REVIEW: Recharging Oxidative Protein Repair: Catalysis by Methionine Sulfoxide Reductases towards Their Amino Acid, Protein, and Model Substrates
L. Tarrago and V. N. Gladyshev*
Brigham and Women’s Hospital and Harvard Medical School, 77 Ave. Louis Pasteur, Boston, MA 02115, USA; fax: (617) 525-5147; E-mail: vgladyshev@rics.bwh.harvard.edu; ltarrago@rics.bwh.harvard.edu* To whom correspondence should be addressed.
Received April 13, 2012; Revision received June 7, 2012
The sulfur-containing amino acid methionine (Met) in its free and amino acid residue forms can be readily oxidized to the R and S diastereomers of methionine sulfoxide (MetO). Methionine sulfoxide reductases A (MSRA) and B (MSRB) reduce MetO back to Met in a stereospecific manner, acting on the S and R forms, respectively. A third MSR type, fRMSR, reduces the R form of free MetO. MSRA and MSRB are spread across the three domains of life, whereas fRMSR is restricted to bacteria and unicellular eukaryotes. These enzymes protect against abiotic and biotic stresses and regulate lifespan. MSRs are thiol oxidoreductases containing catalytic redox-active cysteine or selenocysteine residues, which become oxidized by the substrate, requiring regeneration for the next catalytic cycle. These enzymes can be classified according to the number of redox-active cysteines (selenocysteines) and the strategies to regenerate their active forms by thioredoxin and glutaredoxin systems. For each MSR type, we review catalytic parameters for the reduction of free MetO, low molecular weight MetO-containing compounds, and oxidized proteins. Analysis of these data reinforces the concept that MSRAs reduce various types of MetO-containing substrates with similar efficiency, whereas MSRBs are specialized for the reduction of MetO in proteins.
KEY WORDS: enzyme catalysis, methionine oxidation, methionine sulfoxide, methionine sulfoxide reductase, sulfenic acid, selenenic acid, protein oxidation, oxidative protein repairDOI: 10.1134/S0006297912100021
Among the 22 amino acids used for protein synthesis, cysteine (Cys) and methionine (Met) are the only two that contain a sulfur atom in their side chain. Cysteine can be synthesized by humans and other mammals, whereas Met is an essential amino acid that is produced by microorganisms and plants and provided to mammals with food [1]. Methionine plays major roles in a variety of cellular processes, serving as a central factor in sulfur metabolism and a precursor for several important compounds, such as taurine, carnitine, and S-adenosylmethionine. Methionine is also fundamental for protein translation as an initiator residue in protein synthesis. In cells, approximately 99% of Met is present in the form of proteins [2]. One of the least abundant residues, accounting for 2.4% of amino acids in proteins [3], Met is a nonpolar residue, typically buried in the hydrophobic core of proteins [3]. It is also enriched in the hydrophobic areas involved in protein–protein interactions [4]. The presence of sulfur in their side chains renders Cys and Met sensitive to oxidation. The conversion of Cys to the cystine form leads to the formation of intra- or intermolecular disulfide bonds, many of which are required for protein structure and function. Reactions of Cys with reactive oxygen species can also result in the generation of oxidized derivatives, such as sulfenic acid (Cys-SOH), sulfinic acid (Cys-SO2H), or sulfonic acid (Cys-SO3H), which participate in catalytic processes and regulation of enzymes, such as peroxiredoxins [5] and protein-tyrosine phosphatases [6].
The reaction of Met with an oxidant leads to the formation of two diastereomers, the R and S forms, of methionine sulfoxide (MetO), and further oxidation can produce Met sulfone (MetO2). A racemic mixture of R- and S-diastereomers of MetO is formed by oxidation of free Met in vitro [7]. Little information is available regarding the amount of MetO in proteins in vivo, and virtually nothing is known about the proportion of each diastereomer in oxidized cellular proteins. The oxidation of Met in MetO can be reversed by the catalytic action of methionine sulfoxide reductases (MSRs) A (MSRA) and B (MSRB), which are specific for the S- and R-diastereomers, respectively [8-10]. The first evidence of MetO reduction activity was obtained in 1960 by fractionation of Saccharomyces cerevisiae cells. The authors demonstrated that three cellular fractions were required for the reduction of both R- and S-diastereomers of MetO at the expense of NADPH, and that the mechanism involved disulfide exchange [11]. In 1979, the in vivo reduction of MetO was shown in an Escherichia coli strain auxotrophic for Met, which grew in media containing MetO as a sole source of Met [12]. MSRA was then isolated from E. coli [8], whereas MSRB was discovered 20 years later in bacteria [9] and eukaryotes [10]. MSR genes have been found in almost all organisms with the exception of some parasites and hyperthermophiles [10, 13-15]. More recently, a third class of MSRs was discovered, fRMSR, which is specific for the reduction of the R-diastereomer of free MetO. It is present only in prokaryotes and lower eukaryotes, such as the yeast S. cerevisiae [16, 17]. In addition, it was shown that bacterial biotin sulfoxide reductase BisC can reduce specifically the S-diastereomer of free MetO [18].
The use of genetically modified organisms with knocked-out or overexpressed MSR genes allowed defining the two main roles of MSRs in cells, i.e. protection against oxidative stress and regulation of lifespan. However, because of the lack of clearly identified target proteins for either MSRA or MSRB, the specific cellular processes involved remain unclear. Due to its early discovery, MSRA received much more attention than other MSRs. It was found that knockout of MSRA genes increased susceptibility to oxidative stress in bacteria [19-21], yeast [22, 23], Caenorhabditis elegans [24], mice [25], and plants [26, 27]. Conversely, overexpression of MSRA increased resistance to oxidative stress in Drosophila [28], mammalian cells [29, 30], and plants [26]. Deletion of MSRA often reduces lifespan, whereas its overexpression was shown to increase lifespan of fruit flies by 70% [28]. The involvement of MSRBs in protection against oxidative stress was similarly shown in knockout plants [31, 32] or human cells overexpressing the enzyme [33], but their roles remain somewhat less clear due to weaker phenotypes. For example, deletion of MSRA in yeast results in a stronger sensitivity to hydrogen peroxide than the deletion of MSRB [17, 22, 34], and MSRB overexpression did not affect the lifespan of fruit flies [35]. Moreover, deletion of MSRB did not noticeably affect yeast lifespan, although strains deficient in both MSRA and MSRB exhibited a greater reduction in lifespan compared to cells deficient in MSRA only [22, 34]. A role in the host/pathogen interaction was also identified for MSRs, whose genes were highly expressed during pathogen invasion in both the host and the pathogen in response to the high levels of reactive oxygen species produced by both organisms [21, 36-38].
DIVERSITY OF MSRs AND THEIR REGENERATION MECHANISMS
MSRs belong to the family of thiol oxidoreductases, which possess redox-active Cys and/or selenocysteines (Sec) residues. Although both MSRA and MSRB catalyze the reduction of MetO, they do not share sequence similarity [39]. Interestingly, determination of protein structures from various organisms indicates a mirror-like relationship between the MSRA and MSRB active sites [39], in which a Trp faces the catalytic Cys and allows the docking of the substrate in the optimal position for its reduction (Figs. 1a and 1b). MSRA has a G[C/U]FW motif, located in the N-terminal part of the protein, that includes the catalytic residue (Fig. 1c). The great majority of known MSRAs possess a catalytic Cys, with a few Sec-containing protein forms found in ticks, spiders, some marine organisms, and certain unicellular algae such as Chlamydomonas reinhardtii [15, 40]. Extensive characterization of bacterial MSRAs [41-43] as well as determination of three-dimensional structures of prokaryotic and eukaryotic enzymes [44-48] yielded a mechanism of MetO reduction wherein a sulfenic acid is formed on the catalytic Cys after the formation of a sulfurane-type transition state [42, 43]. Although the existence of the sulfenic acid was established for E. coli [41], P. trichocarpa [47], and mouse enzymes [49], the mechanism leading to its formation is still unclear. Indeed, it was proposed that the oxygen atom forming the sulfenic acid comes directly from MetO [43], but the data based on the use of water labeled with isotopic 18-oxygen (H218O) argue that the sulfenic acid is derived from a water molecule [49].
The majority of MSRBs possess a catalytic Cys as part of the RxCxN motif located in the C-terminal region. Mammals express a Sec-containing form of MSRB displaying a slightly different active site in which the asparagine conserved in all identified Cys-containing isoforms is replaced with phenylalanine [50] (Fig. 1d). As in the case of MSRA, theoretical and biochemical studies give conflicting models for the MetO reduction step catalyzed by MSRB. Both models agree on the fact that a sulfonium cation is the initial intermediate, but an in silico analysis indicates that the formation of a sulfenic acid is not enzymatically feasible [51-53]. On the other hand, biochemical analyses using sulfenic acid-specific reagents and mass spectrometry demonstrated the formation of a sulfenic acid in archaeal [54], plant [55, 56], and fruit fly [57] MSRBs, although the mechanism leading to its formation was not addressed.Fig. 1. Mirror-like relationship between MSRA (a) and MSRB (b) active sites and the multiple sequence alignment of representative MSRAs (c) and MSRBs (d). The residues of the Bos taurus MSRA [44] (Protein Data Bank accession 1FVA) (a) and of the MSRB domain of the Neisseria gonorrhoeae pilB [39] (Protein Data Bank accession 1L1D) (b) involved in catalysis are shown in stick representation. The figure was created using PyMol v0.99 (Delano Scientific LLC). Strictly conserved amino acids of typical MSRAs (c) and MSRBs (d) are shown in white on black, and other conserved amino acids are shaded. Cysteine residues are highlighted by black boxes. Catalytic Cys or Sec are indicated by black arrows. Biochemically validated resolving Cys are indicated by gray arrows. Black dots correspond to the conserved Cys involved in the coordination of Zn in MSRB sequences. UniProt accession numbers: Populus trichocarpa, Q6QPJ5; Bos taurus, P54149; Saccharomyces cerevisiae, C8Z745; Synechocystis, P72800; Bacillus subtilis, P54154; Chlamydomonas reinhardtii, Q8H6T1; Escherichia coli, P0A744; Xanthomonas campestris, B0RWG5; Arabidopsis thaliana, Q9C8M2, Homo sapiens, Q9NZV6. Poorly conserved N-terminal regions are not shown.
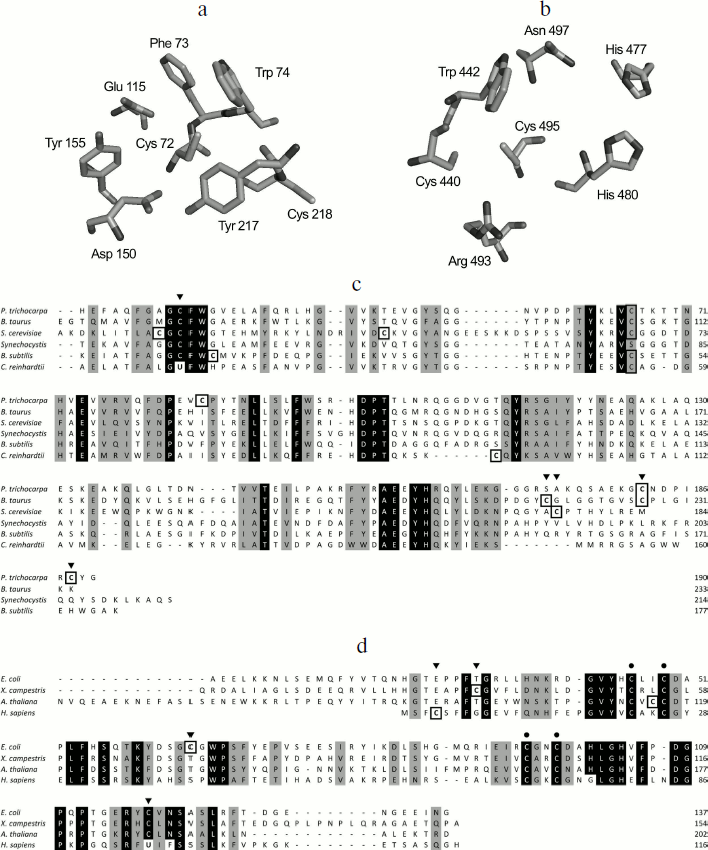
In the case of both MSRA and MSRB forms containing Sec, the formation of a selenenic acid was proposed as an intermediate [40, 50], but no biochemical evidence has been reported. Although not entirely confirmed for all MSR forms, the formation of sulfenic acid (or selenenic acid) on the catalytic Cys (or Sec) appears to be the first step in their mechanism, concomitant with the reduction of the substrate (Fig. 2a). For a majority of identified MSRs, the second step is the reduction of the sulfenic (selenenic) acid by an intramolecular resolving Cys leading to the formation of a disulfide (or selenenylsulfide) bond. The last step is the regeneration of the reduced form of MSR by a disulfide exchange reaction with thioredoxin (Trx) (Fig. 2a). In this review, the MSRs containing only the catalytic and resolving Cys are termed 2-Cys MSRs, and the enzymes containing a catalytic Sec and a resolving Cys, 1-Sec/1-Cys MSRs. In the case of MSRAs, the characterized 2-Cys enzymes, such as the S. cerevisiae [58, 59] or Neisseria meningitidis [60] proteins, possess the resolving Cys in the C-terminal region (Fig. 1c). Several other Cys are present in the sequences of these 2-Cys MSRAs, but site-directed mutagenesis approaches demonstrated that they are neither involved in the reduction of the substrate nor in the regeneration of enzyme activity.
Another type of MSRA, represented by the Bacillus subtilis enzyme, possesses only one potential resolving Cys located three amino acids after the catalytic Cys. Although not biochemically proven, proximity with the catalytic Cys and the protein structure similar to other thiol oxidoreductases, such as Trx, suggest that this Cys could reduce the sulfenic acid formed on the catalytic Cys after the reduction of the substrate, and thus can act as the resolving Cys [61]. Some MSRAs possess two resolving Cys residues, termed 3-Cys MSRAs, such as the E. coli [41, 62], Bos taurus [44], and P. trichocarpa [47] enzymes (Fig. 1c). For these proteins, the first disulfide bond formed between the catalytic Cys and the first resolving Cys is further reduced by the second resolving Cys, leading to the formation of a disulfide bond between the two resolving Cys, which is then reduced by Trx (Fig. 2b). Interestingly, these resolving Cys residues are located in the C-terminal part of the protein, but they show poor conservation and exhibit variations in the order in which they participate in the reduction of catalytic Cys. Indeed, in plant 3-Cys MSRAs, the first disulfide bond is formed between the catalytic Cys and the most C-terminal Cys, and then it is reduced by the other resolving Cys [47], whereas in the E. coli and Bos taurus enzymes, the most C-terminal Cys acts as the second resolving residue [41, 44, 62].Fig. 2. Regeneration mechanisms of different forms of MSRs. a) 2-Cys MSRA and 1-Sec/1-Cys MSRB. Reduction of MetO by an MSR leads to the formation of sulfenic acid or selenenic acid on the catalytic residue, Cys or Sec (1), which is followed by the formation of disulfide (or selenenylsulfide) and the release of one molecule of water (2). The disulfide bond is then reduced by Trx through disulfide exchange, and thereby a transient intermolecular covalent complex is formed (3) and then reduced, leading to the release of the reduced form of MSR and the oxidized form of Trx (4). b) 3-Cys MSRA. The steps (1) and (2) are identical to those shown in (a). A second resolving Cys reduces the first disulfide bond, leading to the formation of a disulfide bond between the two resolving Cys (3), which is then reduced by Trx (4) leading to the release of the reduced 3-Cys MSRA and oxidized Trx (5). c) 1-Cys MSRA, 1-Sec MSRA, and 1-Cys MSRB. A sulfenic or selenenic acid is formed after the reduction of MetO (1). Pathway 1: the catalytic Cys of Trx directly reduces the sulfenic (selenenic) acid leading to the release of a water molecule and the formation of a transient intermolecular disulfide between MSR and Trx (2), which is then reduced by the resolving Cys of Trx, leading to the release of the reduced MSR and oxidized Trx (3). Pathway 2: the sulfenic (selenenic) acid reacts with reduced glutathione (GSH) leading to glutathionylation of the catalytic Cys of MSR and release of water (2′). The MSR-glutathione adduct is then reduced by Grx through a monothiol mechanism, leading to the release of the reduced MSR and glutathionylated Grx (3′).
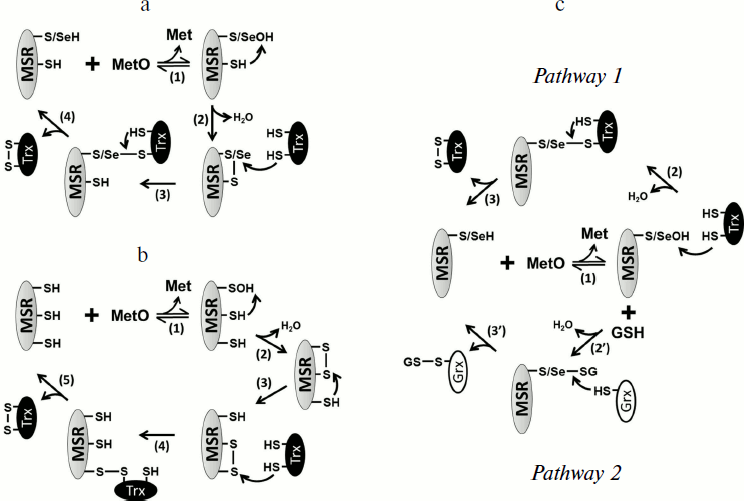
Some prokaryotic MSRAs, illustrated by the Synechocystis sequence and termed 1-Cys MSRA, contain only the catalytic Cys (Fig. 1c), indicating the use of an alternative mechanism for the regeneration of their activity. Similarly, the Sec-containing form in C. reinhardtii possesses two Cys in addition of the catalytic Sec, but mutagenesis experiments showed that their Cys residues are not redox active, and that the enzyme can be defined as a 1-Sec MSRA [40]. Similarities with other characterized MSRs suggest that their regeneration mechanism involves the direct reduction of the sulfenic/selenenic acid by Trx (Fig. 2c, pathway 1). Recently, it was shown that glutaredoxin (Grx) and glutathione (GSH) can also regenerate the activity of 1-Cys MSRAs [63]. By analogy with the mechanism of known 1-Cys MSRBs (see below), these findings suggest that the regeneration of 1-Cys MSRA by Grx/GSH proceeds through glutathionylation/deglutathionylation (Fig. 2c, pathway 2).
Based on the similarity of mechanisms of MSRs and other thiol oxidoreductases, such as peroxiredoxins (Prxs) and glutathione peroxidases (GPxs), another potential mechanism could involve the formation of a sulfenyl–amide or selenenyl–amide bond, formed by the reaction of the sulfenic or selenenic acid with a nitrogen atom of an adjacent residue, which could be directly reduced by Trx or another thiol compound, such as GSH [64]. The mechanisms of regeneration of 1-Cys and 1-Sec MSRA have not been thoroughly characterized.
The typical MSRBs, such as the E. coli enzyme, possess a unique resolving Cys located in the well-conserved GCGWP motif and are defined as 2-Cys MSRBs [15, 65] (Fig. 1d). For some proteins, the position of the resolving Cys is not conserved, as shown for the X. campestris enzyme, in which the resolving Cys is in the N-terminal part of the protein [66] (Fig. 1d). The main mammalian form, MSRB1, possesses a catalytic Sec and a resolving Cys located in position 4 of the sequence (Fig. 1d) and can be defined as a 1-Sec/1-Cys MSRB [50]. In all cases, the regeneration process is similar to that of 2-Cys MSRAs (Fig. 2a). Among all identified MSRBs, around 40% do not possess a potential resolving Cys. The best-characterized form is the plastidial 1-Cys MSRB1 from plants. Two mechanisms for the regeneration of its activity were delineated (Fig. 2c). The first is the direct reduction of the sulfenic acid by the plant-specific Trx CDSP32 [56] (Fig. 2c, pathway 1), and the second is the reduction of the oxidized catalytic Cys by the Grx/GSH system [55, 67] (Fig. 2c, pathway 2). Mammalian genomes code for two 1-Cys MSRBs, MSRB2 and MSRB3, which are likely reduced similarly to the A. thaliana 1-Cys MSRB1 as it was shown that Trx, and particularly the plant Trx CDSP32, can regenerate their activities [50, 68, 69].
The fRMSR enzyme belongs to a family of GAF-domain proteins and is structurally unrelated to either MSRA or MSRB. However, it uses a similar thiol-based chemistry for the reduction of the R-diastereomer of free MetO. Sequence alignments, three-dimensional structure analyses and biochemical assays demonstrated that fRMSRs possess a catalytic Cys on which a sulfenic acid is formed after the reduction of the substrate and that the regeneration involves a unique resolving Cys, similar to 2-Cys MSRs [16, 17, 70, 71] (Fig. 2a).
CATALYTIC PARAMETERS OF MSRs
Several lines of evidence argue for the role of Trxs as in vivo reductants of MSRA, MSRB, and fRMSR. Indeed, deletion of Trx genes in the context of Met auxotrophy in E. coli [72, 73] and yeast [74] renders these genetically modified organisms unable to use MetO as the sole source of Met. Moreover, MSRs were found in proteomic searches for Trx targets in algae [75] and higher plants [76-78]. The fact that some prokaryotic genomes encode fusion proteins containing MSR and Trx domains [39, 79] also strongly argues for the role of Trx as MSR reductant. Interestingly, the alga Gracilaria gracilis possesses a gene encoding a fusion of two Grx domains and a MSRA domain (NCBI accession: AAD43253.1), suggesting that Grx could act as an in vivo reductant in some cases, as proposed for 1-Cys MSRs [55, 63, 80]. This hypothesis is reinforced by the fact that overexpression of Grx1 allowed the growth of the E. coli Trx mutant auxotroph for Met on MetO as the source of Met [72, 81].
In vitro, the MetO reduction activity of MSRs can be determined using dithiothreitol (DTT) as a reductant and dabsyl (dimethylaminoazosulfonate)-MetO as substrate, following the formation of the product, dabsyl-Met, by HPLC. This method allowed determining catalytic constants (kcat) and specific activities for all types of MSRA and MSRB. It should be noted that fRMSR does not reduce dabsyl-MetO [16, 17]. For MSRAs, the kcat varies from 0.03 s–1 for yeast 2-Cys MSRA to 4 s–1 for 1-Sec MSRA from C. reinhardtii (Table 1). This selenoprotein shows the highest activity, and mutation of its Sec to Cys decreased its activity 10-fold. Such decrease was similarly observed for GPx [64], indicating that, in the context of thiol oxidoreductases, Sec has higher reactivity than Cys. In the case of MSRBs, the kcat was around 0.05 s–1, which can be compared with 0.004 s–1 for yeast 2-Cys MSRB and 0.14 s–1 for human 1-Cys MSRB3 (Table 1). As for 1-Sec MSRB, mutation of its catalytic Sec in the mouse 1-Sec/1-Cys enzyme dramatically decreased MetO reductase activity (Table 1).
Table 1. Catalysis of dabsyl-MetO reduction
by MSRs using DTT as a reducing agent
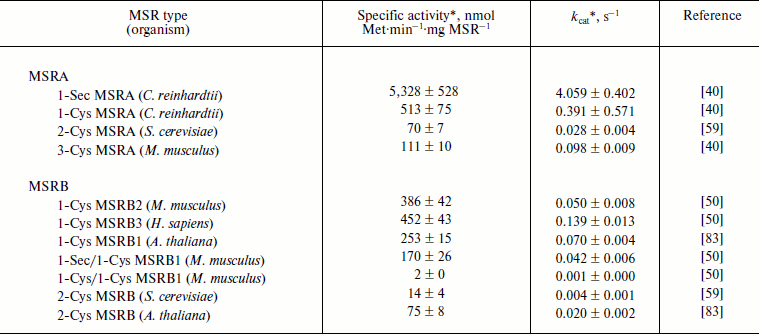
* The activities were recorded for a single concentration of
substrate.
The MSR activity can also be determined using a reconstituted Trx system composed of NADPH, Trx reductase (TR), and Trx. Alternatively, the GSH/Grx system composed of NADPH, GSH, GSH reductase (GR), and Grx can be used, particularly for 1-Cys MSRBs not efficiently reduced by Trx [55, 67, 80]. In both cases, the consumption of NADPH is directly proportional to MetO reduction [9, 55, 59, 82] and can be monitored spectrophotometrically following absorbance at 340 nm. This presents an advantage in that such assay is physiologically relevant and allows the use of any MetO-containing substrate. Indeed, such system was used to determine the kinetic parameters for MSRs using free MetO, N-acetyl-MetO, dabsyl-MetO, or oxidized proteins as substrates (Tables 2-4). In the Trx-dependent reaction, the oxidized MSR produced by the reduction of MetO is the substrate of Trx. Their interaction follows Michaelis–Menten kinetics, and the KM values for various MSRs using Trx as a substrate were found to be in the low micromolar range [47, 62, 80, 83-85]. Similarly, in the case of the Grx-dependent reaction, the glutathionylated MSR was the substrate for Grx, and its KM values were in the low micromolar range [55, 63, 80, 86]. In general, MSRs display Michaelis–Menten kinetics with their substrates using Trx and GSH/Grx systems [47, 55, 59, 62, 63, 65, 80, 83, 84, 86]. In the complete enzymatic system, three enzymes, MSR, Trx (or Grx), and TR (or GR), are successively reduced and oxidized, and they follow a ping-pong mechanism [62, 65]. This is a feature of many thiol oxidoreductases, as shown for peroxiredoxins and other peroxidases [87]. The catalytic parameters of free MetO reduction were determined with the Trx system for typical MSRA, MSRB, and fRMSR forms (Table 2). For MSRAs, the catalytic activities varied from 0.3 s–1 for P. trichocarpa 3-Cys MSRA to ~8 s–1 for yeast 2-Cys MSRA. The KM values for free MetO were in the range of 0.24-2.1 mM, giving catalytic efficiencies (kcat/KM) from 1,200 to 13,770 M–1·s–1 for plant and yeast MSRAs, respectively. The catalytic efficiencies recorded for the tested MSRBs were far lower than those for MSRAs due to lower activities and higher KM for free MetO. This is particularly the case for E. coli enzymes, with MSRB having kcat/KM ~1,300-fold lower than that of MSRA. Catalytic efficiencies of fRMSRs are in the range of those recorded for MSRAs (Table 2).
Table 2. Kinetic parameters of free MetO
reduction by MSRs using the NADPH-coupled Trx system
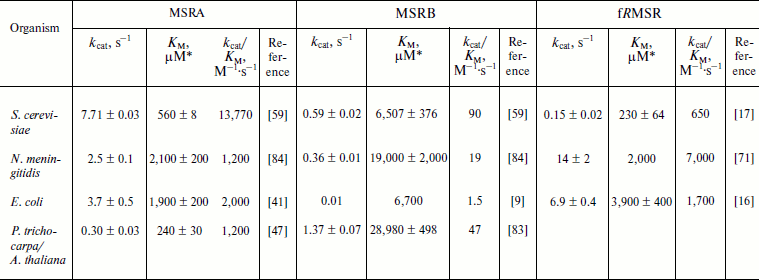
* Catalytic parameters were determined using R- and
S-diastereomers of free MetO, or a racemic mixture. In the
latter case, for accurate comparison, the KM values
presented correspond to the half of the measured values, as only one
diastereomer could be reduced by each MSR type and inhibition of MSR
activity by the other diastereomer was not demonstrated.
The reductase activity of all typical MSRAs and MSRBs was also determined using the chemical variants of free MetO, i.e. N-acetyl-MetO and dabsyl-MetO (Table 3). Addition of another group to the oxidized amino acid increases its volume and, thus, these modified forms of MetO are considered as peptide-bond mimics of MetO-containing substrates. In the case of MSRAs, the catalytic activities and KM values, when determined, were in the range of those recorded with free MetO (Table 2). As observed using DTT as a reductant, the Sec-to-Cys form of C. reinhardtii MSRA had a 10-fold lower activity, indicating that the Sec-containing MSRA was more efficient in the reduction of the substrate than the Cys mutant independent of the reductant used. MSRBs showed catalytic activities with N-acetyl-MetO and dabsyl-MetO similar to those recorded with free MetO, but the KM values were lower. The resulting kcat/KM values varied from ~170 for A. thaliana 1-Cys MSRB and bacterial 2-Cys MSRBs to ~2,700 M–1·s–1 for plant 2-Cys MSRBs (Table 3).
Table 3. Kinetic parameters of
N-acetyl-MetO or dabsyl-MetO reduction by MSRs using the
NADPH-coupled Trx or GSH/Grx system
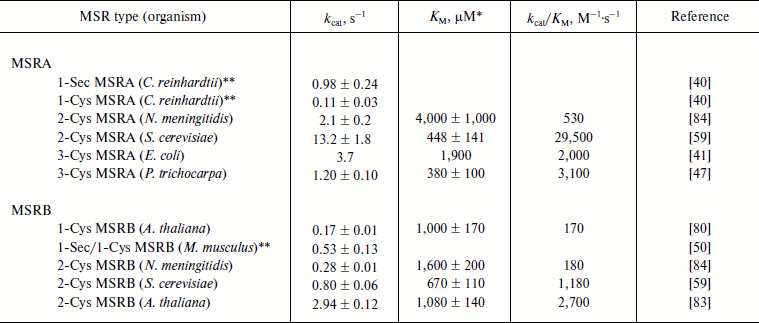
* Catalytic parameters were determined using R- and
S-diastereomers of N-acetyl-MetO or dabsyl-MetO, or their
racemic mixtures. In the latter case, for accurate comparison, the
KM values presented correspond to the half of the
measured values, as only one diastereomer could be reduced by each MSR
type and inhibition of MSR activity by the other diastereomer was not
demonstrated.
** In this study [40], activities were recorded for
a single concentration of the substrate.
NADPH-dependent Trx and GSH/Grx systems also support the use oxidized proteins as substrates. Catalytic parameters for the reduction of MetO-containing proteins were determined for the oxidized form of calmodulin (CaMox) in a reaction catalyzed by bovine MSRA [88] and for several model proteins for reactions catalyzed by yeast MSRA and MSRB [59] (Table 4). In the case of the reduction of oxidized proteins by MSRAs (Table 4), catalytic efficiencies were in the range of those recorded with the low molecular weight MetO-containing molecules (Tables 2 and 3). However, the KM values were at least 10-fold lower for oxidized proteins. Interestingly, the use of the same set of oxidized proteins in reactions catalyzed by the yeast MSRB demonstrated that MSRB is as efficient as MSRA [59] (Table 4). CaMox was also used to demonstrate the capacity of E. coli MSRs for the reduction of MetO in oxidized proteins. With 30 µM CaMox, the kcat values of ~0.08 and ~0.05 s–1 were recorded for MSRA and MSRB, respectively [9] (Table 4). In the case of MSRA, this value was in excellent agreement with the theoretical kcat of CaMox reduction by bovine MSRA, calculated for the 30 µM substrate to be ~0.086 s–1, using Michaelis–Menten parameters. By comparison, the theoretical kcat of the bacterial MSRA using 30 µM free MetO should be ~0.06 s–1, which is similar to those recorded for CaMox. However, the theoretical kcat calculated for MSRB using 30 µM free MetO was only ~4.5·10–5 s–1, which is more than 1,000-fold lower. These data indicate that E. coli MSRA reduces free MetO and CaMox with equivalent efficiencies, whereas MSRB is specialized for the reduction of MetO in protein substrates. The activity of E. coli MSRs was also tested using the Ffh protein, a component of the signal recognition particle [82]. This protein contains 28 Met and, after oxidation, becomes a very good substrate for prokaryotic MSRs as shown by the extremely high activities recorded with 10 µM substrate (Table 4). Indeed, for both MSRs the values were more than 300-fold higher with 10 µM oxidized Ffh than with 30 µM CaMox (Table 4). The activities of plastidial 1-Cys MSRB1 and 2-Cys MSRB2 of A. thaliana were tested using several potential substrates [32, 89] (Table 4). For all, with the concentration of the oxidized protein between 10 and 24 µM, the activities were ~0.3 to 2.6 s–1 (Table 4). Similar to E. coli MSRB, the activity was, at an equivalent concentration of free MetO, ~10,000-fold lower with 2-Cys MSRB2 (Table 2), indicating that the plant enzyme is specialized for the reduction of MetO in proteins.
Table 4. Catalysis of reduction of oxidized
proteins by MSRs using the NADPH-coupled Trx or GSH/Grx system
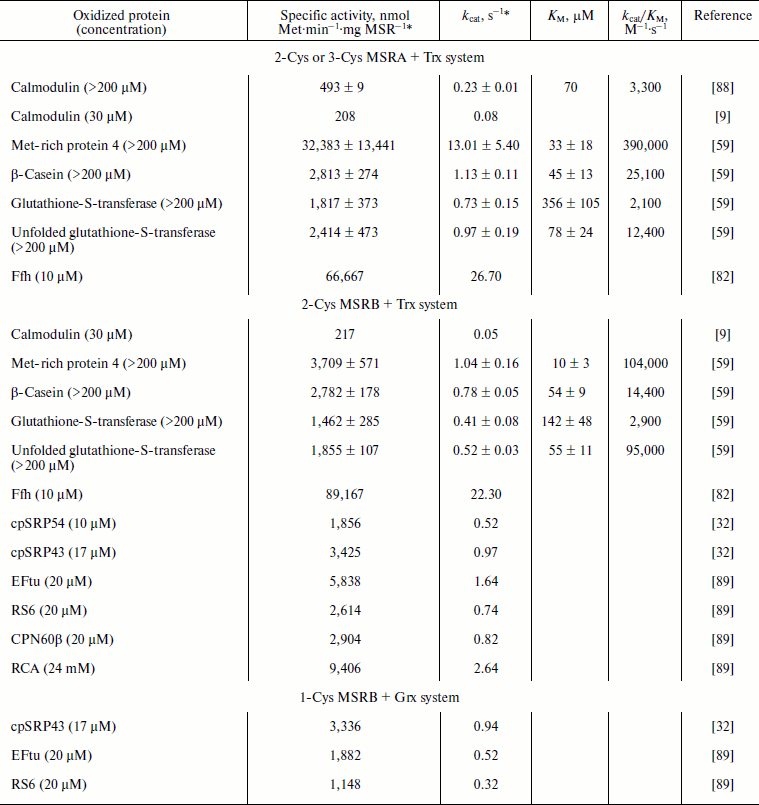
* Except in references [59] and [88], the activities were recorded for a single
substrate concentration.
MSRs are ubiquitous enzymes found in organisms across the three domains of life, and they are involved in fundamental processes such as resistance to abiotic and biotic stresses. They also regulate lifespan of various organisms. Although studied for many years, catalysis by MSRAs, MSRBs, and fRMSRs is incompletely understood. For instance, the mechanisms leading to the formation of the sulfenic acid require further studies.
Activity assays involving recombinant enzymes allowed delineation of regeneration mechanisms for most of the typical MSRs; however, some details remain unclear. For example, the regeneration of 1-Cys MSRs is not fully clear, and the mechanism proposed for plant 1-Cys MSRB1 has not been verified in the physiological setting. Similarly, information on the mechanisms of Sec-containing forms is scarce. Comparison of catalytic properties of different MSR types, discussed in this review, sheds light on their use of substrates. Whereas MSRAs show similar catalytic efficiency for all MetO-containing substrates, MSRBs are very poor reductants of free MetO and low molecular weight MetO compounds, whereas they efficiently reduce MetO in proteins. In excellent agreement with this finding, mammalian MSRAs reduce the S-diastereomers of various methylsulfinyl-containing drugs and compounds, whereas MSRBs are inactive on the R-diastereomers [90]. Moreover, human cells cannot reduce the R-diastereomer of free MetO, despite the presence of three MSRBs in these cells [91]. Analyses of reduced and oxidized structures of MSRA and MSRB demonstrated high flexibility of these enzymes [39, 48, 92-94]. These properties should be important for the interaction with their substrates, which could hardly be mimicked by small molecules. In the reduced state, MSR should have high affinity for oxidized proteins, whereas the oxidized enzymes, after releasing the product, should have high affinity for the reductant. Determination of precise catalytic parameters of MSRA and MSRB using oxidized proteins would bring substantial information on the functions of these enzymes. For instance, in a recent study [59] we showed that yeast MSRs are more efficient in reducing unfolded oxidized proteins, and thus could have a major function in rescuing and repairing oxidized nascent polypeptides as well as proteins en route for subcellular compartments or unfolded by oxidative stress. It was also found that MetO in oxidized proteins in human cells is mainly surrounded by polar amino acids (Asp, Glu, Arg, Lys, Gln) and very likely localized on protein surface [95]. The use of synthetic peptides showed that the nature of the amino acid in the –1 position relative to MetO is important; this amino acid influenced the reduction capacity, with Arg favoring the reduction, and Glu, Asp, or Pro decreasing the MSR activity [95]. Another recent characterization of the mouse MSRA demonstrated that the enzyme could be myristoylated [96], and this modification strongly increases its capacity for reduction of MetO in a potential protein substrate, but does not affect its activity with small peptides [97]. The use of model proteins and known MSR targets should help to better define MSR reaction mechanisms and could bring new information on the in vivo functions of these enzymes.
REFERENCES
1.Fukagawa, N. K. (2006) J. Nutr., 136,
1676S-1681S.
2.Luo, S., and Levine, R. L. (2009) FASEB J.,
23, 464-472.
3.Marino, S. M., and Gladyshev, V. N. (2010) J.
Mol. Biol., 404, 902-916.
4.Kochanczyk, M. (2011) BMC Struct. Biol.,
11, 34.
5.Rhee, S. G., and Woo, H. A. (2011) Antioxid.
Redox. Signal., 15, 781-794.
6.Tanner, J. J., Parsons, Z. D., Cummings, A. H.,
Zhou, H., and Gates, K. S. (2011) Antioxid. Redox Signal.,
15, 77-97.
7.Lee, B. C., and Gladyshev, V. N. (2011) Free
Radic. Biol. Med., 50, 221-227.
8.Brot, N., Weissbach, L., Werth, J., and Weissbach,
H. (1981) Proc. Natl. Acad. Sci. USA, 78, 2155-2158.
9.Grimaud, R., Ezraty, B., Mitchell, J. K., Lafitte,
D., Briand, C., Derrick, P. J., and Barras, F. (2001) J. Biol.
Chem., 276, 48915-48920.
10.Kryukov, G. V., Kumar, R. A., Koc, A., Sun, Z.,
and Gladyshev, V. N. (2002) Proc. Natl. Acad. Sci. USA,
99, 4245-4250.
11.Black, S., Harte, E. M., Hudson, B., and
Wartofsky, L. (1960) J. Biol. Chem., 235, 2910-2916.
12.Ejiri, S. I., Weissbach, H., and Brot, N. (1979)
J. Bacteriol., 139, 161-164.
13.Delaye, L., Becerra, A., Orgel, L., and Lazcano,
A. (2007) J. Mol. Evol., 64, 15-32.
14.Zhang, X-H., and Weissbach, H. (2008) Biol.
Rev. Camb. Philos. Soc., 83, 249-257.
15.Tarrago, L., Laugier, E., and Rey, P. (2009)
Mol. Plant, 2, 202-217.
16.Lin, Z., Johnson, L. C., Weissbach, H., Brot, N.,
Lively, M. O., and Lowther, W. T. (2007) Proc. Natl. Acad. Sci.
USA, 104, 9597-9602.
17.Le, D. T., Lee, B. C., Marino, S. M., Zhang, Y.,
Fomenko, D. E., Kaya, A., Hacioglu, E., Kwak, G. H., Koc, A., Kim, H.
Y., and Gladyshev, V. N. (2009) J. Biol. Chem., 284,
4354-4364.
18.Ezraty, B., Bos, J., Barras, F., and Aussel, L.
(2005) J. Bacteriol., 187, 231-237.
19.St John, G., Brot, N., Ruan, J.,
Erdjument-Bromage, H., Tempst, P., Weissbach, H., and Nathan, C. (2001)
Proc. Natl. Acad. Sci. USA, 98, 9901-9906.
20.Alamuri, P., and Maier, R. J. (2004) Mol.
Microbiol., 53, 1397-1406.
21.Lee, W. L., Gold, B., Darby, C., Brot, N., Jiang,
X., de Carvalho, L. P., Wellner, D., St John, G., Jacobs, W. R., Jr.,
and Nathan, C. (2009) Mol. Microbiol., 71, 583-593.
22.Koc, A., Gasch, A. P., Rutherford, J. C., Kim,
H-Y., and Gladyshev, V. N. (2004) Proc. Natl. Acad. Sci. USA,
101, 7999-8004.
23.Kaya, A., Koc, A., Lee, B. C., Fomenko, D. E.,
Rederstorff, M., Krol, A., Lescure, A., and Gladyshev, V. N. (2010)
Biochemistry, 49, 8618-8625.
24.Minniti, A. N., Cataldo, R., Trigo, C., Vasquez,
L., Mujica, P., Leighton, F., Inestrosa, N. C., and Aldunate, R. (2009)
Aging Cell, 8, 690-705.
25.Zhang, C., Jia, P., Jia, Y., Weissbach, H.,
Webster, K. A., Huang, X., Lemanski, S. L., Achary, M., and Lemanski,
L. F. (2010) J. Cell Biochem., 111, 94-103.
26.Romero, H. M., Berlett, B. S., Jensen, P. J.,
Pell, E. J., and Tien, M. (2004) Plant Physiol., 136,
3784-3794.
27.Guo, X., Wu, Y., Wang, Y., Chen, Y., and Chu, C.
(2009) Planta, 230, 227-238.
28.Ruan, H., Tang, X. D., Chen, M. L., Joiner, M.
L., Sun, G., Brot, N., Weissbach, H., Heinemann, S. H., Iverson, L.,
Wu, C. F., and Hoshi, T. (2002) Proc. Natl. Acad. Sci. USA,
99, 2748-2753.
29.Picot, C. R., Petropoulos, I., Perichon, M.,
Moreau, M., Nizard, C., and Friguet, B. (2005) Free Radic. Biol.
Med., 39, 1332-1341.
30.Haenold, R., Wassef, R., Brot, N., Neugebauer,
S., Leipold, E., Heinemann, S. H., and Hoshi, T. (2008) Free Radic.
Res., 42, 978-988.
31.Kwon, S. J., Kwon, S. I., Bae, M. S., Cho, E. J.,
and Park, O. K. (2007) Plant Cell. Physiol., 48,
1713-1723.
32.Laugier, E., Tarrago, L., Vieira Dos Santos, C.,
Eymery, F., Havaux, M., and Rey, P. (2010) Plant J., 61,
271-282.
33.Cabreiro, F., Picot, C. R., Friguet, B., and
Petropoulos, I. (2006) Ann. N. Y. Acad. Sci., 1067,
37-44.
34.Koc, A., and Gladyshev, V. N. (2007) Ann. N.
Y. Acad. Sci., 1100, 383-386.
35.Shchedrina, V. A., Vorbruggen, G., Lee, B. C.,
Kim, H. Y., Kabil, H., Harshman, L. G., and Gladyshev, V. N. (2009)
Mech. Ageing Dev., 130, 429-443.
36.Achilli, C., Ciana, A., Rossi, A., Balduini, C.,
and Minetti, G. (2008) J. Leukoc. Biol., 83, 181-189.
37.Rosen, H., Klebanoff, S. J., Wang, Y., Brot, N.,
Heinecke, J. W., and Fu, X. (2009) Proc. Natl. Acad. Sci. USA,
106, 18686-18691.
38.Mahawar, M., Tran, V., Sharp, J. S., and Maier,
R. J. (2011) J. Biol. Chem., 286, 19159-19169.
39.Lowther, W. T., Weissbach, H., Etienne, F., Brot,
N., and Matthews, B. W. (2002) Nat. Struct. Biol.,
9, 348-352.
40.Kim, H-Y., Fomenko, D. E., Yoon, Y-E., and
Gladyshev, V. N. (2006) Biochemistry, 45,
13697-13704.
41.Boschi-Muller, S., Azza, S., Sanglier-Cianferani,
S., Talfournier, F., Van Dorsselear, A., and Branlant, G. (2000) J.
Biol. Chem., 275, 35908-35913.
42.Antoine, M., Gand, A., Boschi-Muller, S., and
Branlant, G. (2006) J. Biol. Chem., 281,
39062-39070.
43.Boschi-Muller, S., Gand, A., and Branlant, G.
(2008) Arch. Biochem. Biophys., 474, 266-273.
44.Lowther, W. T., Brot, N., Weissbach, H., and
Matthews, B. W. (2000) Biochemistry, 39, 13307-13312.
45.Tete-Favier, F., Cobessi, D., Boschi-Muller, S.,
Azza, S., Branlant, G., and Aubry, A. (2000) Structure,
8, 1167-1178.
46.Taylor, A. B., Benglis, D. M., Jr.,
Dhandayuthapani, S., and Hart, P. J. (2003) J. Bacteriol.,
185, 4119-4126.
47.Rouhier, N., Kauffmann, B., Tete-Favier, F.,
Palladino, P., Gans, P., Branlant, G., Jacquot, J. P., and
Boschi-Muller, S. (2007) J. Biol. Chem., 282,
3367-3378.
48.Ranaivoson, F. M., Antoine, M., Kauffmann, B.,
Boschi-Muller, S., Aubry, A., Branlant, G., and Favier, F. (2008) J.
Mol. Biol., 377, 268-280.
49.Lim, J. C., You, Z., Kim, G., and Levine, R. L.
(2011) Proc. Natl. Acad. Sci. USA, 108, 10472-10477.
50.Kim, H-Y., and Gladyshev, V. N. (2005) PLoS
Biol., 3, e375.
51.Robinet, J. J., Dokainish, H. M., Paterson, D.
J., and Gauld, J. W. (2011) J. Phys. Chem. B, 115,
9202-9212.
52.Neiers, F., Sonkaria, S., Olry, A.,
Boschi-Muller, S., and Branlant, G. (2007) J. Biol. Chem.,
282, 32397-32405.
53.Neiers, F., Boschi-Muller, S., and Branlant, G.
(2011) J. Phys. Chem. B, 115, 10775; 10776-10777.
54.Carella, M., Becher, J., Ohlenschlager, O.,
Ramachandran, R., Guhrs, K. H., Wellenreuther, G., Meyer-Klaucke, W.,
Heinemann, S. H., and Gorlach, M. (2011) Mol. Microbiol.,
79, 342-358.
55.Tarrago, L., Laugier, E., Zaffagnini, M.,
Marchand, C., Le Marechal, P., Rouhier, N., Lemaire, S. D., and Rey, P.
(2009) J. Biol. Chem., 284, 18963-18971.
56.Tarrago, L., Laugier, E., Zaffagnini, M.,
Marchand, C. H., Le Marechal, P., Lemaire, S. D., and Rey, P. (2010)
J. Biol. Chem., 285, 14964-14972.
57.Kumar, R. A., Koc, A., Cerny, R. L., and
Gladyshev, V. N. (2002) J. Biol. Chem., 277,
37527-37535.
58.Ma, X. X., Guo, P. C., Shi, W. W., Luo, M., Tan,
X. F., Chen, Y., and Zhou, C. Z. (2011) J. Biol. Chem.,
286, 13430-13437.
59.Tarrago, L., Kaya, A., Weerapana, E., Marino, S.
M., and Gladyshev, V. N. (2012) J. Biol. Chem., 287,
24448-24459.
60.Olry, A., Boschi-Muller, S., and Branlant, G.
(2004) Biochemistry, 43, 11616-11622.
61.Gladyshev, V. N. (2002) Proteins,
46, 149-152.
62.Boschi-Muller, S., Azza, S., and Branlant, G.
(2001) Protein Sci., 10, 2272-2279.
63.Kim, H-Y. (2012) Acta Biochim. Biophys.
Sin., 44, 623-627.
64.Flohe, L., Toppo, S., Cozza, G., and Ursini, F.
(2011) Antioxid. Redox Signal., 15, 763-780.
65.Boschi-Muller, S., Olry, A., Antoine, M., and
Branlant, G. (2005) Biochim. Biophys. Acta, 1703,
231-238.
66.Neiers, F., Kriznik, A., Boschi-Muller, S., and
Branlant, G. (2004) J. Biol. Chem., 279, 42462-42468.
67.Chibani, K., Tarrago, L., Gualberto, J. M.,
Wingsle, G., Rey, P., Jacquot, J. P., and Rouhier, N. (2012) Plant
Physiol., 159, 592-605.
68.Ding, D., Sagher, D., Laugier, E., Rey, P.,
Weissbach, H., and Zhang, X. H. (2007) Biochem. Biophys. Res.
Commun., 361, 629-633.
69.Kim, H-Y., and Kim, J-R. (2008) Biochem.
Biophys. Res. Commun., 371, 490-494.
70.Bong, S. M., Kwak, G. H., Moon, J. H., Lee, K.
S., Kim, H. S., Kim, H. Y., and Chi, Y. M. (2010) J. Biol.
Chem., 285, 25044-25052.
71.Gruez, A., Libiad, M., Boschi-Muller, S., and
Branlant, G. (2010) J. Biol. Chem., 285, 25033-25043.
72.Ritz, D., and Beckwith, J. (2001) Annu. Rev.
Microbiol., 55, 21-48.
73.Jacob, C., Kriznik, A., Boschi-Muller, S., and
Branlant, G. (2011) FEBS Lett., 585,
1905-1909.
74.Mouaheb, N., Thomas, D., Verdoucq, L., Monfort,
P., and Meyer, Y. (1998) Proc. Natl. Acad. Sci. USA, 95,
3312-3317.
75.Lemaire, S. D., Guillon, B., Le Marechal, P.,
Keryer, E., Miginiac-Maslow, M., and Decottignies, P. (2004) Proc.
Natl. Acad. Sci. USA, 101, 7475-7480.
76.Motohashi, K., Kondoh, A., Stumpp, M. T., and
Hisabori, T. (2001) Proc. Natl. Acad. Sci. USA, 98,
11224-11229.
77.Rey, P., Cuine, S., Eymery, F., Garin, J., Court,
M., Jacquot, J. P., Rouhier, N., and Broin, M. (2005) Plant J.,
41, 31-42.
78.Montrichard, F., Alkhalfioui, F., Yano, H.,
Vensel, W. H., Hurkman, W. J., and Buchanan, B. B. (2009) J.
Prot., 72, 452-474.
79.Ezraty, B., Aussel, L., and Barras, F. (2005)
Biochim. Biophys. Acta, 1703, 221-229.
80.Vieira Dos Santos, C., Laugier, E., Tarrago, L.,
Massot, V., Issakidis-Bourguet, E., Rouhier, N., and Rey, P. (2007)
FEBS Lett., 581, 4371-4376.
81.Stewart, E. J., Aslund, F., and Beckwith, J.
(1998) EMBO J., 17, 5543-5550.
82.Ezraty, B., Grimaud, R., El Hassouni, M.,
Moinier, D., and Barras, F. (2004) EMBO J., 23,
1868-1877.
83.Vieira Dos Santos, C., Cuine, S., Rouhier, N.,
and Rey, P. (2005) Plant Physiol., 138, 909-922.
84.Olry, A., Boschi-Muller, S., Marraud, M.,
Sanglier-Cianferani, S., Van Dorsselear, A., and Branlant, G. (2002)
J. Biol. Chem., 277, 12016-12022.
85.Chibani, K., Tarrago, L., Schurmann, P., Jacquot,
J.-P., and Rouhier, N. (2011) FEBS Lett., 585,
1077-1081.
86.Couturier, J., Stroher, E., Albetel, A. N.,
Roret, T., Muthuramalingam, M., Tarrago, L., Seidel, T., Tsan, P.,
Jacquot, J. P., Johnson, M. K., Dietz, K. J., Didierjean, C., and
Rouhier, N. (2011) J. Biol. Chem., 286, 27515-27527.
87.Trujillo, M., Ferrer-Sueta, G., Thomson, L.,
Flohe, L., and Radi, R. (2007) Subcell. Biochem., 44,
83-113.
88.Xiong, Y., Chen, B., Smallwood, H. S., Urbauer,
R. J., Markille, L. M., Galeva, N., Williams, T. D., and Squier, T. C.
(2006) Biochemistry, 45, 14642-14654.
89.Tarrago, L., Kieffer-Jaquinod, S., Lamant, T.,
Marcellin, M. N., Garin, J. R., Rouhier, N., and Rey, P. (2012)
Antioxid. Redox Signal., 16, 79-84.
90.Lee, B. C., Fomenko, D. E., and Gladyshev, V. N.
(2011) ACS Chem. Biol., 6, 1029-1035.
91.Lee, B. C., Le, D. T., and Gladyshev, V. N.
(2008) J. Biol. Chem., 283, 28361-28369.
92.Ranaivoson, F. M., Neiers, F., Kauffmann, B.,
Boschi-Muller, S., Branlant, G., and Favier, F. (2009) J. Mol.
Biol., 394, 83-93.
93.Aachmann, F. L., Kwak, G. H., Del Conte, R., Kim,
H. Y., Gladyshev, V. N., and Dikiy, A. (2011) Proteins,
79, 3123-3131.
94.Aachmann, F. L., Sal, L. S., Kim, H. Y., Marino,
S. M., Gladyshev, V. N., and Dikiy, A. (2010) J. Biol. Chem.,
285, 33315-33323.
95.Ghesquiere, B., Jonckheere, V., Colaert, N., Van
Durme, J., Timmerman, E., Goethals, M., Schymkowitz, J., Rousseau, F.,
Vandekerckhove, J., and Gevaert, K. (2011) Mol. Cell.
Proteomics, 10, M110.006866.
96.Kim, G., Cole, N. B., Lim, J. C., Zhao, H., and
Levine, R. L. (2010) J. Biol. Chem., 285,
18085-18094.
97.Lim, J. C., Gruschus, J. M., Ghesquiere, B., Kim,
G., Piszczek, G., Tjandra, N., and Levine, R. L. (2012) J. Biol.
Chem., 287, 25589-25595.