Mild Uncoupling of Respiration and Phosphorylation as a Mechanism Providing Nephro- and Neuroprotective Effects of Penetrating Cations of the SkQ Family
E. Y. Plotnikov1,2*, D. N. Silachev1,2, S. S. Jankauskas2,3, T. I. Rokitskaya1,2, A. A. Chupyrkina1,2, I. B. Pevzner2,3, L. D. Zorova2,4, N. K. Isaev1,2, Y. N. Antonenko1,2, V. P. Skulachev1,2,3, and D. B. Zorov1,2*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: plotnikov@genebee.msu.su; zorov@genebee.msu.ru2Mitoengeneering Institute, Lomonosov Moscow State University, 119991 Moscow, Russia
3Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia
4International Laser Center, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received May 15, 2012; Revision received May 21, 2012
It is generally accepted that mitochondrial production of reactive oxygen species is nonlinearly related to the value of the mitochondrial membrane potential with significant increment at values exceeding 150 mV. Due to this, high values of the membrane potential are highly dangerous, specifically under pathological conditions associated with oxidative stress. Mild uncoupling of oxidative phosphorylation is an approach to preventing hyperpolarization of the mitochondrial membrane. We confirmed data obtained earlier in our group that dodecylrhodamine 19 (C12R1) (a penetrating cation from SkQ family not possessing a plastoquinone group) has uncoupling properties, this fact making it highly potent for use in prevention of pathologies associated with oxidative stress induced by mitochondrial hyperpolarization. Further experiments showed that C12R1 provided nephroprotection under ischemia/reperfusion of the kidney as well as under rhabdomyolysis through diminishing of renal dysfunction manifested by elevated level of blood creatinine and urea. Similar nephroprotective properties were observed for low doses (275 nmol/kg) of the conventional uncoupler 2,4-dinitrophenol. Another penetrating cation that did not demonstrate protonophorous activity (SkQR4) had no effect on renal dysfunction. In experiments with induced ischemic stroke, C12R1 did not have any effect on the area of ischemic damage, but it significantly lowered neurological deficit. We conclude that beneficial effects of penetrating cation derivatives of rhodamine 19 in renal pathologies and brain ischemia may be at least partially explained by uncoupling of oxidation and phosphorylation.
KEY WORDS: ischemia, rhabdomyolysis, kidney, brain, stroke, oxidative stress, mitochondria-targeted compounds, SkQ, mitochondriaDOI: 10.1134/S0006297912090106
Abbreviations: AKI, acute kidney injury; C12R1, dodecylrhodamine 19; DNP, 2,4-dinitrophenol; I/R, ischemia/reperfusion; ROS, reactive oxygen species; SkQ, cationic derivatives of plastoquinone; SkQR1, 10-(6′-plastoquinonyl)decylrhodamine 19; SkQR4, 10-(6′-plastoquinonyl)decylrhodamine B.
Structural and functional changes in mitochondria [1], in particular the induction of nonspecific
permeability of mitochondria coupled to excessive generation of
reactive oxygen species (ROS) [2], release of a
number of proapoptotic factors from mitochondria, and as a
result cell death [1, 3],
are known to be the basis of the mechanisms of cell damage and death in
nephrons that have experienced oxidative stress. Myoglobinuria (crush
syndrome or rhabdomyolysis) is also a nephrological pathology coupled
to oxidative stress with a typical significant damage to mitochondria
and excessive ROS generation in them [4]. As in
kidneys, oxidative damage is one of the major causes of nervous cells
death in ischemia/hypoxia/brain reoxygenation [5,
6], and metabolic characteristics of nervous tissue
make it inherently more vulnerable to the damaging effects of ROS.
Thus, the central role of mitochondria in launching and enhancing the
pathological cascades connected to oxidative stress allows us to see
them as a possible target for pharmacological treatment. In addition to
the strategy of targeted delivery of antioxidants to mitochondria [7, 8], the reduction of the rate
of ROS generation in mitochondria due to mild uncoupling [9, 10] seems to be another
promising avenue for prevention of oxidative damage to components of
cells. It is the positive correlation between mitochondrial membrane
potential and ROS production that it the basis for this phenomenon.
Even a small increment in membrane potential exceeding the level of
approximately 150 mV was shown to increase H2O2
generation by mitochondria, this effect being disproportionately high
[9, 11, 12]. Accordingly, mild uncoupling, i.e. a slight
decrease in membrane potential, which does not lead to a decrease in
ATP synthesis in mitochondria, may have a useful antioxidant effect [10]. Since a number of clinically significant
diseases of kidney [1, 3, 4] and brain [5, 6] are associated with oxidative stress, it seems
reasonable to try to apply the strategy of partial uncoupling in these
cases, so as to protect these organs from damage.
Hydrophobic derivatives of rhodamine 19, a representative of a new class of cationic uncouplers, seem to be particularly promising in this respect. As shown in our laboratory [13], such compounds, namely SkQR1 and C12R1 (see Fig. 1), are accumulated in mitochondria due to the membrane potential in these organelles. Their accumulation reduces this potential, a fact preventing further excessive accumulation (and thus uncoupling). In other words, SkQR1 and C12R1 act as “mild” uncouplers – their moderate concentrations can prevent mitochondrial hyperpolarization and the associated ROS generation without the risk of disabling the main mitochondrial function, ATP synthesis, which depends on membrane potential, but does not require very high values of it. It seems quite significant that SkQR4 molecule, where rhodamine 19 is substituted for rhodamine B, does not carry groups with dissociating H+ ion (in contrast to rhodamine 19 and its derivatives), and of itself it has no protonophorous properties and requires free fatty acids for the manifestation of uncoupling activity [14].
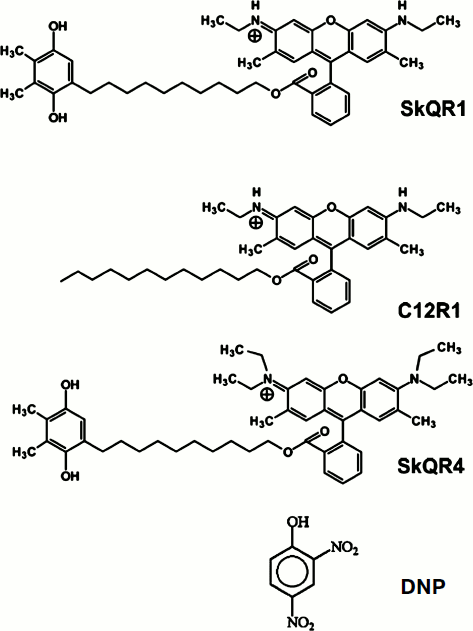
In this study we compared the properties of the derivatives of rhodamine 19 (SkQR1 and C12R1) and rhodamine B (SkQR4), as well as the conventional uncoupler, the protonophore 2,4-dinitrophenol [15] as protective agents in ischemia and reperfusion (I/R) of kidneys, rhabdomyolysis, and experimental stroke.Fig. 1. Structural formulas of the studied compounds: 10-(6′-plastoquinonyl)decyltriphenylphosphonium (SkQ1); 10-(6′-plastoquinonyl)decylrhodamine 19 (SkQR1); dodecylrhodamine 19 (C12R1); 10-(6′-plastoquinonyl)decylrhodamine B (SkQR4); 2,4-dinitrophenol (DNP).
MATERIALS AND METHODS
Modeling of renal ischemia. Experiments were performed on male outbred white rats (200-250 g) on an ad libitum diet. A 40-min ischemia of the left kidney was conducted as previously described [3]. Right-sided nephrectomy was performed along with the ischemia. Blood samples were taken from the animals on the second day after ischemia. The concentration of urea and creatinine in blood was determined using a CellTac blood analyzer (Nihon Kohden, Italy).
Modeling of rhabdomyolysis. Rhabdomyolysis was induced according to a conventional procedure by injecting 50% aqueous solution of glycerol (ICN, USA) into the paw muscles of rats as previously described [4]. Blood samples were taken from the animals on the second day after rhabdomyolysis. The concentration of urea and creatinine in blood was determined.
All the experiments on the ischemia and rhabdomyolysis models were performed on at least eight animals in each group. Data are presented as average ± SEM.
Modeling of brain ischemia. Ischemia of rat brain was induced by the introduction of a silicone-coated nylon thread into the middle cerebral artery [16, 17]. The blood flow was occluded for 60 min; then the thread was removed from the vessel, restoring the blood flow in the basin of the middle cerebral artery. The animal’s body temperature was maintained at 37 ± 0.5°C during and after the operation. The sham-operated animals were subjected to the same procedures except for the cutting of blood vessels and introduction of the thread. The area of cerebral infarction was determined on the first day for the studies of neuroprotective effect of the compounds or within 7 days for studies of the dynamics of development of ischemic damage. This was evaluated by morphometric analysis of digital images obtained by magnetic resonance imaging (MRI). All the MRI experiments were performed as previously described [18] on a BioSpec 70/30 instrument (Bruker, Germany) with the magnetic field induction of 7 T and the gradient system 105 mT/m.
Behavioral tests were carried out 1 day before surgery and on the first day after ischemia. A 14-point scale [19] modified as in [20] was used to estimate neurological disorders caused by the occlusion of the middle cerebral artery. The final score is formed in this scale as the sum of the scores in seven tests evaluating the response of the hindlimbs and forelimbs to tactile and proprioceptive stimulation. The following counting system was used to estimate the disturbances in the function of the limbs: 2 points, the rat fully performed the test; 1 point, the rat performed the test with a delay of more than 2 sec or incompletely; 0 points, the rat did not respond to the stimulation of the limb. The animals were treated and all the experiments in accordance with generally accepted international guidelines for experimentation on animals.
The results are expressed as the mean value ± standard error of mean. Normality of the characteristic distribution in the sample was evaluated using the Shapiro–Wilk W-test. The Mann–Whitney U-test was used to compare the data in the behavioral tests (for independent samples). The t-test with a significance level p < 0.05 was used to estimate the statistical significance of differences in infarct size.
Measurement of mitochondrial membrane potential. Mitochondria were isolated from rat liver by differential centrifugation according to a conventional procedure. The extraction medium (pH 7.4) contained 250 mM sucrose, 20 mM MOPS, 1 mM EGTA, and 1.2 mg/ml bovine serum albumin (BSA). Protein concentration was determined using bicinchoninic acid with BSA as a standard [21].
Mitochondrial membrane potential was measured by fluorescence of the dye DiS-C3-(5) (3,3′-dipropylthiadicarbocyanine iodide) (Molecular Probes, USA) according to [22]. The fluorescence signal at 690 nm (excitation 622 nm) was measured on a Panorama Fluorat 02 spectrofluorimeter (Lumex, Russia). The incubation medium contained 150 mM KCl, 5 mM succinate, 1 μM rotenone, 0.5 mM EGTA, 1 μM DiS-C3-(5), and 20 mM HEPES, pH 7.4. The concentration of mitochondrial protein was 0.4 mg/ml.
RESULTS
Influence of penetrating cations on mitochondrial membrane potential. The penetrating cationic dye DiS-C3-(5) with absorption and fluorescence in the red region (excitation 622 nm, emission 690 nm), i.e. with longer wavelengths when compared to rhodamine, was used to study the influence of C12R1 and SkQR4 on the membrane potential of isolated mitochondria, as well as to compare the effect with the conventional uncoupler DNP. Energization of mitochondria leads to the quenching of fluorescence of DiS-C3-(5) because of the aggregation of the dye in the matrix in the course of its accumulation due to electrochemical potential [22].
Figure 2a shows a recording of DiS-C3-(5) fluorescence in the presence of the respiratory substrate succinate. Addition of 5 μM C12R1 and 10 μM DNP to isolated mitochondria causes an increase in fluorescence, indicating membrane potential decrease. In contrast to C12R1, the effect of SkQR4 on membrane potential is much weaker, which is consistent with our previous findings [13]. Some SkQR4-induced decrease in membrane potential may be explained by the increase in proton conductivity of the mitochondrial membrane mediated by endogenous fatty acids [14]. Data on the effect of different concentrations of C12R1 on the mitochondrial membrane potential are presented on Fig. 2b. According to these data, C12R1 reduces the potential in the micromolar concentration range, and decrease is evolving over time. The concentration dependences of the effects of C12R1 and DNP are presented in Fig. 2c. The effective concentrations of C12R1 were found to be about an order of magnitude less than those of DNP.
Therapy of renal functions after unilateral ischemia. The I/R kidney (from ischemia/reperfusion) is one of the most common models of acute kidney injury (AKI) [3, 23]. Unilateral 40-min ischemia of the left kidney with simultaneous removal of the right kidney was used to study the effect of penetrating cations and DNP on I/R kidney. The following protocol of treatment was used for the therapy: a preliminary intraperitoneal injection of the drug 3 h before ischemia; 1 h after ischemia, and then 3 times at 12-h intervals. Summarized data are presented in Table 1, where the doses of SkQR1, SkQR4, C12R1, or DNP per injection are indicated. Acute renal failure was shown to take place on the second day after I/R left without treatment: the levels of creatinine and urea in the blood serum rose more than 4-fold. In the case of SkQR1-treatment following the above-described protocol at the concentration of 100 nmol/kg per injection, more than twofold decrease in creatinine and urea levels in blood 48 h after I/R could be observed (Table 1). Almost as significant effects could be observed when C12R1 was injected at the same doses, whereas injection of lesser (20 nmol/kg) or higher (500 nmol/kg) doses was ineffective (see Table 1). No reliable reduction of renal failure was observed when DNP was administered at the doses of 5500 and 27,500 nmol/kg, whereas the dose of 275 nmol/kg caused a strong decrease in creatinine and urea concentration comparable to the effects of SkQR1 and C12R1. In similar experiments, the use of 100 nmol/kg SkQR4 per injection resulted in a slight (statistically insignificant) decrease in creatinine and urea concentration in the blood (see Table 1).Fig. 2. Effects of rhodamine derivatives and DNP on membrane potential of isolated rat liver mitochondria measured by fluorescence of DiS-C3-(5). a, b) Kinetics of fluorescence changes of DiS-C3-(5) caused by addition of C12R1, DNP, and SkQR4. c) Dependence of the decrease in DiS-C3-(5) fluorescence on concentration of C12R1 and DNP.
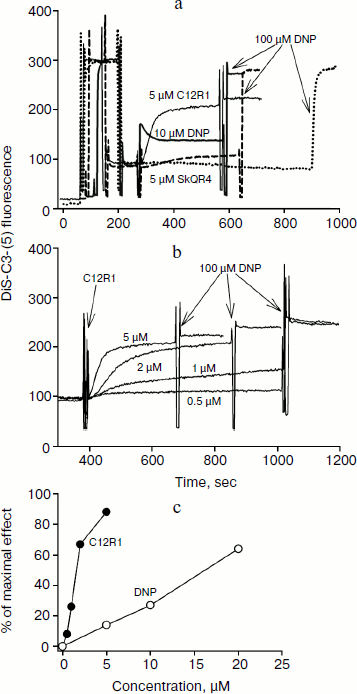
Table 1. Influence of penetrating cations
and uncoupler on development of AKI (increase in creatinine and urea
concentration in blood)
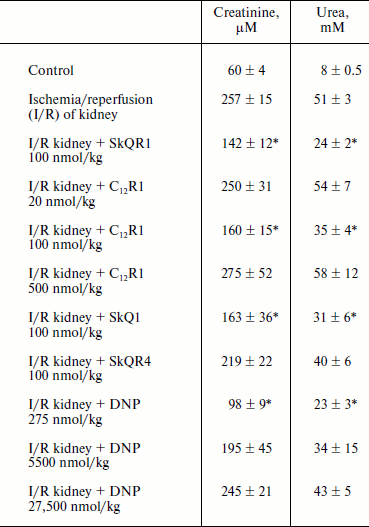
Note: The compounds were administered in the stated doses according to
the protocol described in “Materials and Methods”. Blood
tests were performed 48 h after the end of renal ischemia.
* p < 0.05 compared to ischemia without treatment.
Therapy of renal functions after induced rhabdomyolysis. The studies of dynamics of development of AKI in rhabdomyolysis showed that this pathology causes severe renal failure similar to ischemic AKI, resulting in an increase in creatinine and urea concentration in the blood (it reaches a peak on the second day, see Table 2). The treatment protocol used in this therapy was similar to the one used for ischemic AKI: intraperitoneal injection of the drug 1 h after induction of rhabdomyolysis and then three times at 12-h intervals at a dose of 100 nmol/kg per injection. In the case of SkQR1 treatment (which was conducted according to the above-described protocol), a 2.5-fold decrease in creatinine and urea concentrations in blood (in comparison with rhabdomyolysis without treatment) 48 h after glycerol injection could be observed (see Table 2). C12R1 also had a protective effect on renal functions, although it was less pronounced than that of SkQR1 (see Table 2). SkQR4 had a slight protective effect in rhabdomyolysis as well as in kidney ischemia (see Table 2).
Table 2. Effect of penetrating cations on
increase in creatinine and urea concentration in blood in experimental
rhabdomyolysis
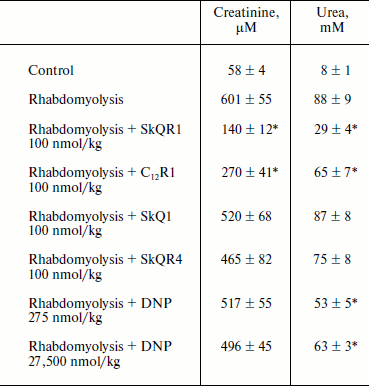
Note: The compounds were injected 1, 12, 24, and 36 h after induction of
rhabdomyolysis. Blood tests were performed 48 h after induction of
rhabdomyolysis.
* p < 0.05 compared to rhabdomyolysis without treatment.
Protective effect of penetrating cations in cerebral ischemia. The tested compounds were administered intraperitoneally at a dose of 1 µmol/kg 24 h before induction of ischemia. MRI study of brain was performed 24 h after reperfusion so as to evaluate the size of the lesion area; neurological deficit was determined by using the “limb-placing” test. The volume of the ischemic damage in the brains of animals receiving no treatment was taken as 100%. Single injections of SkQR1 and C12R1 decreased the damaged area to 53.4 ± 10.5 and 82 ± 6.0% (p = 0.051), respectively (see Table 3). Middle cerebral artery occlusion disrupted sensomotoric functions, and the animals in the control group received an average score of 1.5 ± 0.2 points in the limb-placing test. SkQR1 and C12R1 reduced the severity of neurological deficit in a statistically significant way, to 5.5 ± 0.7 and 5.0 ± 0.6 points, respectively. Injection of SkQR4 did not significantly decrease the brain damage or improve neurological outcome (see Table 3). Thus, these experiments show that among tested drugs SkQR1 had the best neuroprotective properties. However, C12R1 reduced the neurological deficit in animals, although the reduction of ischemic damage was less than in case of SkQR1. As to SkQR4, the effect of this compound, if observed, should be described as a trend rather than a reliable change.
Table 3. Influence of penetrating cations on
size of ischemic brain damage and neurological deficit (decrease in
neurological status) in experimental stroke
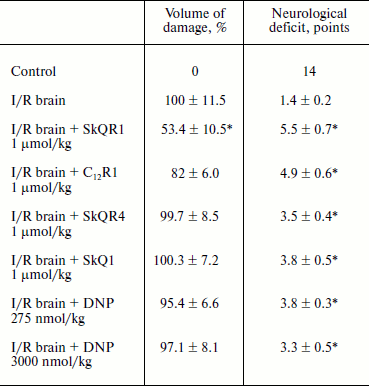
Note: The compounds were administered one day before the induction of
cerebral ischemia. Evaluation of the size of the ischemic area by MRI
and neurological status in the limb-placing test was conducted 24 h
after the experimental stroke.
* p < 0.05 compared to ischemia without treatment.
DISCUSSION
Protective effects of penetrating cations C12R1 and SkQR4 in vivo and their activity as uncouplers of oxidative phosphorylation in mitochondria in vitro was comparatively analyzed in this work. We evaluated the nephro- and neuroprotective effects of these compounds in comparison with the mitochondria-targeted antioxidant SkQR1, the protective effect of which has been previously demonstrated on a large number of renal [24-26] and brain [24, 27, 28] pathologies. In addition, we used DNP as a conventional protonophorous uncoupler for comparison; the beneficial effects of DNP had been shown for the aging of rats [29], flies [30], and mice [31] and for several pathologies accompanied by oxidative stress [32-34]. While DNP has already been used for several decades as an uncoupler of oxidative phosphorylation in mitochondria and as a medicine [35, 36], the use of mitochondria-targeted compounds of the SkQ family as cytoprotectors began only in 2007-2008 [8, 27, 37-39], and their uncoupling effects have been demonstrated quite recently [13].In this study we confirmed that C12R1 has a pronounced uncoupling effect, and the C12R1-induced decrease in mitochondrial potential is achieved at very low concentrations, an order of magnitude lower than those shown for DNP. SkQR1 was shown to have similar effects on mitochondrial membrane potential and the rate of respiration in mitochondria [13]. At the same time, the effect of SkQR4 on the reduction of mitochondrial membrane potential was significantly less. It can be assumed that the uncoupling effect of mitochondria-targeted compounds that contain rhodamine 19 as the cation is mediated by the possibility of protonation/deprotonation of the tertiary nitrogen of the heterocycle, whereas the quaternary nitrogen of rhodamine B in the SkQR4 molecule (see Fig. 1) is virtually incapable of protonation/deprotonation at physiological pH values. We previously showed [13] that SkQR1 and C12R1 have practically equal abilities to accelerate mitochondrial respiration in state 4, which indicates their uncoupling effect. On the other hand, the favorable effect of SkQ-type compounds in vivo may be linked also to their ability not only to prevent ROS formation, but also to quench already formed ROS [14].
In this regard, it should be noted that there are several ways to uncouple oxidative phosphorylation [40, 41]. In addition to classical uncoupling protonophores such as DNP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), and propofol directly engaged in proton transfer across a bilayer membrane containing no proteins, there is another uncoupling mechanism that depends on free fatty acids. The mechanism involves several processes: protonation of fatty acid anions in the intermembrane space of mitochondria, diffusion of protonated fatty acid molecules across the membrane, their deprotonation in the matrix to form fatty acid anions, and return of these anions to the intermembrane space. It is the return of fatty acid anions to the outside of the inner mitochondrial membrane that is the limiting step of this cycle. The transport of the anionic form of fatty acids was shown to be substantially accelerated by a variety of protein carriers (uncoupling proteins (UCP), adenine nucleotide translocator, and dicarboxylate carrier) [40, 42-44].
Finally, passive leak of K+ ions into the matrix across the inner mitochondrial membrane or their transfer through the ATP-dependent K+-channel may serve as another mechanism of uncoupling. The accumulation of K+ in the matrix can cause the reduction of transmembrane potential, swelling of mitochondria, activation of K+/H+-antiporter, and as a result the pumping of K+ from mitochondria at the expense of ΔpH. It seems quite interesting that most of these mechanisms involved in the uncoupling of oxidative phosphorylation are in some way involved in the induction of tolerance of cells and tissues to oxidative damage, especially in ischemic preconditioning [45-47]. It should be noted though that there are two opposing perspectives on this phenomenon, each confirmed by experimental evidence, that activation of a K+ channel is accompanied by either a decrease in ROS generation [46, 48, 49] or an increase in their production [47, 50-53].
When not focusing on the contradictions and discrepancies relevant to partial uncoupling caused by potassium ion transfer to the mitochondrial matrix, the majority of researchers agree that, in general, mitochondrial uncouplers (endogenous or exogenous) can reduce ROS production by decreasing mitochondrial membrane potential, especially in hyperpolarized mitochondria. These facts led to the development of a new positive understanding of “mild” uncoupling, that is, a limited decrease in membrane potential that may have beneficial effects through the reduction of ROS generation in the respiratory chain while having no substantial negative impact on ATP synthesis. This indicates the possible use of different types of uncouplers for the prevention and treatment of pathologies caused by excessive ROS generation in mitochondria [41]. It should be noted though that there exists another perspective, that the phenomenon of “mild” uncoupling is mainly characteristic for in vitro systems, when isolated mitochondria oxidizing succinate and generating ROS by reverse electron transport are studied, whereas in vivo such states are unlikely and, therefore, the uncouplers will have no positive effect under these conditions [54].
This view directly contradicts the data obtained in our group in studies of mitochondrial membrane potential in yeast cells in vivo [55]. In these experiments, the induction of a phenoptosis (suicide) mechanism in yeast was accompanied by increase in membrane potential, and its partial reduction with the uncoupler halts phenoptosis.
However, substantial evidence of the positive impact of various types of uncouplers in pathologies of the cardiovascular system [34, 56] and brain [32, 33, 57] have already been accumulated, although at this point there is no similar data for ROS-mediated renal pathologies. Therefore, this work has expanded the spectrum of possible applications of the conventional uncoupler DNP to reduce the severity of AKI that is widespread in clinical practice. However, the small difference between toxic and protective DNP doses [58] remains the principal problem of using DNP, since the doses required for “mild” uncoupling are close to those causing fatal reduction of ATP synthesis in mitochondria. In this respect, mitochondria-targeted compounds with limited uncoupling effect, such as SkQ1, SkQR1, and C12R1 studied in this paper, seem to be far more promising.
Three possible mechanisms of antioxidant action of the studied compounds are listed in Table 4. Two of them prevent the generation of ROS by the respiratory chain. This is “mild” uncoupling of respiration and membrane potential generation either by direct protonophorous action of the studied compound [13], or through the stimulation of such action by endogenous fatty acids [14]. In addition, plastoquinone-containing compounds can quench already formed ROS [8, 25]. As seen from Table 1, low concentrations of the conventional protonophore DNP (the first antioxidant mechanism in Table 4) and SkQR1 (all three antioxidant mechanisms in Table 4) are the most effective in alleviating the symptoms of kidney failure (increased levels of creatinine and urea in blood) after kidney I/R. C12R1 (the first two mechanisms) and SkQ1 (the last two mechanisms) have somewhat weaker effect. SkQR4 shows even smaller effect (also the last two mechanisms). In the case of rhabdomyolysis, the order of activities of the compounds remained roughly the same as in I/R, if their action is considered at the level of urea concentration. With regard to creatinine level, the spread is too large to analyze the entire series of the studied compounds (see Table 2). In case of experimental brain stroke, a reliable reduction of the damaged area was observed only with one compound (SkQR1), while the reduction of neurological deficit was similar for all five studied compounds (see Table 3).
Table 4. Possible mechanisms of antioxidant
effects of the studied compounds

When comparing the studied antioxidants, one should take into account the possible role of factors other than those specified in Table 4. For instance, these compounds are very different in their affinity to membranes, which is measured on the basis of distribution in the octanol–water system. This parameter is the highest for C12R1 and the lowest for DNP. In addition, our data [26] suggest that SkQ derivatives can raise the erythropoietin level in blood, which mobilizes antioxidant mechanisms of cells and the organism.
This work was supported by the Mitoengineering Institute, Lomonosov Moscow State University, by RFBR grants 11-04-00771 and 11-04-01307, and by President’s grant MK-729.2012.4.
REFERENCES
1.Plotnikov, E. Y., Vasileva, A. K., Arkhangelskaya,
A. A., Pevzner, I. B., Skulachev, V. P., and Zorov, D. B. (2008)
FEBS Lett., 582, 3117-3124.
2.Zorov, D. B., Filburn, C. R., Klotz, L. O., Zweier,
J. L., and Sollott, S. J. (2000) J. Exp. Med., 192,
1001-1014.
3.Plotnikov, E. Y., Kazachenko, A. V., Vyssokikh, M.
Y., Vasileva, A. K., Tcvirkun, D. V., Isaev, N. K., Kirpatovsky, V. I.,
and Zorov, D. B. (2007) Kidney Int., 72, 1493-1502.
4.Plotnikov, E. Y., Chupyrkina, A. A., Pevzner, I.
B., Isaev, N. K., and Zorov, D. B. (2009) Biochim. Biophys.
Acta, 1792, 796-803.
5.Lipton, P. (1999) Physiol. Rev., 79,
1431-1568.
6.Amantea, D., Marrone, M. C., Nistico, R., Federici,
M., Bagetta, G., Bernardi, G., and Mercuri, N. B. (2009) Int. Rev.
Neurobiol., 85, 363-374.
7.Kelso, G. F., Porteous, C. M., Hughes, G.,
Ledgerwood, E. C., Gane, A. M., Smith, R. A., and Murphy, M. P. (2002)
Ann. N. Y. Acad. Sci., 959, 263-274.
8.Skulachev, V. P. (2007) Biochemistry
(Moscow), 72, 1385-1396.
9.Korshunov, S. S., Skulachev, V. P., and Starkov, A.
A. (1997) FEBS Lett., 416, 15-18.
10.Skulachev, V. P. (1997) Biosci. Rep.,
17, 347-366.
11.Hansford, R. G., Hogue, B. A., and Mildaziene, V.
(1997) J. Bioenerg. Biomembr., 29, 89-95.
12.Pistor, K., Scharer, K., Olbing, H., and
Tamminen-Mobius, T. (1985) Clin. Nephrol., 23,
278-284.
13.Antonenko, Y. N., Avetisyan, A. V., Cherepanov,
D. A., Knorre, D. A., Korshunova, G. A., Markova, O. V., Ojovan, S. M.,
Perevoshchikova, I. V., Pustovidko, A. V., Rokitskaya, T. I., Severina,
I. I., Simonyan, R. A., Smirnova, E. A., Sobko, A. A., Sumbatyan, N.
V., Severin, F. F., and Skulachev, V. P. (2011) J. Biol. Chem.,
286, 17831-17840.
14.Severin, F. F., Severina, I. I., Antonenko, Y.
N., Rokitskaya, T. I., Cherepanov, D. A., Mokhova, E. N., Vyssokikh, M.
Y., Pustovidko, A. V., Markova, O. V., Yaguzhinsky, L. S., Korshunova,
G. A., Sumbatyan, N. V., Skulachev, M. V., and Skulachev, V. P. (2010)
Proc. Natl. Acad. Sci. USA, 107, 663-668.
15.Skulachev, V. P., Sharaf, A. A., Yagujzinsky, L.
S., Jasaitis, A. A., Liberman, E. A., and Topali, V. P. (1968) Curr.
Mod. Biol., 2, 98-105.
16.Longa, E. Z., Weinstein, P. R., Carlson, S., and
Cummins, R. (1989) Stroke, 20, 84-91.
17.Koizumi, J., Yoshida, Y., Nakazawa, T., and
Ooneda, G. (1986) Jpn. J. Stroke, 8, 1-8.
18.Silachev, D. N., Uchevatkin, A. A., Pirogov, Yu.
A., Zorov, D. B., and Isaev, N. K. (2009) Byul. Eksp. Biol.
Med., 147, 232-237.
19.De Ryck, M., van Reempts, J., Borgers, M.,
Wauquier, A., and Janssen, P. A. (1989) Stroke, 20,
1383-1390.
20.Jolkkonen, J., Puurunen, K., Rantakomi, S.,
Harkonen, A., Haapalinna, A., and Sivenius, J. (2000) Eur. J.
Pharmacol., 400, 211-219.
21.Smith, P. K., Krohn, R. I., Hermanson, G. T.,
Mallia, A. K., Gartner, F. H., Provenzano, M. D., Fujimoto, E. K.,
Goeke, N. M., Olson, B. J., and Klenk, D. C. (1985) Anal.
Biochem., 150, 76-85.
22.Laris, P. C., Bahr, D. P., and Chaffee, R. R.
(1975) Biochim. Biophys. Acta, 376, 415-425.
23.Cadenas, E., and Sies, H. (1985) Adv. Enzyme
Regul., 23, 217-237.
24.Plotnikov, E. Y., Silachev, D. N., Chupyrkina, A.
A., Danshina, M. I., Jankauskas, S. S., Morosanova, M. A., Stelmashook,
E. V., Vasileva, A. K., Goryacheva, E. S., Pirogov, Y. A., Isaev, N.
K., and Zorov, D. B. (2010) Biochemistry (Moscow), 75,
145-150.
25.Skulachev, M. V., Antonenko, Y. N., Anisimov, V.
N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov, M.
V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov, E.
Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F. F.,
Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M. Y.,
Zamyatnin, A. A., Jr., Zorov, D. B., and Skulachev, V. P. (2011)
Curr. Drug Targets, 12, 800-826.
26.Plotnikov, E. Y., Chupyrkina, A. A., Jankauskas,
S. S., Pevzner, I. B., Silachev, D. N., Skulachev, V. P., and Zorov, D.
B. (2011) Biochim. Biophys. Acta, 1812, 77-86.
27.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. B., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashook, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasilyeva, A. K., Victorov, I. V., Zorov, D. B., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1288-1299.
28.Kapay, N. A., Isaev, N. K., Stelmashook, E. V.,
Popova, O. V., Zorov, D. B., Skrebitsky, V. G., and Skulachev, V. P.
(2011) Biochemistry (Moscow), 76, 1367-1370.
29.Tainter, M. L. (1938) J. Pharmacol. Exp.
Ther., 63, 51-57.
30.Padalko, V. I. (2005) Biochemistry
(Moscow), 70, 986-989.
31.Caldeira da Silva, C. C., Cerqueira, F. M.,
Barbosa, L. F., Medeiros, M. H., and Kowaltowski, A. J. (2008) Aging
Cell, 7, 552-560.
32.Pandya, J. D., Pauly, J. R., Nukala, V. N.,
Sebastian, A. H., Day, K. M., Korde, A. S., Maragos, W. F., Hall, E.
D., and Sullivan, P. G. (2007) J. Neurotrauma, 24,
798-811.
33.Korde, A. S., Pettigrew, L. C., Craddock, S. D.,
and Maragos, W. F. (2005) J. Neurochem., 94,
1676-1684.
34.Rodrigo, G. C., Lawrence, C. L., and Standen, N.
B. (2002) J. Mol. Cell Cardiol., 34, 555-569.
35.Parascandola, J. (1974) Mol. Cell
Biochem., 5, 69-77.
36.Colman, E. (2007) Regul. Toxicol.
Pharmacol., 48, 115-117.
37.Anisimov, V. N., Bakeeva, L. E., Egormin, P. A.,
Filenko, O. F., Isakova, E. F., Manskikh, V. N., Mikhelson, V. M.,
Panteleeva, A. A., Pasyukova, E. G., Pilipenko, D. I., Piskunova, T.
S., Popovich, I. G., Roshchina, N. V., Rybina, O. Y., Saprunova, V. B.,
Samoylova, T. A., Semenchenko, A. V., Skulachev, M. V., Spivak, I. M.,
Tsybul’ko, E. A., Tyndyk, M. L., Vyssokikh, M. Y., Yurova, M. N.,
Zabezhinsky, M. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1329-1342.
38.Agapova, L. S., Chernyak, B. V., Domnina, L. V.,
Dugina, V. B., Efimenko, A. Y., Fetisova, E. K., Ivanova, O. Y.,
Kalinina, N. I., Khromova, N. V., Kopnin, B. P., Kopnin, P. B.,
Korotetskaya, M. V., Lichinitser, M. R., Lukashev, A. L., Pletjushkina,
O. Y., Popova, E. N., Skulachev, M. V., Shagieva, G. S., Stepanova, E.
V., Titova, E. V., Tkachuk, V. A., Vasiliev, J. M., and Skulachev, V.
P. (2008) Biochemistry (Moscow), 73, 1300-1316.
39.Neroev, V. V., Archipova, M. M., Bakeeva, L. E.,
Fursova, A., Grigorian, E. N., Grishanova, A. Y., Iomdina, E. N.,
Ivashchenko, Zh. N., Katargina, L. A., Khoroshilova-Maslova, I. P.,
Kilina, O. V., Kolosova, N. G., Kopenkin, E. P., Korshunov, S. S.,
Kovaleva, N. A., Novikova, Y. P., Philippov, P. P., Pilipenko, D. I.,
Robustova, O. V., Saprunova, V. B., Senin, I. I., Skulachev, M. V.,
Sotnikova, L. F., Stefanova, N. A., Tikhomirova, N. K., Tsapenko, I.
V., Shchipanova, A. I., Zinovkin, R. A., and Skulachev, V. P. (2008)
Biochemistry (Moscow), 73, 1317-1328.
40.Skulachev, V. P. (1998) Biochim. Biophys.
Acta, 1363, 100-124.
41.Cunha, F. M., Caldeira da Silva, C. C.,
Cerqueira, F. M., and Kowaltowski, A. J. (2011) Curr. Drug
Targets, 12, 783-789.
42.Skulachev, V. P. (1991) FEBS Lett.,
294, 158-162.
43.Wojtczak, L., and Wieckowski, M. R. (1999) J.
Bioenerg. Biomembr., 31, 447-455.
44.Samartsev, V. N., Marchik, E. I., and
Shamagulova, L. V. (2011) Biochemistry (Moscow), 76,
217-224.
45.Nadtochiy, S. M., Tompkins, A. J., and Brookes,
P. S. (2006) Biochem. J., 395, 611-618.
46.Facundo, H. T., de Paula, J. G., and Kowaltowski,
A. J. (2005) J. Bioenerg. Biomembr., 37, 75-82.
47.Juhaszova, M., Zorov, D. B., Kim, S. H., Pepe,
S., Fu, Q., Fishbein, K. W., Ziman, B. D., Wang, S., Ytrehus, K.,
Antos, C. L., Olson, E. N., and Sollott, S. J. (2004) J. Clin.
Invest., 113, 1535-1549.
48.Ferranti, R., da Silva, M. M., and Kowaltowski,
A. J. (2003) FEBS Lett., 536, 51-55.
49.Kulawiak, B., Kudin, A. P., Szewczyk, A., and
Kunz, W. S. (2008) Exp. Neurol., 212, 543-547.
50.Rasmusson, I. (2006) Exp. Cell Res.,
312, 2169-2179.
51.Andrukhiv, A., Costa, A. D., West, I. C., and
Garlid, K. D. (2006) Am. J. Physiol. Heart Circ. Physiol.,
291, H2067-2074.
52.Heinen, A., Aldakkak, M., Stowe, D. F., Rhodes,
S. S., Riess, M. L., Varadarajan, S. G., and Camara, A. K. (2007)
Am. J. Physiol. Heart Circ. Physiol., 293,
H1400-1407.
53.Thuc, L. C., Teshima, Y., Takahashi, N.,
Nagano-Torigoe, Y., Ezaki, K., Yufu, K., Nakagawa, M., Hara, M., and
Saikawa, T. (2010) Apoptosis, 15, 669-678.
54.Shabalina, I. G., and Nedergaard, J. (2011)
Biochem. Soc. Trans., 39, 1305-1309.
55.Pozniakovsky, A. I., Knorre, D. A., Markova, O.
V., Hyman, A. A., Skulachev, V. P., and Severin, F. F. (2005) J.
Cell Biol., 168, 257-269.
56.Modriansky, M., and Gabrielova, E. (2009) J.
Bioenerg. Biomembr., 41, 133-136.
57.Patel, S. P., Sullivan, P. G., Pandya, J. D., and
Rabchevsky, A. G. (2009) J. Neurosci. Res., 87,
130-140.
58.McFee, R. B., Caraccio, T. R., McGuigan, M. A.,
Reynolds, S. A., and Bellanger, P. (2004) Vet. Hum. Toxicol.,
46, 251-254.