement of ADP with phosphoenol pyruvate and pyruvate kinase. The photometric registration of pH change using Phenol Red as indicator was found unsatisfactory in the assay system employed (data will be reported elsewhere). ATP synthesis measured as H+ consumption was completely sensitive to the uncouplers, malonate (excess), and venturicidin.
Respiratory activity. Respiratory rates were measured amperometrically with a covered platinum electrode.
Membrane potential was followed by safranine response [24] (intact mitochondria) or Oxonol VI response [25] (inside-out vesicles). The standard reaction mixture was composed of 0.25 M sucrose, 20 mM potassium chloride, 1 mM Hepes (pH 8.0), 5 mM MgCl2, 0.1 mM EDTA, and 2.5 mM succinate. Other additions are indicated in the legends to the figures and table. All experiments were performed at 30°C.
Note should be made on the statistics. The procedure employed for P. denitrificans coupled vesicles is well reproducible, although the absolute values for the respiratory activities and, particularly, for the respiratory control and rates of oxidative phosphorylation were slightly variable among different batches. The data shown in the figures should be considered as representative ones, and they were qualitatively reproducible when repeated using preparations derived from several culture batches. We feel that statistical treatment of the data is not required for the conclusion made.
RESULTS
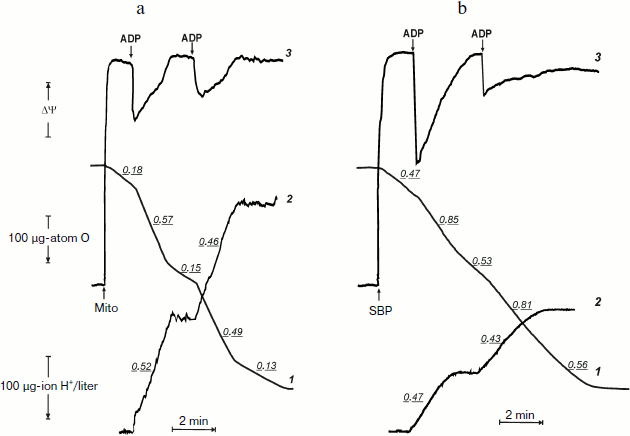
Before the kinetics of oxidative phosphorylation in the system where F1·Fo is directly accessible for the substrates will be discussed, it seems worthwhile to compare actual tracing of respiration, phosphorylation, and membrane potential as they appear in the experiments carried out using coupled rat heart mitochondria and inside-out P. denitrificans plasma membrane vesicles. Figures 1a and 1b demonstrate synchronous recording of these parameters for mitochondria (classical well-known pattern) and for P. denitrificans inside-out system, respectively. Mitochondria (Fig. 1a) oxidized succinate (in the presence of rotenone) at a slow rate, which was increased 3-4-fold when 200 µM ADP was added (curve 1). The stimulating effect of ADP was accompanied by a drop in the membrane potential (curve 3) and immediate (within the time resolution scale) initiation of ATP synthesis, as was evident from alkalization of the medium (curve 2). These cyclic responses to the addition of ADP could be repeated after the respiration decreased and the membrane potential returned to the original level when a limited amount of ADP was converted to ATP (State 4). The same pattern was seen for inside-out particles, although the stimulatory effect of ADP on respiration (State 4–State 3–State 4 transition) was not as prominent as in mitochondria (Fig. 1b). It was, however, adequate for analysis of the rate of ATP synthesis and, more importantly, the steady-state level of ATP/ADP during State 4 respiration.
The latter (ATP/ADP) ratio was estimated from the ratio H+ (scalar) consumed per ADP added during the State 3–State 4 transition. The values found for the experiments shown in Fig. 1b (curve 2) at different ADP concentrations were close to 15. Moreover, the same ratio was found for repeated cycles of the State 3–State 4 transition, i.e. under conditions where phosphorylation was initiated by limited ADP when ATP was formed by the previous cycle. The proton consumption assay was then verified by direct determination of remaining ADP in State 4 with the phosphoenolpyruvate/pyruvate kinase trap. A comparison of the data is shown in table. The complete (or almost complete) phosphorylation of added ADP argued by itself against thermodynamic equilibration between p and the external phosphoryl potential. The concentrations of ADP and Pi are equal terms in the equation describing the actual free energy accumulated in ATP (ΔGp = ΔGp0 + 2.3RT·log(ATP/ADP × Pi), where ΔGp0 is the standard free energy of ATP hydrolysis). If p in State 4 is equal to ΔGp, the ratio ATP/ADP is expected to vary as a function of Pi concentration. The results presented in the table show that this was not the case. Equal amounts of ATP were formed at 10-fold different Pi concentrations.
Oxidative phosphorylation catalyzed by P. denitrificans vesicles (pH 8.0, 30°C)*Fig. 1. Oxidative phosphorylation catalyzed by rat heart mitochondria (a) and P. denitrificans plasma membranes (b). a) Rat heart mitochondria (Mito, 0.2 mg/ml) were added to the reaction mixture comprised of 0.25 M sucrose, 10 mM KCl, 5 mM potassium phosphate (pH 7.5), 0.1 mM EDTA, 2.5 mM potassium succinate, 1 µM rotenone, and 10 µM safranine. ADP (200 µM) was added where indicated. Curve 1-3 show oxygen consumption, hydrogen ion consumption, and safranine response, respectively. Underlined figures on curves 1 and 2 indicate the rates of oxygen consumption and ATP synthesis, respectively (µmoles/min per mg protein). b) P. denitrificans plasma membrane vesicles (SBP, 250 µg/ml) were added to the reaction mixture comprised of 0.25 M sucrose, 20 mM KCl, 5 mM potassium phosphate, 1 mM Hepes (pH 8.0), 5 mM MgCl2, 0.1 mM EDTA, 2.5 mM potassium succinate, and 1.5 µM Oxonol VI. ADP (100 µM) was added where indicated. Curve 3 is the Oxonol VI response, other curves as in (a).

* For assay mixture, see “Material and Methods” and legend to Fig. 1b.
** Determined by phosphoenolpyruvate/pyruvate kinase trap.
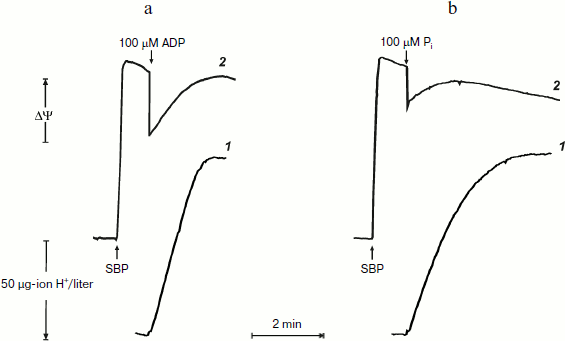
Remarkably similar patterns of ATP synthesis and change in the membrane potential were seen when phosphorylation was initiated by limited amount of either ADP or Pi if the counterpart substrate was present in excess (Fig. 2). The only difference was that when the reaction was initiated by 100 µM Pi, the phosphorylation proceeded in a close to first-order process. This is expected, because the apparent Km for Pi is in the range of 60-120 µM [20].
It was of interest to look more closely at the kinetics of ATP synthesis and change in the membrane potential (Oxonol VI response) as induced by ADP addition. Figure 3 shows that ATP synthesis proceeds as a zero-order reaction during ADP (100 µM) phosphorylation up to about 20% of the remaining ADP. A more complex and unexpected pattern of the membrane potential was seen: the initial drop induced by the ADP addition started to be restored during phosphorylation. In other words, ATP synthesis proceeded at constant rate at variable membrane potential. It should be noted that only the electrical component of p (Δψ) was traced, and we do not know what ΔpH change (if any) occurred during the transition.Fig. 2. ATP synthesis by P. denitrificans vesicles (140 µg/ml) and membrane potential change during ATP synthesis initiated by ADP (a) or inorganic phosphate (b). Potassium phosphate (0.5 mM) (a) and 0.5 mM ADP (b) were present in the reaction mixture (other components were as in Fig. 1b). Curves 1 and 2 are hydrogen ion consumption and Oxonol VI response, respectively.
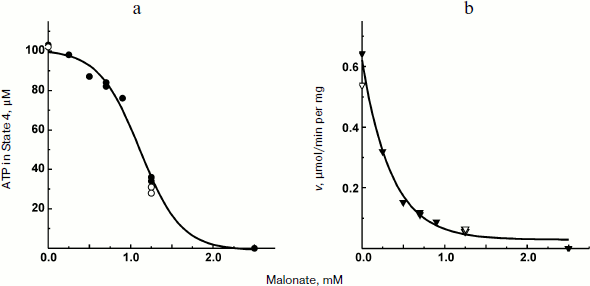
The experiments shown in Figs. 1 and 2a were carried out in the presence of “kinetic” and “thermodynamic” excess of Pi. A peculiar effect of Pi on proton-translocating activity of P. denitrificans F1·Fo was described in our previous publications [17, 19]. It is well known that Pi does not inhibit ATPase activity catalyzed by either F1 or F1·Fo at concentrations, which are orders of magnitude higher than its apparent Km for oxidative phosphorylation (see [26], however, for the opposite). Moreover, the energy-dependent binding of Pi is required for the proton-translocating ATPase activity of F1·Fo [19, 27]. To gain insight into these puzzling phenomena, the effect of Pi on ATP synthesis was further investigated. The rate of ATP synthesis as expected depended on Pi concentration as shown in Fig. 4a, with apparent Km of about 70 µM, in accord with our previously reported data [20]. More interesting, Pi was quantitatively consumed during oxidative phosphorylation: the amount of ATP formed was equal to that of Pi added if ADP was present in excess (Fig. 4b). Considered together, the data shown in the table and Fig. 4 suggest that under the conditions employed, F1·Fo catalyzes p-dependent irreversible synthesis of ATP from ADP and Pi. The possibility exists that the magnitude of p under the experimental conditions employed (aerobic succinate oxidation by coupled vesicles) was always significantly higher than that required to poise a detectable equilibrium between the substrate/product of reversibly operating ATPase/synthase. This possibility was scrutinized by the experiment where the respiration-supported p was gradually decreased by malonate, a competitive inhibitor of succinate oxidation (Fig. 5). No substantial decrease of the State 4 ATP level was seen when respiration was gradually decreased upon increase in malonate concentration up to about 1 mM (Fig. 5a), i.e. within the range where the rate of phosphorylation was significantly inhibited (Fig. 5b).Fig. 3. Hydrogen ion consumption (curve 1) and membrane potential change (Oxonol VI response, curve 2) during oxidative phosphorylation catalyzed by P. denitrificans membranes (110 µg/ml). The reaction mixture composition was as in Fig. 1b.
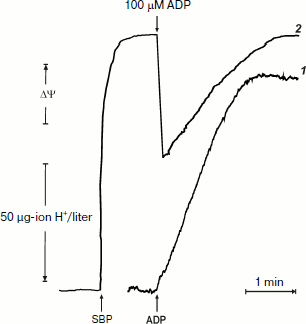
Fig. 4. Oxidative phosphorylation catalyzed by P. denitrificans membranes (150 µg/ml) as a function of Pi concentration. a) Initial rate of ATP synthesis (the insert is a double-reciprocal plot); b) amount of ATP formed in State 4.
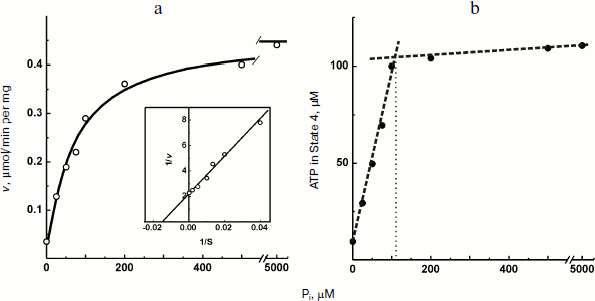
Fig. 5. State 4 ATP level (a) and rate of oxidative phosphorylation (b) during limited respiratory activity. Phosphorylation was initiated by the addition of 113 µM ADP to the reaction mixture comprised as in the Fig. 1b. Closed and open symbols indicate that 5 or 0.5 mM Pi was present, respectively.
DISCUSSION
It was shown many years ago that membrane vesicles derived from P. denitrificans show very low ATPase activity compared with their capacity to synthesize ATP, and irreversibility of F1·Fo operation during NADH oxidation supported ATP synthesis has been suggested [28]. More recently, we have shown that active proton-translocating ATPase in coupled P. denitrificans vesicles can be induced by the energization of the membrane [17, 18]. ATP hydrolysis catalyzed by the energized ATPase rapidly declines if p is abolished by an uncoupler [18]. The requirement of p for ATPase activity was shown to be brought about by energy-linked Pi binding [19] that prevents formation of ADP(Mg2+)-inhibited enzyme, a dead-end intermediate and a central species accounting for very complex kinetics of ATP hydrolysis [29, 30]. We have emphasized that F1·Fo ATPase is both p-generating and p-consuming machinery that uses free energy of the catalyzed reaction to maintain its catalytically competent state [31]. Thus, a priori it seems unlikely that F1·Fo is a reversibly operating enzyme equilibrating p and phosphoryl group transfer potential of ATP as it is widely stated in numerous reviews and in some textbooks. The energy-dependent transformation of F1·Fo is a characteristic phenomenon described for chloroplast [32], bacterial [33], and mammalian [34] enzymes.
The data obtained in his study confirm and extend the original report by Ferguson et al. on ATPase as an irreversible component of electron transfer supported ATP synthesis [28]. A limited amount of ADP was almost completely (more than 90%) transformed into ATP when Pi was present in excess (Fig. 2a) and, a limited amount of Pi was also quantitatively incorporated into ATP when ADP was in excess (Fig. 2b). Moreover, 10-fold decrease in Pi concentration did not change the amount of ATP formed from limited ADP (the table). These results do not agree with a paradigm describing State 4 as a state where p is equilibrated with ATP phosphoryl potential (p = ΔGp0 + RTln(ATP/ADP × Pi)). In agreement with our previously reported data [20], the rate, not the amount of ATP formed, was decreased (in a limited range) when p-generating succinate oxidation was gradually inhibited by malonate (Fig. 5). It worth noting that when the rate of ATP synthesis was slow, the true State 4 value for ADP consumed could not be accurately measured because the vesicles become spontaneously uncoupled if incubated for a long time under p-generating conditions (unpublished observation from this laboratory).
Transitory change of Δψ induced by initiation of ATP synthesis is unexpected and merits brief discussion. As shown in Fig. 2, the addition of ADP causes an immediate drop in the membrane potential followed by its rapid restoration, whereas ATP synthesis started and proceeded at a constant rate up to the level of the remaining ADP close to or less than its Km. This unexpected pattern is hard to explain in terms of a simple model where ATP-synthesizing F1·Fo operates just as a proton-conducting load for the respiration-driven p generator.
If possible artifacts in Δψ quantitation by Oxonol VI response are to be excluded, several explanations can be offered. A drop in Δψ induced by an increased load may be followed by an immediate compensation by a putative ATP-dependent (activated) K+/H+-exchanger, which would increase Δψ along with formation of ATP. Another more likely possibility is that there is a p- and ATP/ADP ratio-dependent interplay between ATP synthase and proton-translocating ATPase conformations of F1·Fo during the time course of oxidative phosphorylation. Accumulating evidence suggests that two interconvertible conformational states of F1·Fo, ATP synthase and ATP hydrolase exist, and that ATP synthesis is not the reversal of ATP hydrolysis [30, 35-37]. A number of effectors modulating the hydrolytic activity are known, these are: tightly bound inhibitory ADP(Mg2+) [29, 30], Pi [19, 27], inhibitory ATP-binding ε-subunit [38], protein inhibitor [39-41], activating p, and recently discovered in P. denitrificans ξ-subunit [42] (see [43] for review). Their interplay in the control of ATP synthase/ATP hydrolase activities of F1·Fo is complex and poorly understood, and further studies in this direction are needed.
Perhaps the most important conclusion from the original [28] and this study is that when p and the substrates of oxidative phosphorylation are available, P. denitrificans F1·Fo catalyzes synthesis of ATP irreversibly. If the binding-change rotary mechanism of F1·Fo is operative (note should be made that no evidence for rotation in the p-dependent ATP synthase reaction are available), the rotation proceeds unidirectionally as a ratchet-and-pawl mechanism, a device widely used in many man-made machines. It remains to be established what particular part(s) of the enzyme serves as a ratchet (specific hydrogen bond arrangements of γ and α-β subunit interaction [44]) and as a pawl (P. denitrificans ξ-subunit [42], protein inhibitor [39-41], inhibitory ADP(Mg2+) [29, 30]). Certainly such a device is not able to violate constrains of thermodynamics as it has been elegantly discussed by R. Feynman [45]. It is conceivable that other than bulk p force(s) is(are) involved in ATP synthesis, as has been discussed many years ago [46]. Also, variation of H+ translocated/ATP synthesized stoichiometry upon variation of p cannot be excluded. Whatever the mechanical arrangement that furnishes the one-way operation of the enzyme are, a simple model of Fo·F1 as an enzyme reversibly equilibrating free energy of ATP hydrolysis and p does not hold at least for the P. denitrificans system. Whether this is true for Fo·F1 from other sources remains to be experimentally tested.
The authors thank Ms. A. V. Kareyeva for her valuable help in preparation of rat heart mitochondria. We thank Dr. T. Yu. Lipskaya for critical reading the manuscript.
This work was supported by the Russian Foundation for Basic Research (grant 11-04-00916).
REFERENCES
1.Haldane, J. S., and Pries, J. G. (1935)
Respiration, New Haven, Yale University Press.
2.Belitzer, V. A. (1939) Biokhimiya, 4,
408-501.
3.Belitzer, V. A., and Tzibakova, E. T. (1939)
Biokhimiya, 4, 516-535.
4.Lardy, H. A., and Wellman, H. (1952) J. Biol.
Chem., 195, 215-224.
5.Chance, B., and Williams, G. R. (1955) J. Biol.
Chem., 217, 383-393.
6.Chance, B., and Williams, G. R. (1955) J. Biol.
Chem., 217, 409-427.
7.Nicholls, D. G., and Ferguson, S. J. (2002)
Bioenergetics 3, Academic Press, London.
8.Wohlrab, H. (1986) Biochim. Biophys. Acta,
853, 115-134.
9.LaNoue, K., Mizani, S. M., and Klingenberg, M.
(1978) J. Biol. Chem., 253, 191-198.
10.Jacobus, W. E., Moreadith, R. W., and Vandegaer,
K. M. (1982) J. Biol. Chem., 257, 2397-2402.
11.Kuster, U., Bohnensack, R., and Kunz, W. (1976)
Biochim. Biophys. Acta, 440, 391-402.
12.Klingenberg, M., and Schollmeyer, P. (1963) in
Proc. 5th Int. Congr. Biochemistry, Moscow, 1961, Pergamon
Press, Oxford, Vol. 5, pp. 46-68.
13.Holian, A., Owen, C. S., and Wilson, D. F. (1977)
Arch. Biochem. Biophys., 181, 164-171.
14.Erecinska, M., and Wilson, D. F. (1982) J.
Membr. Biol., 70, 1-14.
15.John, P., and Whatley, F. R. (1970) Biochim.
Biophys. Acta, 216, 342-352.
16.John, P., and Hamilton, W. A. (1970) FEBS
Lett., 10, 246-248.
17.Zharova, T. V., and Vinogrodov, A. D. (2003)
Biochemistry (Moscow), 68, 1101-1108.
18.Zharova, T. V., and Vinogrodov, A. D. (2004)
J. Biol. Chem., 279, 12319-12324.
19.Zharova, T. V., and Vinogrodov, A. D. (2006)
Biochemistry, 45, 14552-14558.
20.Kegyarikova, K. A. Zharova, T. V., and
Vinogradov, A. D. (2010) Biochemistry (Moscow), 75,
1264-1271.
21.Jacobus, W. E., and Saks, V. A. (1982) Arch.
Biochem. Biophys., 219, 167-78.
22.Gostimskaya, I. S., Grivennikova, V. G., Zharova,
T. V., Bakeeva, L. E., and Vinogradov, A. D. (2003) Anal.
Biochem., 313, 46-52.
23.Chance, B., and Nishimura, M. (1967) Meth.
Enzymol., 10, 641-650.
24.Akerman, K. E., and Wikstrom, M. K. (1976)
FEBS Lett., 68, 191-197.
25.Waggoner, A. S. (1979) Meth. Enzymol.,
55, 689-695.
26.D’Alessandro, M., Turina, P., and Melandri,
B. A. (2011) Biochim. Biophys. Acta, 1807, 130-143.
27.Feniouk, B. A., Suzuki, T., and Yoshida, M.
(2007) J. Biol. Chem., 282, 764-772.
28.Ferguson, S. J., John, P., Lloyd, W. J., Radda,
G. K., and Whatley, F. R. (1976) FEBS Lett., 62,
272-275.
29.Vasilyeva, E. A., Fitin, A. F., Minkov, I. B.,
and Vinogradov, A. D. (1980) Biochem. J., 188,
807-815.
30.Vinogradov, A. D. (2000) J. Exp. Biol.,
203, 41-49.
31.Zharova, T. V., and Vinogradov, A. D. (1997)
Biochim. Biophys. Acta, 1320, 256-264.
32.Junge, W. (1970) Eur. J. Biochem.,
14, 582-592.
33.Fischer, S., Graber, P., and Turina, P. (2000)
J. Biol. Chem., 275, 30157-30162.
34.Galkin, M. A., and Vinogradov, A. D. (1999)
FEBS Lett., 448, 123-126.
35.Syroeshkin, A. V., Vasilyeva, E. A., and
Vinogradov, A. D. (1995) FEBS Lett., 366, 29-32.
36.Suzuki, T., Murakam, T., Iino, R., Suzuki, J.,
Ono, S., Shirakihara, Y., and Yoshida, M. (2003) J. Biol. Chem.,
278, 46840-46846.
37.Haagsma, A. C., Driessen, N. N., Hahn, M. M.,
Lill, H., and Bald, D. (2010) FEMS Microbiol. Lett., 313,
68-74.
38.Feniouk, B. A., Suzuki, T., and Yoshida, M.
(2006) Biochim. Biophys. Acta, 1757, 326-338.
39.Panchenko, M. V., and Vinogradov, A. D. (1989)
Biokhimiya, 54, 569-579.
40.Vasil’eva, E. A., Panchenko, M. V., and
Vinogradov, A. D. (1989) Biokhimiya, 54, 1490-1498.
41.Zanotti, F., Gnoni, A., Mangiullo, R., and Papa,
S. (2009) Biochem. Biophys. Res. Commun., 384, 43-48.
42.Morales-Rios, E., de la Rosa-Morales, F.,
Mendoza-Hernandez, G., Rodriiguez-Zavala, J. S., Celis, H.,
Zarco-Zavala, M., and Garcia-Trejo, J. J. (2010) FASEB J.,
24, 599-608.
43.Feniouk, B. A., and Yoshida, M. (2008) Results
Prob. Cell Differ., 45, 279-308.
44.Stocker, A., Keis, S., Vonck, J., Cook, G. M.,
and Dimroth, P. (2007) Structure, 15, 904-914.
45.Feynman, R. P., Leighton, R. B., and Sands, M.
(1963) in The Feynman Lectures on Physics, Vol. 1, Chap. 46,
Addison-Wesley Inc., Massachusetts, PaloAlto, London.
46.Padan, E., and Rottenberg, H. (1973) Eur. J.
Biochem., 40, 431-437.