REVIEW: Telomerase RNA Biosynthesis and Processing
E. M. Smekalova1, O. S. Shubernetskaya1, M. I. Zvereva1,2*, E. V. Gromenko1, M. P. Rubtsova1,2, and O. A. Dontsova1,2
1Chemical Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia2Belozersky Research Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; E-mail: zvereva@genebee.msu.ru
* To whom correspondence should be addressed.
Received April 13, 2012
Telomerase synthesizes repetitive G-rich sequences (telomeric repeats) at the ends of eukaryotic chromosomes. This mechanism maintains the integrity of the genome, as telomere shortening leads to degradation and fusion of chromosomes. The core components of telomerase are the telomerase catalytic subunit and telomerase RNA, which possesses a small template region serving for the synthesis of a telomeric repeat. Mutations in the telomerase RNA are associated with some cases of aplastic anemia and also cause dyskeratosis congenita, myelodysplasia, and pulmonary fibrosis. Telomerase is active in 85% of cancers, and telomerase activation is one of the first steps in cell transformation. The study of telomerase and pathways where this enzyme is involved will help to understand the mechanism of the mentioned diseases and to develop new approaches for their treatment. In this review we describe the modern conception of telomerase RNA biosynthesis, processing, and functioning in the three most studied systems – yeast, vertebrates, and ciliates.
KEY WORDS: telomerase RNA, telomerase, telomeresDOI: 10.1134/S0006297912100045
Telomeres are specialized DNA–protein structures that are localized at the ends of eukaryotic chromosomes; their main function is to maintain stability of the genome [1]. Telomeric DNA consists of repetitive sequences (telomeric repeats). The classical replication mechanism cannot provide synthesis of a chromosomal end; that leads to telomere shortening after each round of cell division, destabilization of the genome, and senescence. To prevent those processes, cell uses different mechanisms, the main being telomerase activation. Telomerase synthesizes long sequences that consist of telomeric repeats using a small part of telomerase RNA as a matrix for telomere synthesis [2]. The telomerase catalytic subunit (TElomerase Reverse Transcriptase, TERT) and Telomerase RNA (TR) thus constitute a core enzyme that can maintain in vitro telomerase activity in the reconstruction system, while in vivo there are many more factors needed to provide proper functioning of the enzyme [3]. Thus telomerase is an RNA-dependent DNA polymerase, which acts as a reverse transcriptase on the basis of its own RNA component.
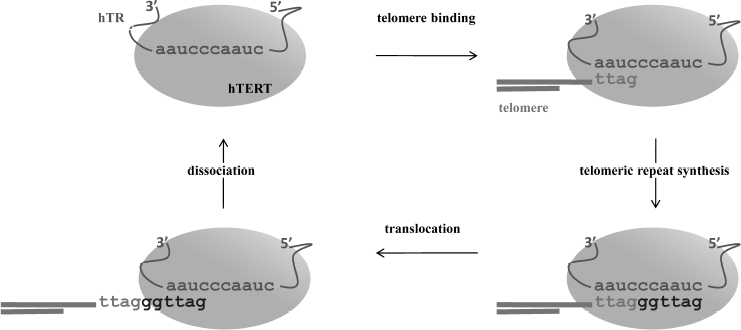
The telomerase catalytic cycle consists of two stages: synthesis of a telomeric repeat on the 3′ terminus of a telomere, and then release of the matrix from the RNA/DNA duplex to allow for synthesis of the next repeat (Fig. 1). Sequential synthesis of the repetitive sequences on the basis of one single matrix is not an inherent property of usual polymerases, and it demands a specific mechanism to release the telomerase RNA matrix region after each round of replication. The reaction starts from 3′-end telomere binding to the region downstream of the matrix, which results in formation of a short hybrid DNA/RNA duplex and then proceeds with the synthesis of one telomeric repeat. In case of human telomerases, six nucleotides are reverse transcribed into the 5′-GGTTAG-3′ sequence [4]. After reaching the end of the matrix region, synthesis stops; newly synthesized DNA can either translocate to the beginning of the matrix region or dissociate from the enzyme. Translocation is known to be a complicated multistage process; its mechanism is poorly understood even now, after decades of extensive studies of telomerase. Translocation involves several stages: unwinding of the DNA/RNA heteroduplex, relocation, and binding of the DNA to the beginning of the template [5]. The ability to synthesize several repeats in a row is termed telomerase processivity. Its efficiency depends on various factors [6].
Telomerase structure and function, TERT in particular, were discussed in detail in a recent review [7]. Here we concentrate on the synthesis and maturation of the second component of the telomerase complex – telomerase RNA. Its functions are not restricted to providing the template for telomeric repeat synthesis. Different parts of the complicated molecule structure shape the telomerase catalytic center together with amino acids of TERT, participate in the catalytic reaction of the nucleotide incorporation, mediate efficiency of the translocation process [8], condition the association of the different telomerase protein factors and complex assembly [9, 10], and play a key role in transport [11, 12] and regulation of telomerase activity [13].Fig. 1. The telomerase reaction cycle. The nucleotide sequence is specified for human telomerase.
Telomerase RNAs of different organisms vary greatly in terms of structure, sequence, and length. The length of ciliate TR is 147-205 nt, vertebrate TR ranges from 312 to 559 nt, and yeast ranges from 779 to 2030 nt [14, 15]. Comparative and phylogenetic analysis of TRs together with structural studies has been used to predict the 2D structure of TR for ciliates and vertebrates [9, 16]. For yeast telomerase RNA, this was a more difficult task due to the large length and evolutionary flexibility of the molecule. However, it has been shown that a large part of yeast TR is not required for the functioning of the complex [17]. Studies have revealed several elements of high order structure that are conserved between telomerase RNAs of different species. In addition to the template region, TRs contain the Template Boundary Element (TBE) situated from the 5′ terminus of the template. It restricts the reaction of reverse transcription in such a way that telomerase would not be able to synthesize sequence beyond the template [18, 19]. Also, one can point out a large region that includes template, pseudoknot, and a hairpin end [20]. In vertebrates this structure is known as the core domain, which, together with the CR4/CR5 domain (trans-activating domain) and TERT allows reconstruction of the active complex. The other elements of TR high order structure are specific for particular classes of organisms. For example, the presence of the Est1 binding hairpin in yeast TRs [21] or the presence of H/ACA scaRNA (small Cajal body RNA) domain in the 3′ region of vertebrate TRs [22].
Stable accumulation of most noncoding cellular RNAs implies processing of the initial transcript. This can involve RNA splicing, modification of nucleotide base and carbohydrate skeleton, and association with proteins, which stabilize the molecule to prevent action of nonspecific nucleases. We will further look closer at the formation of the initial transcript and TR processing for the three most studied groups of telomerase RNA – for yeast, human (which represents vertebrates), and ciliates.
TRANSCRIPTION AND PROCESSING
Transcription and processing of yeast telomerase RNA (Fig. 2a). Studies of model systems showed that yeast TR is transcribed by RNA-polymerase II, capped by monomethylguanosine (m7G), and polyadenylated [23]. During maturation of the molecule, a polyadenylic tail is removed and TR is capped by 2,2,7-trimethylguanosine (TMG) [24]. Telomerase RNA of Saccharomyces cerevisiae (TLC1) is stabilized by association with Sm proteins from its 3′ end. The function of Sm proteins is to protect and stabilize the 3′ end of snRNAs (small nuclear RNAs) [24]. They function as RNA chaperons and can facilitate modification or degradation of the RNA, participate in intracellular transport, and stabilize the RNA molecule [25]. The complex functions as a multimer that gathers from separately associated SmD1-D2, Sm-E-F-G, and SmB-D3. The primary structure of the vast majority of TRs includes the Sm protein binding site AU5-6GPu, which is identical to the Sm site of U4 snRNA. Mutations in this region of TR result in a cell being unable to process the 3′ end of the molecule, accumulation of the polyadenylated form of TR, and transcript degradation [24]. However, cells remain viable, but telomeres shorten in the strains bearing mutations in the Sm site [26]. It was shown that at least 77% of active telomerase interacts with Sm proteins [24]. Co-immunoprecipitation revealed that TR is associated with two out of seven Sm proteins – SmD1 and SmD3. The fact that the Sm complex functions as heptamer suggested that other Sm proteins also take part in the biogenesis of TR [24]. TR is polyadenylated in accordance with the classical cellular mechanism with the participation of polyA polymerase (PAP1) and protein RNA15, which is necessary for the dissection of the initial transcript and polyadenylation [23]. The 5′-regulatory region of TLC1 contains an AATAA sequence, which corresponds to one of the two conserved initiation sites used by polymerase II in S. cerevisiae. TR mRNA holds several cis-elements that provide high efficiency of polyadenylation. In S. cerevisiae, those elements are A-rich positioning element, A–U rich element enhancing the efficiency of the first element, and a polyadenylation site consisting of pyrimidines and adenosines (YA). Mutations in the suggested cis-elements did not decrease cell viability, but the position of polyadenylation was changed and the amount of TR polyadenylated form decreased [23]. Recent data indicate that the polyadenylation process does not participate in producing the functional TR transcript, as removal of the polyadenylation sites does not impact the amount of processed TR [27]. Moreover, it was shown that the transcription termination site occurs upstream of the polyadenylation site and includes binding sites for Nrd1, Nrd3, and Nab3 proteins. Thus, TR is processed in the same way as for snRNA and involves participation of the Nrd1/Nab3 termination pathway [27].
These data are consistent with the fact that polyadenylated TR of Schizosaccharomyces pombe is not active in vivo, as it does not provide association of the two main components of telomerase [26, 28]. This indicates the importance of the correct 3′ end processing for obtaining the active transcript. In the case of the fission yeast S. pombe, 3′-end formation is driven by the splicosome. The classical splicing mechanism implies two tightly connected events – breakage of the 5′ splice site followed by the formation of a “lasso” structure and breakage of the 3′ splice site with the formation of the covalent phosphodiester bond between the two exons. However, only the first reaction takes place during processing of S. pombe TR, and it results in the removal of the 5′ exon [29]. Mutation in the 5′ splice site led to a decrease in TR expression level and to shortening of telomeres, while mutations in the 3′ splice site or branch point that are involved in the second step of splicing do not influence cell viability. Interestingly, completion of both splicing steps led to inactive TR transcript that degraded rapidly. It turned out that binding of the primary transcript to Sm protein complex stimulates splicing [30]. The next step of TR processing is hypermethylation of the 5′ end of the molecule, which is followed by the displacement of the Sm proteins by the Lsm2-8 complex. The latter provides association of the TR and telomerase catalytic subunit. It remains unclear what the factor that divides those two steps in the functioning of the splicosome is. The reason might be enlarged distance between the branch point and 3′ splice site or the sequence of the intron. The fact that a change in intron leads to inability of a cell to produce active TR makes the second supposition quite reasonable [29].
Several TR of the budding yeast Candida possess conserved 5′ splice site and branch point [21]. This suggests that the mechanism of partial splicing is also conserved in Candida species.Fig. 2. Biosynthesis of telomerase RNA and its place in the telomerase complex assembly: a) for ciliates; b) for yeast; differences are specified for the two most studied yeast species – S. cerevisiae (upper arrow) and S. pombe (bottom arrow); c) for human.
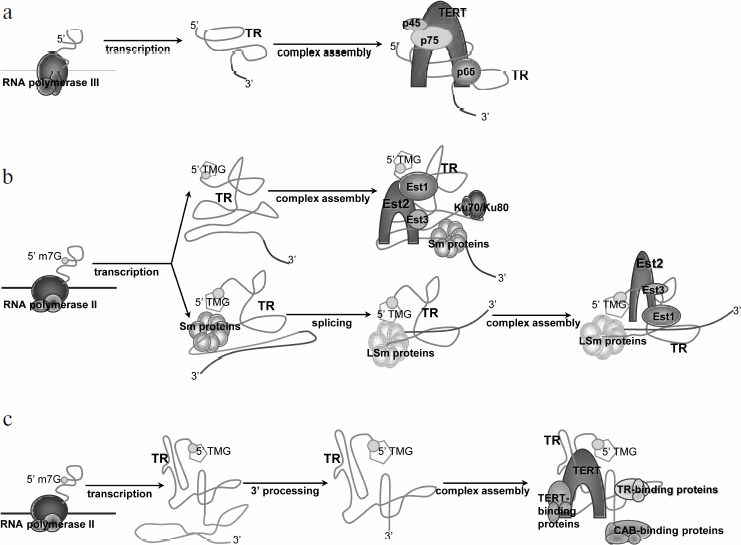
Transcription and processing of ciliate TR (Fig. 2b). Ciliate telomerase RNA (Tetrahymena, Euplotes, Oxytricha) is transcribed by RNA polymerase III, and its 3′ end finishes with heterogeneous tracts of uracils [31, 32]. The telomerase RNA level increases during development of the macronucleus. In accordance with the fact that RNA polymerase III drives the transcription of ciliate TR, there is no polyadenylation of TR in those organisms, and its 3′ end is generated directly as a result of transcription termination. Unlike yeast TR, which is stabilized by proteins characteristic of other snRNAs, stabilization and structure formation of ciliate TR is provided by specific proteins [33]. In Tetrahymena, TERT and TR interact only in the presence of p65 protein, which is part of the enzyme complex. It was shown that p65 and its ortholog from Euplotes aediculatus bind specifically to TR in vitro and in vivo [34, 35]. P65 interacts with the terminal hairpin of TR and its 3′ end that is rich in polyuridines [36], and this interaction increases the affinity of TERT to TR. Reconstruction of the telomerase, which was performed with purified p65, TR, and TERT, showed that p65 is the factor that initiated the assembly of the complex.
Transcription and processing of the human TR (Fig. 2c). Similarly with yeast TR, the human one is transcribed by RNA polymerase II [37]. The length of the maturated transcript is 451 nt. Another common trait of yeast and human TR transcription is the 2,2,7-TMG cap on the 5′ end of the molecule. Artificial transcription of human TR from RNA polymerase III promoter could not provide adequate processing of the molecule, and no functional transcript could be detected in a cell [22]. This finding is consistent with the knowledge that the transcription machinery is different for RNA polymerases II and III, including editing, capping, and splicing factors [38].
Despite the similar transcription enzymes that are used to create yeast and human TR, to date there is no reliable data on the polyadenylation of human TR. Vertebrate TR is believed to be non-polyadenylated [38-41]. Artificial expression of human TR with use of CMV (virus), CAG, and PGK promoters (promoters of the genes which encode different proteins) provides polyadenylation of human TR, but the efficiency of the transcript maturation decreases by tens of times. In contrast, TR transcription with the use of IU1 promoter (gene U1 of snRNA) results in the correct processing [39]. These data suggest that the mechanism that led to TR polyadenylation inhibited TR maturation in vivo. Changes in the 5′ sequence of TR did not influence the ratio of processed/non-processed form of the transcript, which argues against the importance of the 5′ gene region in the processing. The change in the nucleotide sequence in the 3′ region downstream from the TR gene also had no impact on the expression of TR [39].
Several cell types (in particular, LNCaP prostate cancer cells and immortalized VA13 fibroblasts with TR superexpression) contain a TR 3′ end that lacks the core domain [39, 42]. It includes an H/ACA box, which functions to stabilize and transport snoRNAs (small nucleolar RNAs) by interaction with the DKC protein complex [39]. It is likely that in the case of TR, one of the main functions of the H/ACA box is to stabilize the molecule as well. It is still unknown what mechanism leads to the formation of this 3′ fragment. It might be the result of endo- or exonucleolytic digestion or a combination of these processes. The function of this truncated 3′ fragment is also unclear, though it is most likely to be an intermediate product of nonspecific degradation.
One of the factors that determine the 3′ end processing of TR is the sequence in the CR7 element of the H/ACA scaRNA domain. Interestingly, the same hairpin contains the signal of TR localization to Cajal bodies (UGAG nucleotides of CAB box and helix). It has long been thought that those two signals (localization and processing) are connected [38], but it turned out that they act independently. Also, it was shown that the 3′ end processing takes place before the localization of the molecule to Cajal bodies [11]. To date there are no data about the protein factor that is responsible for the processing of the 3′ end of human TR.
LOCALIZATION AND TRANSPORT
Localization and transport of yeast TR. Studies of TR localization include different methods. The one that is used most is ChIP analysis (Chromatin Immunoprecipitation), which gives a clue about the interaction of telomerase and telomeres in a large population of cells. By methods of ChIP analysis, it was shown that in S. cerevisiae TLC1 and Est2 associates with telomeres starting from G1 to late S phase. These results suggested the model in which telomerase is present on telomeric ends from G1 to S phase in an inactive form, and it is activated only in the late S phase specifically on the shortest telomeres [43]. The alternative model proposes that Cdc13 protein, which interacts with single-stranded telomeric DNA, brings telomerase complex to short telomeres in late S phase. This process is driven by the interaction of Cdc13 and Est1 protein (a participant of the yeast telomerase complex). ChIP analysis also showed that there were increased amounts of Cdc13 and Est1 on telomeres in the late S phase. This model is also supported by genetic studies, where Cdc13–Est1 fusion compensates for ΔEst1 phenotype [44].
Recent data on the changes in telomerase localization during the cell cycle also speak in favor of the second model [45]. The system contained a TR sequence with MS2 hairpin on its 3′ end, while GFP reporter was fused to the MS2 binding protein. The results showed that during G1 and G2 phases TLC1 was associated with TERT, but stable association with telomeres was not detected. However, in the late S phase TR molecules gather in clusters on short telomeres, and this event correlates with telomere lengthening. Cluster formation depends on the factors that bring telomerase to telomeres – MRX, Tel1, Rif1, Rif2, and Cdc13. Rif1 and Rif2 act as negative regulators preventing telomere synthesis out of late S phase, and their absence provides telomere lengthening even in G1 phase [45].
Several studies suggest importance of telomerase transport to the cytoplasm for the proper functioning of the complex [37, 46]. Mutations in the MTR10 gene encoding for importin β, which is necessary for the import of mRNA binding proteins to the nucleus, lead to telomere shortening. The cells with mutant MTR10 accumulated superexpressed TLC1 in the cytoplasm, and at the same time TR level of processing and transcription remained the same [47]. Knockout of each of the Est proteins, Ku70, or MRX complex also resulted in the accumulation of TLC1 in the cytoplasm [46]. This effect might reveal the necessity of TLC1 going to the cytoplasm as one of its biogenesis steps, or inability of the nucleus to hold TLC1 in mutant strains due to the lack of the needed components. Use of the heterokaryon model system showed the exchange of TLC1 between nucleus and cytoplasm, thus indicating the correctness of the first model. TR export is mediated by exportin Crm1p, while importin β, Mtr10, and Kap122 are responsible for its import into the nucleus [46].
The described nuclear–cytoplasmic transport is reminiscent of the biogenesis and assembly of snRNP (small nuclear ribonucleoprotein) in multicellular organisms, where snRNA is exported to the cytoplasm with the help of Crm1, gains a TMG (trimethylguanosine) cap, and binds to Sm proteins before going back to the nucleus [37]. Processed TLC1 possesses TMG cap and binds to Sm proteins as other yeast snRNAs. Methyltransferase Tgs1 is responsible for the hypermethylation of TR in yeast; it is localized in the nucleolus. Knockout of the TGS1 gene inhibits TLC1 hypermethylation and results in accumulation of TLC1 in the nucleolus. It is likely that TLC1 transport to the nucleolus is followed by its export, because the strain deficient for Crm1 protein accumulates hypermethylated TLC1 in the nucleolus.
Because TLC1 accumulation in the nucleus requires the presence of all three Est proteins and Ku70/80 heterodimer, those factors are likely to serve for import and/or retention of TLC1 in the nucleus [37]. It is possible that TLC1 and Est proteins form a complex in the cytoplasm that activates telomerase transport into the nucleus. Est1 protein level varies during the cell cycle – it decreases in G1 and increases in G2 and S phases. However, this does not impact TR accumulation in cytoplasm, meaning that at each moment of cell cycle there are enough Est proteins to assemble with TR and export it to the nucleus. Once being in the nucleus, telomerase is attached to telomeres, and this interaction is thought to be mediated by the Ku70/80 proteins [48].
It is possible that the transport of TR to cytoplasm serves as the quality control for the folding of the molecule. The TR structure represents a “flexible platform” [17] that tethers telomerase protein components. It might be that correct conformation of TR is determined in the cytoplasm. TR that was incorrectly folded would not be able to mediate telomere elongation, while retention of such a molecule in cytoplasm would reduce possible harm to a cell [37].
Localization and transport of vertebrate TR. In contrast to yeast TR, the vertebrate one possesses an H/ACA box. The presence of this element changes significantly TR biogenesis in higher organisms. Functional similarity between the H/ACA box of hTR (human TR) and snoRNA was confirmed by mutagenesis. Mutations that impaired H/ACA consensus in TR led to a significant decrease in TR level in HeLa cells [22]. It was also shown that a small amount of TR localizes in the nucleolus. The necessity of the H/ACA domain in vivo and subcellular fractionation profile followed by the detection of TR transcript in different cellular organelles argue that one step of TR processing occurs in nucleolus, and after that TR is transported to the cytoplasm. It was suggested that the nucleolar telomerase complex possesses different properties in comparison with cytoplasmic or nuclear telomerase holoenzyme [22]. Unfortunately, there is no data on the role of TR localization in the nucleolus. The apical loop of the TR 3′ hairpin contains a CAB sequence (5′-UGAG-3′), which is a specific signal of localization to Cajal bodies [49]. In the case of human TR, sequences that could be responsible for modifications are not conserved, so it is unlikely that TR participates in the modification of RNA [50]. The H/ACA domain is thought to mediate TR transport to Cajal bodies. The H/ACA box structure is characterized by two irregular hairpins and an internal loop between them. The internal single-stranded region contains a conserved H box (5′-ANANNA-3′ consensus); the 3′ end of the element possesses a 5′-ACA-3′ motif, which is 3 nt before the end of the processed TR transcript. In addition to the H/ACA motif, scaRNA also contain a CAB box (this one is located in the CR7 element of TR), which is necessary for TR 3′ end processing, accumulation, and transport of TR into Cajal bodies [51, 52]. The change in CR7 element in hTR to U64 snoRNA does not influence RNA processing, but this results in hTR being localized in the nucleolus instead of Cajal bodies. Similarly with yeast TR, hTR (and hTERT) are present at telomeres in S phase to perform telomere replication in cancer cells [40, 53, 54]. By methods of in situ hybridization (FISH), it was shown that in cancer cells (HeLa and Hep2) during the major part of cell cycle hTR is localized in Cajal bodies [40, 53]. Notably, 25% of telomeres associated with TR colocalize with Cajal bodies as well. Moreover, fluorescence microscopy showed the movement of Cajal bodies together with telomeres in S phase for 10-40 min. Taken together, the data suggest that telomerase maturation and assembly with telomeres take place in Cajal bodies [12, 40]. In a system where HeLa cells superexpressed TR, the CAB box (signal of localization to Cajal bodies) was not necessary for telomere maintenance, although its presence accelerated telomeric DNA synthesis and increased telomere length [12].
In spite of the fact that hTR expression occurs is normal cells, it has so far been impossible to trace hTR association with telomeres, Cajal bodies, or other nuclear structures; on the contrary, it is distributed evenly in the nucleus [55]. It was shown that association of hTR with telomeres and Cajal bodies depends on hTERT. hTERT knockdown did not influence total hTR level, but led to the loss of the association between hTR, telomeres, and Cajal bodies. It is notable that hTERT expression in cells that lack telomerase activity (primary cell culture or cancer cell lines with alternative mechanisms of telomere elongation) causes association of hTR with both telomeres and Cajal bodies. It was shown that elevated levels of hTR, Cajal bodies, SV40 T antigen, or oncogene RAS do not impact hTR localization [56]. Therefore, to date the only factor that is known to be responsible for the difference in hTR localization in normal and cancer cells is the telomerase catalytic subunit.
The presence of an H/ACA box in vertebrate TR causes its association with the two core components of H/ACA snRNA – these being proteins hGar1 and dyskerin (human analog of yeast Cbf5 protein). Expression of hTR in S. cerevisiae initiates cooperation of the processed hTR form with proteins of H/ACA snRNP – Cbf5, Nhp2, Nop10, and Gar1. Three of these (except Gar1) are necessary for hTR accumulation in a yeast cell. At the same time, endogenous TLC1 does not require any of these factors for its biogenesis, and it is unlikely that it interacts with them. This experiment demonstrates that yeast TR, in contrast to the vertebrate one, is not related to H/ACA snoRNA class [56].
The same interaction of hTR with the proteins of H/ACA snoRNP takes place in a human cell [50]. First, all three main H/ACA components co-purify with hTR. Dyskerin, Nop10, and Nhp2 were detected by mass spectrometry in the telomerase extract purified from a human cell line [57]. Using tandem affinity purification, the authors determined the stoichiometry of H/ACA snoRNP proteins and hTR. The 3′ end of hTR binds independently to two sets of H/ACA snoRNP proteins (dyskerin, GAR1, NOP10, and NHP2). It was also shown that TCAB1/WDR79 exists in one copy for the telomerase complex, and its association depends on the CAB box consensus in the 3′ hairpin of hTR [50]. The CAB box protein TCAB1 is not necessary for telomerase functioning, but it promotes accumulation of hTR in Cajal bodies. TCAB1 knockdown in a fibrosarcoma cell line caused telomere shortening, and it was therefore suggested that hTR accumulation in Cajal bodies plays an important role in telomere length maintenance [73]. One should remember that those effects were detected in cancer cells. It is so far unclear whether there is any localization of hTR to Cajal bodies in normal cells. The 5′-H/ACA box hairpin does not influence telomerase activity. It might have evolved in such a way that it does not influence TR processing and is not used for other RNA modifications, and its role is currently unclear [50]. There are many mutations in the gene of dyskerin DKC1 that are associated with X-linked recessive dyskeratosis. For some patients, it was shown that mutations in NOLA2 gene, which encodes for NHP2, and NOLA3 gene, which encodes for NOP10, correlate with autosomal recessive dyskeratosis. Dyskerin and proteins associated with this factor participate not only in the telomerase biogenesis, but also in the function of ribosomes [59]. However, those mutations do not influence ribosomal RNA processing in human cells. Two mutations in the WRD79 gene encoding TCAB1 have been described, and these lead to autosomal recessive dyskeratosis. These mutations led to the impairment of TR processing, which resulted in decreased amounts of active telomerase complex [5].
INTERACTION WITH ADDITIONAL PROTEINS
Interaction of TR with additional proteins in yeast. Yeast TR represents a “flexible platform” that serves to assemble proteins, in particular telomerase catalytic subunit Est2 and complementary proteins such as Est1, Est3, Cdc13, and Ku70/80. Proteins Est1 and Est2 bind independently and directly to TLC1 [15, 17], while Est3 protein is attached through the interaction with Est2 that is connected to TR [37]. The telomerase catalytic subunit is a necessary component of the enzyme, whereas Est1 and Est3 are not needed for the function of telomerase in vitro. Est1 protein was shown to activate telomerase depending on the cell cycle phase. It is likely that this process occurs via interaction with Cdc13 protein in late S phase [60, 61]. Est3 protein specifically unbinds DNA/RNA heteroduplexes in a GTPase dependent manner [62, 63]. Therefore, it is possible that Est3 is one of the factors that are responsible for the processivity of yeast telomerase. Heterodimer Ku70/80 interacts with TLC1 hairpin. It mediates interaction of telomerase with telomeres depending on the cell phase [60, 61].
Interaction of TR with additional proteins in vertebrates. Among the protein factors that co-purify with human telomerases, there are Sm proteins, proteins binding to mRNA (hnRNPs), and NTPases [57, 64].
Only two of seven Sm proteins (SmB and SmD3) are associated with hTR. Telomerase associated with these factors is active [64]. It turned out that the other scaRNA represent the same situation: all of them interact with two Sm components out of seven, which are necessary for the classical functioning of Sm [64]. It was shown that telomerase extract co-immunoprecipitated with antibodies against hTERT from HeLa cells contained a small amount of SmD1 protein. Vertebrate TR lacks a distinctive U-rich Sm protein-binding site. It was shown that association of hTR and Sm proteins is mediated by a CAB box, which functions to transport RNA molecules to Cajal bodies. Mutation in a CAB box consensus 5′-UGAG-3′ to 5′-UGAC-3′ leads to lack of interaction between TR and Sm proteins and to TR being accumulated in nucleus, which is in accordance with previous data. However, this mutation does not prevent association of TR and TERT and does not influence telomere length. These data do not confirm that SmB and SmD3 recognizes CAB box directly. It might be that lack of interaction happens due to the impairment of hTR localization to Cajal bodies [64]. Recently published data claim that Sm proteins binding to TR facilitates 5′ end modification with TMG cap. The same mechanism could contribute to the biogenesis of hTR.
Proteins hnRNP C, hnRNP U, NAT10 (N-acetyltransferase No. 10), and GNL3L (guanine-binding protein 3 or nucleostemin) co-purify with telomerase from HeLa cells, where telomerase was superexpressed [57, 65]. hTR is shown to contain a consensus sequence (nt 38-43) that binds to hnRNP C protein [57]. Removal of this sequence leads to lack of interaction between hnRNP C and hTR but does not impact hTR accumulation in a cell. Previous studies suggested a direct role of hnRNP C in telomere synthesis [66]. However, in the system where primary fibroblasts from patients carrying X-linked dyskeratosis congenita do not possess functional endogenous TR [67], telomerase activity was restored after the transfection of hTR with deleted region 38-43 or G414C mutation (the one that is responsible for hTR localization to Cajal bodies and interaction with Sm proteins) [57]. This indicates that telomerase transport to Cajal bodies, interaction with Sm proteins, or hnRNP C do not play an important role in telomere elongation, at least in primary cells. On the other hand, superexpression of hnRNP C1 or hnRNP U in fibrosarcoma cell line results in extensive shortening of telomeres. Superexpression of hnRNP A1 elongated telomeres [68]. Telomeres in a system where GNL3L or NAT10 were superexpressed also became shorter, and notably telomerase activity was twice as increased under the superexpression of NAT10 protein [57].
NONCANONICAL FUNCTIONS OF TELOMERASE RNA
A number of studies indicate that elevation of hTR expression level is an early step in the development of cancer. Also, hTR expression level correlates better with cancer malignancy than the level of hTERT expression or telomerase activity [69]. Mice with a deleted TR gene are viable for six generations, till the moment of critical erosion of telomeres. It is notable that the first generation of those mice, where telomeres are long enough to maintain the stability of the genome, is less susceptible to the development of skin cancer [70].
These experiments suggest that TR functions not only as a part of the telomerase complex, but that it might possess some additional functions [71]. Inhibition of hTR expression causes cell cycle arrest that is associated with activation of p53 and CHK1; this effect is not dependent on the presence of hTERT. In this system, hTR suppressed activation of p53 and CHK1-dependent pathways in response to genotoxic stress. These effects are explained by the interaction of hTR with ATR kinase. Therefore, increase in hTR expression is a sufficient factor to impair ATR-dependent response to DNA damage. It was also shown that the interaction between TR and ATR-kinase is specific and leads to the inhibition of TR activity in vitro and in vivo [71]. Further studies revealed that UV radiation, which leads to ATR activation, also increased the endogenous level of TR, and this happened independently of telomerase status.
The concept of TR functioning as a mediator of cellular response correlates well with the knowledge about TR biogenesis. This function explains the constant expression of hTR in somatic cells, while hTERT and telomerase activity could be detected in a limited number of cell types with increased cell proliferative potential. It also provides evidence for the independent role of TR in development of cancer and explains superexpression of hTR in cancer cell lines [13, 71].
In this review, we have tried to generalize recent data about TR maturation and functioning in different organisms. It is obvious that the aspects of TR regulation and biogenesis are tightly linked to the function of telomerase complex, though there are evidences about TR-independent functioning in a cell. There is much more to learn in this field. One of the most interesting questions is the difference in telomerase functioning in normal and cancer cells (apart from the impairment of telomerase gene regulation). A good example of such a difference is telomerase localization to Cajal bodies – this was detected reliably only in cancer cells; in normal cells this process is still questionable. It is thought that Cajal bodies serve as a place for the assembly of TERT and TR into the active complex, but apparently this interaction takes place without Cajal bodies in normal cells. So what is the difference? Unfortunately, the interaction of TERT and TR is one of the least studied aspects in the structural chemistry of telomerase. Further studies on telomerase RNA functioning will clearly be a new step in understanding the bases of the development of many diseases and new approaches for their treatment.
This work was supported by the Russian Federation Ministry of Education and Science (P1390 No. 02.740.11.07.06, 16.512.11.2108) and Russian Foundation for Fundamental Research (grants No. 11-04-013-10-a and 11-04-12051-ofi-m-2011, PNR 5.13).
REFERENCES
1.Greider, C. W., and Blackburn, E. H. (1987)
Cell, 51, 887-898.
2.Blackburn, E. H. (2000) Nat. Struct. Biol.,
10, 847-850.
3.Shcherbakova, D. M., Zvereva, M. I., Shpanchenko,
O. V., and Dontsova, O. A. (2006) Mol. Biol. (Moscow),
40, 580-594.
4.Morin, G. B. (1989) Cell, 59,
521-529.
5.Podlevsky, J. D., and Chen, J. J. (2012) Mutat.
Res., 730, 3-11.
6.Chen, J. L., and Greider, C. V. (2003) EMBO
J., 22, 304-314.
7.Rubtsova, M. P., Vasilkova, D. P., Maliavko, A. N.,
Naraikina, Y. V., Zvereva, M. I., and Dontsova, O. A. (2012) Acta
Naturae, 4, 6-23.
8.Qiao, F., and Cech, T. R. (2008) Nat. Struct.
Mol. Biol., 15, 634-640.
9.Theimer, C. A., and Feigon, J. (2006) Curr.
Opin. Struct. Biol., 16, 307-318.
10.Robart, A. R., O’Connor, C. M., and
Collins, K. (2010) RNA, 16, 563-571.
11.Theimer, C. A., Jady, B. E., Chim, N., Richard,
P., Breece, K. E., Kiss, T., and Feigon, J. (2007) Mol. Cell,
27, 869-881.
12.Cristofari, G., Adolf, E., Reichenbach, P.,
Sikora, K., Terns, R. M., and Lingner, J. (2007) Mol. Cell,
27, 882-889.
13.Cairney, C. J., and Keith, W. N. (2008)
Biochimie, 90, 13-23.
14.Zhang, Q., Kim, N-K., and Feigon, J. (2011)
Proc. Natl. Acad. Sci. USA, 108, 20325-20332.
15.Dandjinou, A. T., Levesque, N., Larose, S.,
Lucier, J.-F., Elela, S. A., and Wellinger, R. J. (2004) Curr.
Biol., 14, 1148-1158.
16.Chen, J. L., Blasco, M. A., and Greider, C. W.
(2000) Cell, 100, 503-514.
17.Zappulla, D. C., and Cech, T. R. (2004) Proc.
Natl. Acad. Sci. USA, 101, 10024-10029.
18.Moriarty, T. J., Marie-Egyptienne, D. T., and
Autexier, C. (2005) RNA, 11, 1448-1460.
19.Box, J. A., Bunch, J. T., Zappulla, D. C., Glynn,
E. F., and Baumann, P. (2008) JBC, 283, 24224-24233.
20.Wyatt, H. D., West, S. C., and Beattie, T. L.
(2010) Nucleic Acids Res., 38, 5609-5622.
21.Gunisova, S., Elboher, E., Nosek, J., Gorkovoy,
V., Brown, Y., Lucier, J.-F., Laterreur, N., Wellinger, R. J., Tzfati,
Y., and Tomaska, L. (2009) RNA, 4, 546-559.
22.Mitchell, J. R., Cheng, J., and Collins, K.
(1999) Mol. Cell Biol., 19, 567-576.
23.Chapon, C., Cech, T. R., and Zaug, A. J. (1997)
RNA, 3, 1337-1351.
24.Seto, A. G., Zaug, A. J., Sobei, S. G., Wolin, S.
L., and Cech, T. R. (1999) Nature, 402, 898.
25.Khusial, P., Plaag, R., and Zieve, G. W. (2005)
Trends Biochem. Sci., 30, 522-528.
26.Leonardi, J., Box, J. A., Bunch, J. T., and
Baumann, P. (2008) Nat. Struct. Mol. Biol., 15,
26-33.
27.Noel, J. F., Larose, S., Abou Elela, S., and
Wellinger, R. J. (2012) Nucleic Acids Res., doi:
10.1093/nar/gks200.
28.Wilusz, J. E., and Spector, D. L. (2010)
RNA, 16, 259-266.
29.Box, J. A., Bunch, J. T., Tang, W., and Baumann,
P. (2008) Nature, 456, 910-914.
30.Tanq, W., Kannan, R., Blanchette, M., and
Baumann, P. (2012) Nature, 484, 260-264.
31.Greider, C. W., and Blackburn, E. H. (1989)
Nature, 337, 331-337.
32.Lingner, J., Hendrick, L. L., and Cech, T. R.
(1994) Genes Dev., 8, 1984-1998.
33.Witkin, K. L., and Collins, K. (2004) Genes
Dev., 18, 1107-1118.
34.Aigner, S., Lingner, J., Goodrich, K. L.,
Grosshans, C. A., Shevchenko, A., Mann, M., and Cech, T. R. (2000)
EMBO J., 19, 6230-6239.
35.Aigner, S., Postberg, J., Lipps, H. J., and Cech,
T. R. (2003) Biochemistry, 42, 5736-5747.
36.O’Connor, C. M., and Collins, K. (2006)
Mol. Cell Biol., 26, 2029-2036.
37.Gallardo, F., and Chartrand, P. (2008) RNA
Biol., 5, 212-215.
38.Collins, K., and Mitchell, J. R. (2002)
Oncogene, 21, 564-579.
39.Li, S., and Blackburn, E. H. (2006) Cold
Spring Harb. Symp. Quant. Biol., 71, 211-216.
40.Jady, B. E., Richard, P., Bertran, E., and Kiss,
T. (2006) Mol. Biol. Cell, 17, 944-954.
41.Fu, D., and Collins, K. (2003) Mol. Cell,
11, 1361-1372.
42.Kim, M. M., Rivera, M. A., Botchkina, I. L.,
Shalaby, R., Thor, A., and Blackburn, E. H. (2001) Proc. Natl. Acad.
Sci. USA, 98, 7982-7987.
43.Hug, N., and Lingner, J. (2006)
Chromosoma, 115, 413-425.
44.Takata, H., Tanaka, Y., and Matsuura, A. (2005)
Mol. Cell, 17, 573-583.
45.Gallardo, F., Laterreur, N., Cusanelli, E.,
Quenzar, F., Querido, E., Wellinger, R. J., and Chartrand, P. (2011)
Mol. Cell, 44, 819-827.
46.Gallardo, F., Olivier, C., Dandjinou, A. T.,
Wellinger, R. J., and Chartrand, P. (2008) EMBO J.,
27, 748-757.
47.Ferrezuelo, F., Steiner, B., Aldea, M., and
Futcher, B. (2002) Mol. Cell Biol., 22, 6046-6055.
48.Stellwagen, A. E., Haimberger, Z. W., Veatch, J.
R., and Gottschling, D. E. (2003) Genes Dev., 17,
2384-2395.
49.Kiss, T., Favet, E., Jady, B. E., Richard, P.,
and Weber, M. (2006) Cold Spring Harb. Symp. Quant. Biol.,
71, 407-417.
50.Egan, E. D., and Collins, K. (2010) Mol. Cell
Biol., 30, 2775-2786.
51.Ganot, P., Caizergues-Ferrer, M., and Kiss, T.
(1997) Genes Dev., 11, 941-956.
52.Dez, C., Henras, A., Faucon, B., Lafontain, D.,
Caizergues-Ferrer, M., and Henry, Y. (2001) Nucleic Acids Res.,
29, 598-603.
53.Tomlinson, R. L., Ziegler, T. D., Supakorndej,
T., Terns, R. M., and Terns, M. P. (2006) Mol. Biol. Cell,
17, 955-965.
54.Wright, W. E., Tesmer, V. M., Liao, M. L., and
Shay, J. W. (1999) Exp. Cell Res., 251, 492-499.
55.Zhu, Y., Tomlinson, R. L., Lukowiak, A. A.,
Terns, R. M., and Terns, M. P. (2004) Mol. Biol. Cell,
15, 81-90.
56.Tomlinson, R. L., Abreu, E. B., Ziegler, T., Ly,
H., Counter, C. M., Terns, R. M., and Terns, M. P. (2008) Mol. Biol.
Cell, 19, 3793-3800.
57.Fu, D., and Collins, K. (2007) Mol. Cell,
28, 773-785.
58.Venteicher, A. S., and Artandi, S. E. (2009)
Cell Cycle, 8, 1329-1331.
59.Dokal, I. (2011) Hematol. Am. Soc. Hematol.
Educ. Progr., 2011, 480-486.
60.Evans, S. K., and Lundblad, V. (1999)
Science, 286, 117-120.
61.Taggart, A. K., Teng, S. C., and Zakian, V. A.
(2002) Science, 297, 1023-1026.
62.Sharanov, Y. S., Zvereva, M. I., and Dontsova, O.
A. (2006) FEBS Lett., 580, 4683-4690.
63.Shubernetskaya, O., Logvina, N., Sharanov, Y.,
and Zvereva, M. (2011) Biochimie, 93, 202-206.
64.Fu, D., and Collins, K. (2006) Genes
Dev., 20, 531-536.
65.Lee, C. L., Hsiao, H. H., Lin, C. W., Wu, S. P.,
Huang, S. Y., Wu, C. Y., Wang, A. H., and Khoo, K. H. (2003)
Proteomics, 3, 2472-2486.
66.Ford, L. P., Suh, J. M., Wright, W. E., and Shay,
J. W. (2000) Mol. Cell Biol., 20, 9084-9091.
67.Wong, J. M., and Collins, K. (2006) Genes
Dev., 20, 2848-2858.
68.LaBranche, H., Dupuis, S., Ben-David, Y., Bani,
M. R., Wellinger, R. J., and Chabot, B. (1998) Nat. Genet.,
19, 199-202.
69.Blasco, M. A., Lee, H. W., Hande, M. P., Samper,
E., Lansdorp, P. M., DePinho, R., and Greider, C. W. (1997)
Cell, 91, 25-34.
70.Gonzalez-Suarez, E., Samper, E., Flores, J. M.,
and Blasco, M. A. (2000) Nat. Genet., 26, 114-117.
71.Kedde, M., le Sage, C., Duursma, A.,
Zlotorynski, E., van Leeuwen, B., Nijkamp, W., Beijersbergen, R., and
Agami, R. (2006) J. Biol. Chem., 281, 40503-40514.