REVIEW: Octaheme Nitrite Reductases: Structure and Properties
T. V. Tikhonova1, A. A. Trofimov1,2, and V. O. Popov1,3*
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; fax: (495) 954-2732; E-mail: ttikhonova@inbi.ras.ru2Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; fax: (495) 135-1405
3Russian National Research Center “Kurchatov Institute”, pl. Kurchatova 1, 123182 Moscow, Russia; fax: (499) 196-1704; E-mail: nrcki@nrcki.ru
* To whom correspondence should be addressed.
Received June 8, 2012; Revision received June 22, 2012
Octaheme oxidoreductases are widespread among various bacterial taxa involved in the biogeochemical nitrogen cycle. The evolution of octaheme oxidoreductases of the nitrogen cycle from the evolutionarily more ancient pentaheme nitrite reductases was accompanied by changes in function from reduction of nitrogen oxides to their oxidation under changing environmental conditions. Octaheme nitrite reductases, which are the subject of the present review, are of a transitional form that combines structural and functional characteristics of pentaheme reductases and octaheme oxidases and possesses a number of unique features typical of only this family of enzymes. The review summarizes data on structure–function investigations of the family of octaheme nitrite reductases. Emphasis is given to comparison of the structures and functions of octaheme nitrite reductases and other multiheme oxidoreductases of the nitrogen cycle.
KEY WORDS: multiheme cytochrome c, octaheme nitrite reductase, structure, properties, evolutionDOI: 10.1134/S0006297912100057
Abbreviations: HAO, octaheme hydroxylamine oxidoreductase; MCC, multiheme cytochrome c; NrfA, pentaheme cytochrome c nitrite reductase encoded by the nrfA gene (nitrite reduction with formate); ONR, octaheme nitrite reductase; OTR, octaheme tetrathionate reductase; rmsd, root mean square deviation; TvNiR, octaheme nitrite reductase from Thioalkalivibrio nitratireducens; TvPaR, octaheme nitrite reductase from Thioalkalivibrio paradoxus.
In recent years, understanding of the role of multiheme cytochromes c
(MCC) in the lifecycle of eukaryotes and prokaryotes has greatly
progressed. In addition to the well-known role of MCC in respiratory
electron transfer in cells, their involvement in the catalysis of the
most important metabolic processes related to the global cycles of
nitrogen and sulfur by bacteria has been shown. The most important
processes include nitrification, respiratory ammonification of nitrate
and nitrite, anaerobic ammonium oxidation, reduction of Fe(III), and
reduction of sulfite, thiosulfate, and tetrathionate [1].
One of the most extensive and most studied families of MCC with catalytic function are pentaheme cytochrome c nitrite reductases (NrfA), which catalyze the six electron reduction of nitrite and sulfite, as well as the reduction of nitrogen oxides in intermediate oxidation states (NO, NH2OH) [2-16]. Such broad substrate specificity allows NrfA to perform a variety of functions in cells, participating in respiratory nitrite reduction during anaerobic growth of bacteria with nitrate or nitrite as an electron acceptor and in dissimilative reduction of nitrite, nitric oxide, and hydroxylamine in detoxification of cells [1, 2, 16].
NrfA from different classes of proteobacteria have a number of structural features that allow them to be merged into a single family [2, 17-23]. Monomeric NrfA contains five c-type hemes. Four of the five hemes are coordinated by two histidine residues in the proximal and distal positions, and the fifth heme (catalytic) is coordinated via a lysine residue in the proximal position, and the distal position is occupied by the water molecule or hydroxide ion that are replaced by the substrate molecule during the course of the reaction [23]. There is a heme-binding amino acid sequence motif CXXCK corresponding to the catalytic heme, which is unique for cytochrome c nitrite reductases. Hemes 3 and 4 are parallel to each other, while hemes in pairs 2/3 and 4/5 are almost perpendicular to each other (Fig. 1; see color insert). The catalytic heme 1 is almost parallel to hemes 3 and 4 and is located at a Fe–Fe distance of 9.5 Å from heme 3. The Fe–Fe distance between the other neighboring hemes in the monomer varies from 9 to 12.8 Å, which suggests the possibility of direct electron transfer between these hemes [24, 25].
The NrfA active site is located at the distal side of the catalytic heme and includes conserved catalytic residues such as His, Tyr, and Arg. Two channels for transport of the substrate (nitrite) and the reaction product (ammonium ion), which differ in charge of the inner surface, connect the active site to the surface of the molecule. Each NrfA subunit contains a Ca2+ ion, which is located at the distance of about 11 Å from the catalytic heme iron [17-22]. It is believed that the role of this Ca2+ ion is to stabilize the catalytic conformation of the active site. In some structures of NrfA a second Ca2+ ion has been found; it is located near hemes 3 and 4 [20, 22]. The role of this calcium ion in the catalysis and stability of the enzyme is unclear.Fig. 1. Structure of the NrfA dimer from W. succinogenes (PDB 1FS7). The hemes are numbered according to the position of the heme-binding motif in the amino acid sequence. The catalytic heme 1 is shown in black, and the others are shown in red. A calcium ion is shown in gray.
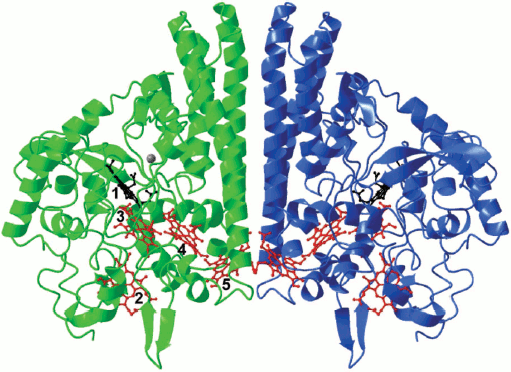
NrfA exists in solution as a functional dimer (Fig. 1). The distance between the hemes 5 from each of two subunits located in the intersubunit contact is 11.7 Å, which leads to form a 10-heme electron transport chain and enables the exchange of electrons between the catalytic centers in the dimer. The formation of NrfA complexes with physiological donors of electrons – tetraheme cytochrome c NrfH or pentaheme NrfB – leads to the formation of even more extended intramolecular electron transport chains consisting of 28 hemes in the 4NrfA–2NrfH complex from the bacterium Desulfovibrio vulgaris [21] or 20 hemes in the 2NrfA–2NrfB complex of Escherichia coli [26]. In each of these chains there is conservative packing of hemes, consisting of alternating diheme motifs formed by parallel and perpendicular oriented hemes. The same organization of hemes was noted in other multiheme cytochromes, and it seems to be optimal for electron transfer in proteins over long distances [27].
Based on the analysis of kinetic and structural data and quantum mechanical calculations, a hypothetical mechanism of six-electron reduction of nitrite catalyzed by NrfA has been proposed [23], which includes the formation of nitric oxide and hydroxylamine as intermediates.
OCTAHEME OXIDOREDUCTASES
The second largest group of MCC with catalytic activity consists of octaheme oxidoreductases, which are widespread in different taxa of bacteria involved in the biogeochemical cycle of nitrogen. In contrast to NrfA, this group of MCC is heterogeneous and includes several subgroups (families) of enzymes that differ in the structure of the active site, substrate specificity, and the type of catalyzed reactions. The following octaheme oxidoreductases were structurally characterized: octaheme nitrite reductases (ONR), hydroxylamine oxidoreductase (HAO) of the nitrifying bacteria Nitrosomonas europaea [28], and octaheme tetrathionate reductase (OTR) from the bacterium Shewanella oneidensis [29, 30].
The physiological function of HAO in cells of nitrifying bacteria is the four-electron oxidation of hydroxylamine to nitrite.
The physiological function of OTR from S. oneidensis has not been determined, but it was shown in vitro that in addition to reduction of tetrathionate to thiosulfate OTR effectively reduces nitrite and hydroxylamine to ammonium, which may suggest the participation of this enzyme in nitrogen metabolism [30].
The evolution of multiheme oxidoreductases of the nitrogen cycle from the evolutionarily more ancient pentaheme nitrite reductases (NrfA) to octaheme oxidases (HAO) with changes in function from the reduction of nitrogen oxides to their oxidation under changing environmental conditions was postulated [31, 32]. Octaheme nitrite reductases, which are the subject of the present review, are considered as a transitional form that combines structural and functional characteristics of pentaheme nitrite reductases (NrfA) and octaheme oxidases (HAO) and has a number of unique features typical of only this family of enzymes.
OCTAHEME NITRITE REDUCTASES
Genes of homologous ONRs have been found in the genomes of different classes of Proteobacteria (β, γ, and δ) including sulfur-oxidizing haloalkaliphilic bacteria of the genus Thioalkalivibrio (γ-proteobacteria) and sulfur-reducing non-halophilic neutrophilic bacteria of the genus Geobacter (δ-proteobacteria) that are characterized by high adaptive flexibility [1, 33]. Biochemical and structural characterization has been reported for ONR isolated from the bacteria Thioalkalivibrio paradoxus (TvPaR) [33] and Thioalkalivibrio nitratireducens (TvNiR) [34, 35]. Both bacteria live in soda lakes with high pH (up to 10) and high salinity (up to 380 g/liter), which is due to high concentration of sodium carbonate in solution [36-38].
These proteins have high similarity of amino acid sequences (81% identical residues), of spatial structure (root mean square deviation (rmsd) is 0.49 Å when aligned by Cα-atoms of subunits TvPaR and TvNiR), and of catalytic properties [33], so from now on we will discuss the general properties of both proteins, calling them ONR.
Catalytic properties. Among all octaheme oxidoreductases, ONR are the closest to pentaheme nitrite reductases both in catalytic properties and in structure. ONR and NrfA both catalyze the six-electron reduction of nitrite (1) and sulfite (2) as well as two-electron reduction of hydroxylamine (3). Reduction is not accompanied by the release of intermediate products into the solution [33, 34].
NO2– + 6 e– + 8 H+ = NH4+ + 2 H2O (1)
HSO3– + 6 e– + 6 H+ = HS– + 3 H2O (2)
NH2OH + 2 e– + 3 H+ = NH4+ + H2O (3)
Catalytic constants of nitrite reduction (kcat) are higher for ONR than for NrfA (see table). However, the second-order rate constants (kcat/Km) (so-called efficiency constants) are comparable for ONR and NrfA from E. coli. The low value of the efficiency constant for NrfA from D. desulfuricans is due to a high Km value, which can be explained by the fact that the experiments were carried out in the presence of sodium dithionite – an effective competitive inhibitor of ONR and NrfA [12, 13].
Kinetic parameters of nitrite,
hydroxylamine, and sulfite reduction catalyzed by NrfA, ONR, OTR, and
HAO
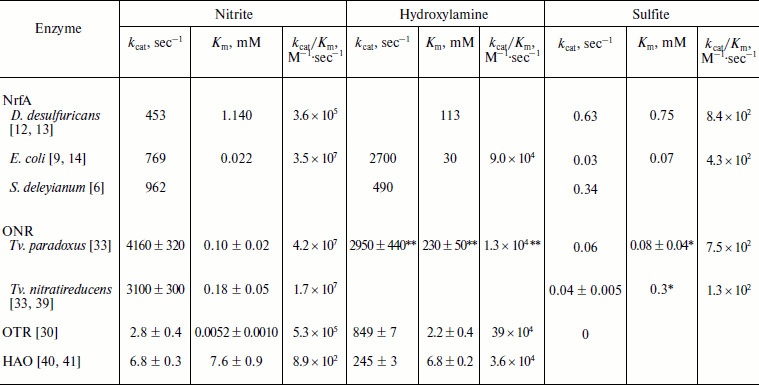
* Instead of Km values, Ki values
for competitive inhibition of nitrite reduction by sulfite are
given.
** Our unpublished data.
The catalytic constant of reduction of hydroxylamine by ONR is comparable to that of nitrite, as well as to kcat of hydroxylamine reduction by NrfA. However, the high Michaelis constant values indicate that hydroxylamine is not a physiological substrate for ONR and NrfA.
In cellular metabolism, HAO acts as an oxidase catalyzing the oxidation of hydroxylamine to nitrite in accordance with the scheme NH2OH → HNO → NO → NO+ → HNO2 [39, 40] to form the same intermediates that are formed during the oxidation of nitrite by NrfA. In vitro, HAO is capable to catalyze the reverse reaction of nitrite, nitric oxide and hydroxylamine reduction to ammonium using reduced methyl viologen as an electron donor (table) [41, 42]. The catalytic constant and the efficiency constant of nitrite reduction by HAO are several orders of magnitude lower than that of NrfA and ONR. The low value of the efficiency constant is due to high Km of HAO for nitrite, which is consistent with the fact that nitrite is the product of the physiological reaction of hydroxylamine oxidation and should easily dissociate from the active site to avoid inhibition by the product.
As already noted, OTR is also able to reduce nitrite and hydroxylamine to ammonium (table). The rate of reduction of nitrite by OTR is comparable to that of HAO and significantly lower than in the case of NrfA and ONR. However, due to the low Km value, nitrite reduction efficiency (kcat/Km) of OTR is comparable to that of nitrite reductases NrfA and ONR, which confirms the opinion of the authors [30] suggesting the possible involvement of OTR in the reduction of nitrite in cells.
In addition to the reduction of compounds of nitrogen, ONR, like NrfA, can reduce sulfite. The rate of reduction of sulfite by ONR is lower than by NrfA (except NrfA from E. coli has comparable sulfite reductase activity). As a result, the ratio of nitrite reductase and sulfite reductase activities for ONR is higher than that for NrfA ((6-7)·104 against the (0.6-5.5)·103), indicating more fine-tuning of the active site for nitrite reduction. Nevertheless, sulfite reductase activity of ONR is comparable to the activity of natural siroheme-containing sulfite reductases [33, 43] and may have physiological significance.
ONR also shows peroxidase activity, but it is significantly lower than the activity of natural peroxidases [34].
A slowly reacting substrate – sulfite – is an effective competitive inhibitor of the nitrite reductase reaction [33]. The inhibition constant (Ki = 0.08 ± 0.04 mM) for sulfite is comparable to the Michaelis constant for nitrite (Km = 0.10 ± 0.02 mM for TvPaR), so an excess of sulfite in the cell may lead to a significant inhibition of nitrite reduction. In addition to sulfite, the reduction of nitrite is inhibited by cyanide and azide [34].
3D structure of ONR. Structure of the monomer. The ONR subunit consists of two domains. The N-terminal domain contains hemes 1-3, the second domain (catalytic) contains hemes 4-8. The structure of the ONR catalytic domain is similar to that of the NrfA monomer [33, 35]. There are about 200 Cα-atoms of the catalytic domain of TvNiR and monomer NrfA that can be superimposed with rmsd less than 1 Å. For this, the five hemes of TvNiR are superimposed with the NrfA hemes, and 19 out the 30 α-helices of TvNiR including the three characteristic long helices at the C-terminus are superimposed with the corresponding helices of NrfA.
Seven hemes c of the ONR monomer (hemes 1-3 and 5-8 in Fig. 2a; see color insert) are associated with the polypeptide chain via classic heme(c)-binding motifs CXXCH and are coordinated in the distal and proximal positions by histidine residues. The catalytic heme 4 is coordinated in the proximal position by the Lys188 residue from the unique heme-binding motif CXXCK, which was previously found only in pentaheme nitrite reductases, and in the distal position – by a water molecule [33, 35].
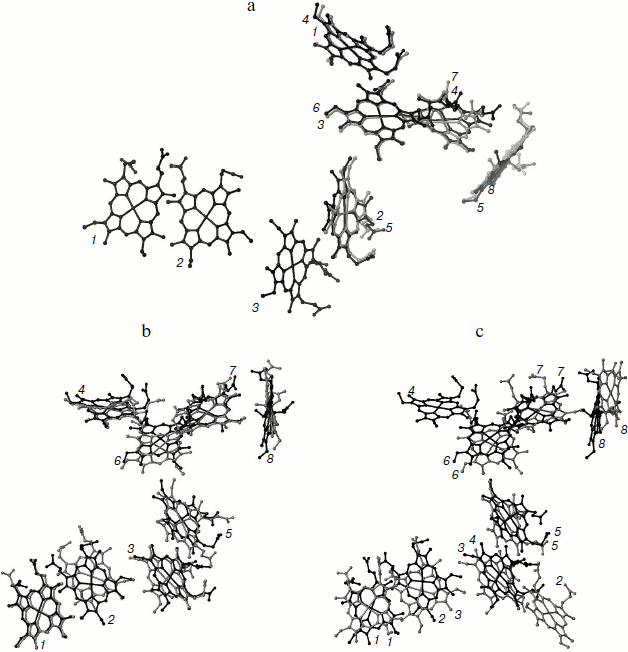
Bis-histidine-coordinated hemes are arranged in diheme motifs, which are characteristic of multiheme cytochromes c. Hemes in the pairs 1/2, 3/5, and 6/7 are parallel to each other with distances between iron atoms of about 9.1-9.6 Å. The planes of hemes 2/3, 5/6, and 7/8 are almost perpendicular to each other, with distances between iron atoms in these pairs of about 11.5-12.9 Å (Fig. 3). Six hemes are organized into two branches that converge to heme 6. Heme 6 is located at Fe–Fe distance of 9.6 Å and is almost parallel to the catalytic heme 4. Arrangement of the eight hemes of ONR coincides with the arrangement of eight hemes of HAO, and the five hemes of the catalytic domain of ONR coincide with the five hemes of NrfA including the catalytic hemes (Fig. 3). Seven hemes of ONR can be superimposed with seven hemes of OTR [35] such that the catalytic hemes are not involved in this superposition and appear on opposite sides of the line of superimposed hemes. Thus, the packing of the hemes in all octaheme oxidoreductases coincides, while the packing of five hemes in NrfA can be regarded as a fragment of a more general octaheme chain.Fig. 2. a) Spatial structure of the Tv. nitratireducens ONR hexamer (PDB 2OT4 [35]). Subunits in the trimer are shown in different colors, and adjacent subunits (dimers) of the different trimers are shown in the same color. The location of hemes in the dimer is shown, the hemes being numbered according to the position of the heme-binding motifs in the amino acid sequence, and the catalytic heme 4 is shown in black. b) Arrangement of the hemes in the trimer, the catalytic hemes being highlighted in black.
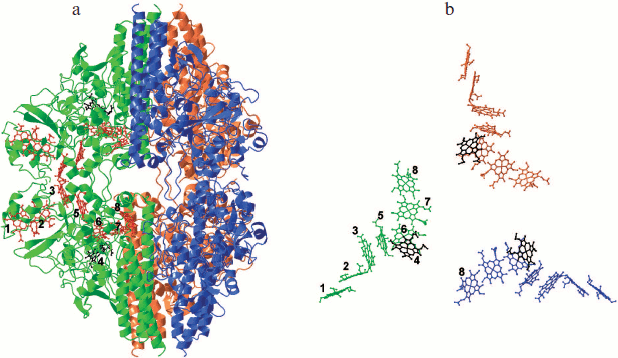
Structure of the hexamer. X-Ray diffraction and chromatographic data as well as small-angle X-ray scattering data showed that ONR in a crystal and in solution forms a stable homohexamer [33-35, 44]. The hexamer shape is close to a triangular bipyramid with a height of ~150 Å and a length of a basis of ~120 Å (Fig. 2a). The ONR hexamer can be described as a “dimer of trimers”. Each ONR monomer forms 36 hydrogen bonds with neighboring monomers in the trimer (the surface area of contact is 3200 Å2 per monomer) and 13 hydrogen bonds with the monomers of neighboring trimer in hexamer (contact surface area 1650 Å2). The total surface area of the monomer inaccessible to solvent is 4850 Å2 [35], which ensures high stability of the hexamer in solution. Dissociation of hexamers was not observed upon dilution to nanomolar concentrations. Hexamers were stable in the temperature range of 20-70°C, at pH 4.5-9.7, and in solutions with high (up to 2 M NaCl) salt concentrations. A presumed role of oligomerization of the ONR from haloalkaliphiles is stabilization at high pH and salinity.Fig. 3. Superimposition of hemes: a) TvNiR (black) and NrfA (gray); b) TvNiR (black) and HAO (gray); c) TvNiR (black) and OTR (gray). Hemes are numbered in accordance with position of the heme-binding motifs in the corresponding amino acid sequence. Heme numbering in TvNiR and HAO coincides.
NrfA dimers are much less stable. The dissociation constant is 1-3 nM for NrfA from Sulfospirillum deleyianum [6] and 4 μM for NrfA from E. coli [9] (area of intersubunit contact is 2150 and 1530 Å2, respectively). The HAO trimer is much more stable than NrfA dimers or ONR hexamers due to the greater area of intersubunit contacts (5430 Å2) [28] as well as through a covalent bonding between the subunits.
The formation of the ONR hexamer structure leads to the formation of a large cavity (38 × 46 Å) inside the hexamer filled with solvent and connected with surrounding solution through three outlets of size 9 × 16 Å, which are located in the trimer–trimer contact site near heme 8 (Fig. 2a) [35].
The forty-eight hemes in the ONR hexamer do not form a common electron transport chain, in contrast to HAO and NrfA. Direct electron transfer is possible only between hemes 3 of the subunits forming the dimer, where the Fe–Fe distance is 13.1 Å (Fig. 2a). The minimum distance between iron atoms of the closest hemes 6 and 8 from the neighboring subunits in the trimer is about 30 Å (Fig. 2b). The absence of a common electron transport chain indirectly confirms the hypothesis that the primary role of oligomerization is the stabilization of the enzyme.
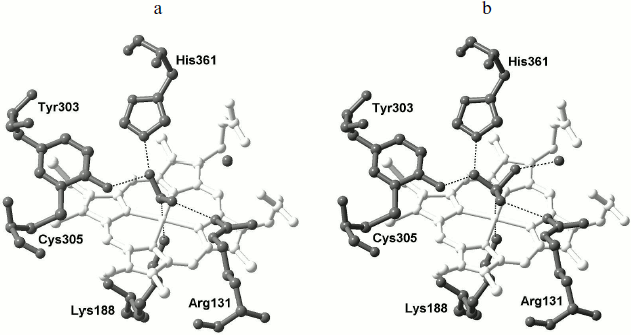
Active site. As in the case of NrfA, the active site of ONR is located on the distal side of the catalytic heme coordinated in the proximal position by the Lys188 residue and includes conserved residues Tyr303, His361, and Arg131. In the free form of the enzyme, residues Tyr303 and His361 form hydrogen bonds with a water molecule coordinated to the heme Fe(III) ion [33, 35]. It is assumed that in ONR, as well as in NrfA, tyrosine and histidine residues are directly involved in catalysis, whereas the arginine provides the correct orientation of the substrate and intermediate products during the reaction [17, 18, 35]. The active sites of NrfA and ONR are very similar [22, 33, 35], but unlike NrfA, the active site of ONR contains a Cys residue that forms a covalent bond with catalytic Tyr303 (Fig. 4a; see color insert). This bond does not affect the overall architecture of the active site, but it causes shifting of the hydroxyl group of the tyrosine residue by 1.2 Å in the direction of the catalytic heme [35] and compaction of the active site. As a result, the hydrogen bonds formed between residues Tyr303 and His361 and the substrate are shorter than in NrfA. The second presumed consequence of this bond formation is a decrease in pKa of the hydroxyl group of Tyr303 by 0.5-1.0 unit, which facilitates the transfer of a proton from the hydroxyl to an oxygen atom in the substrate molecule (nitrite). It has been suggested that the formation of this bond is responsible for the increase in nitrite reductase activity of ONR compared with that of NrfA [35, 45].
In ONR as well as in NrfA there is a Ca2+ ion located at a distance of 10.5 Å from the catalytic heme iron atom. This Ca2+ ion is coordinated with residues Glu302, Tyr303, Lys358, and Gln360 and two water molecules (Fig. 4a). The Gln residue is conserved in multiheme nitrite reductases. Its substitution in NrfA by Glu, which is more common in Ca2+-binding proteins, led to a 10-fold decrease in nitrite binding [22, 46], the catalytic constant being unaffected. Due to the presence of the Ca2+ ion and the charged residues Arg131 and His361, the active site of ONR has a positive charge, which promotes the binding of negatively charged substrates (nitrite, sulfite) and the dissociation of the positively charged product (ammonium ion).Fig. 4. a) Structure of the active site of ONR from Tv. paradoxus (PDB 3SXQ [33]). A map of 2Fo-Fc electron density (1σ) for Tyr303 and Cys305 is shown. The catalytic heme iron and cobalt ions are shown in brown, and the calcium ion is shown in gray. Coordination and hydrogen bonds are shown by dashed lines. b) Transport channels of substrate (orange), product (yellow), and protons (blue) in the structure of the ONR. The side chains of residues forming the channels are shown. Hemes 4, 6, and 7 are shown in gray, water molecules as blue spheres, the calcium ion in gray, and cobalt ions in brown.
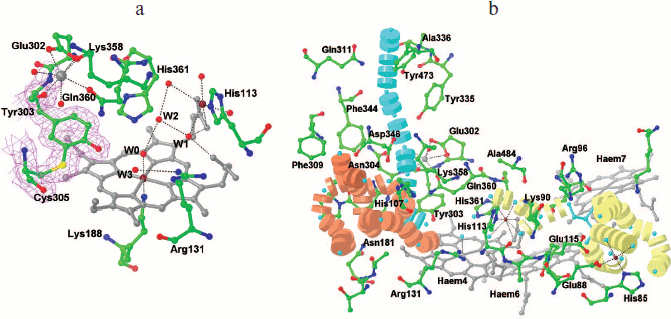
A number of structures of ONR have a second binding site for a metal ion (Ca2+) [44, 45]. A Ca2+ ion is located in the channel of the product transport and is coordinated by carboxyl groups of hemes 6 and 7, by the oxygen atom of the main chain of Pro116, and by three water molecules. A similar binding site for a second Ca2+ ion is found in NrfA molecules [17]. The role of this calcium ion for activity and stability of ONR is unclear. It is possible that the binding of the second Ca2+ ion affects the redox potentials of the coordinated hemes 6 and 7 [22], and thus affects the rate of intramolecular electron transfer.
Substrate and reaction product transport channels. As in the case of NrfA, the active site of ONR is connected with the surface of the subunit via two channels for the transport of substrate and reaction product molecules (Fig. 4b) [33, 35]. The entrance to the substrate channel has a positive charge due to the presence of residues Arg171, Arg316, and Arg348 in TvNiR and Lys213 and Arg171 in TvPaR, which promotes transport of negatively charged nitrite ions to the active site. The central part of the channel is characterized by almost neutral charge with the area of a negative charge around Asp346, which is conserved for ONR and NrfA. This residue seems to contribute to pushing the substrate directly into the active site.
The product transport channel is negatively charged, which promotes the leaving of the positively charged ammonium ion from the active site. The negative charge of the channel is due to the presence of conserved residues Glu83 and Glu115 (TvPaR also has Glu88) and propionate groups of hemes 6 and 7. Due to hexameric structure of ONR, the channel does not transport the product to the surface of the molecule as in NrfA, but leads it into the inner cavity of the hexamer. The exit of the channel into the cavity inside the hexamer has a positive charge. Three of the four residues (Arg96, Arg404, and Lys431) responsible for this charge are conserved in the sequences of other octaheme nitrite reductase, which confirms their importance for the functioning of these enzymes. The reaction product is released from the hexamer inner cavity into the solution via the mentioned three outlets located at sites of trimer–trimer contact near heme 8 (Fig. 2a) [35].
The product ands substrate transport channels are filled with water molecules linked by hydrogen bonds [35] and thus can perform an additional function of transport of protons into the active site. The water molecule of the substrate channel closest to the active site is linked by hydrogen bonds with the amide group of Gln360 and the hydroxyl group of catalytic residue Tyr303, thus providing the transfer of protons to the hydroxyl linked in turn by a hydrogen bond with the oxygen atom of the substrate (nitrite or sulfite). Because the nitrite reduction reaction is accompanied by the binding of eight protons, efficient transport of protons into the active site can have a significant effect on the catalytic process.
In the case of ONR, efficiency of proton transport to the active site can be much higher than in the case of NrfA due to the presence of an additional channel through which protons from the surface of the protein can be transferred to water molecule linked by hydrogen bond with the hydroxyl group of residue Tyr303 (Fig. 4b) [45]. The channel contains a chain of five water molecules linked by hydrogen bonds with each other and the amino acid residues forming the channel wall. The width of the channel excludes the possibility of transport of the substrate or product through it. In NrfA structure, this channel is closed on the surface of the protein by the side chain of an arginine residue, and inside the channel – by the side chains of tyrosine and leucine (isoleucine) residues. In ONR these positions are occupied by Tyr473, Ala339, and Pro357, respectively.
Structure of complexes of ONR with substrates and inhibitors. Localization of the active site at heme 4 was confirmed by obtaining the structures of complexes of ONR with substrates (sulfite, nitrite, and hydroxylamine) and inhibitors (azide [35], cyanide [45], and phosphate [47]), where the ligands bind at the distal side of heme 4 to form a coordination bond with the heme iron atom. In all cases, ligand binding was not accompanied by significant structural changes (rmsd between all Cα atoms of one subunit in apo- and holo-forms was about 0.2 Å) [35].
Binding of nitrite was accompanied by formation of a coordination bond between the nitrogen atom of nitrite and the iron atom of heme 4 (Fig. 5a). One oxygen atom of nitrite forms hydrogen bonds with the catalytic groups Tyr303 and His361, and the other one forms a hydrogen bond with Arg131. As already noted, the length of the hydrogen bonds formed by nitrite with histidine and tyrosine residues is shorter [35, 45] than that in the complex of NrfA with nitrite [14, 17], which may be a reason for the increased activity of ONR in nitrite reduction.
In ONR complexes with sulfite (Fig. 5b) [33, 43, 45], sulfite is coordinated with the catalytic heme iron atom through a sulfur atom. One of the oxygen atoms forms two hydrogen bonds with residues Tyr303 and His361. The second oxygen atom forms a hydrogen bond with the side chain of Arg131, and the third forms a hydrogen bond with a conserved water molecule associated with the carboxyl groups of the heme. Reduction of the complex with sulfite does not lead to significant structural changes in the active site of the enzyme [45]. An oxygen atom of sulfite linked to a conserved water molecule is cleaved first in the reduction process, and this leads to the formation of an intermediate that is bound similarly to the nitrite ion in the corresponding complex.Fig. 5. Binding of nitrite [45] (a) and sulfite [43] (b) in the active site of ONR from Tv. nitratireducens. The active site residues are labeled; the catalytic heme 4 is shown in white. The conservative water molecule linked by hydrogen bonds with the catalytic heme carboxylates is shown by gray sphere. Coordination and hydrogen bonds are shown by dashed lines.
Hydroxylamine is a substrate of NrfA and ONR as well as a possible intermediate in the reduction of nitrite. The location of the hydroxylamine molecule in the structure of the ONR complex [45] is similar to its position in the NrfA complex [23]: the nitrogen atom of NH2OH forms a coordination bond with the catalytic heme iron ion and the molecule is tilted toward residue Arg131. The direction of the N–O bond in the NH2OH molecule coincides with the direction of one of the N–O bonds in the nitrite ion in the corresponding complex (Fig. 5a). This orientation of the hydroxylamine molecule suggested [23] that during the reduction of nitrite the first bond to be broken is N–O, which is directed toward His361 and Tyr303.
During co-crystallization of ONR with hydroxylamine, a modification of the enzyme with the formation of an additional covalent bond between the C-atom of the aromatic ring of Tyr303 located in ortho-position toward the hydroxyl group and the Cγ atom of Gln360 coordinating Ca2+ ion was observed. The formation of this bond leads to the rotation of the Tyr aromatic ring by 12° and changing the conformation of the side chain of Gln360. These structural changes do not affect the position of substrates in the active site of ONR.
Reduced nitrite reductase activity of the modified enzyme was explained [45] by the possible increase of pKa of the hydroxyl of the modified Tyr303, which serves as a primary proton donor during catalysis. The importance of the hydroxyl group of the Tyr303 residue for catalysis was confirmed in [14], where mutation of the corresponding residue Tyr218 in the active site of NrfA to Phe led to almost complete loss of nitrite reductase activity despite the fact that the overall structure of the active site remained virtually unchanged.
On the whole, the arrangement of substrates and inhibitors in the active site of ONR coincides with that in the NrfA active site, which correlates with a similar structure of the active sites, explains the similarity of the catalytic properties of both classes of enzymes, and suggests the same mechanism of catalysis, which has been described for NrfA [23]. According to the proposed mechanism, in the first stage the catalytic heme iron ion is reduced to the oxidation state +2, then the water molecule, which occupies the place of the distal ligand, is replaced by nitrite to form a N–Fe coordination bond. In the next stage heterolytic cleavage of one of N–O bonds of nitrite ion occurs, and this is followed by two one-electron reductions of the formed {FeNO}6 complex to the {FeNO}8 complex. The NO ligand in the {FeNO}8 complex is nucleophilic and is readily protonated to form HNO, which leads to the formation of Fe(II)-HNO. The latter is then reduced by two electrons with the formation of Fe(II)-NH2OH. In the last stage, hydroxylamine undergoes reductive cleavage to form ammonium ion, which dissociates from the active site and is pushed by electrostatic forces to the subunit surface through the product output channel. Formation of NO and NH2OH as the possible intermediate products is consistent with the fact that both compounds are substrates of NrfA and ONR.
Physiological relevance. The role of ONR in the cells of sulfur-oxidizing bacteria of the genus Thioalkalivibrio is unclear. It is known that the bacterium Tv. paradoxus uses thiocyanate, thiosulfate, and sulfide as electron donors for growth. Thioalkalivibrio paradoxus does not grow under anaerobic conditions with nitrate or nitrite as an electron acceptor and does not use oxygen compounds of nitrogen in assimilatory processes [48].
The bacterium Tv. nitratireducens can use sulfur compounds such as sulfide, polysulfide, and thiosulfate, as well as hydrogen, as electron donors and under anaerobic conditions – nitrate (not nitrite) as an electron acceptor in respiratory processes. Reduction of nitrate in anaerobic culture growth is accompanied by accumulation of nitrite in the medium [37, 38].
These metabolic features of both bacteria suggest that the physiological function of ONR is not a respiratory reduction of nitrite. The participation of ONR in the detoxification of cells from intracellular nitrite is also unlikely because in Tv. paradoxus endogenous nitrite is not formed. In addition, most dissimilatory nitrite reductases are not constitutive and are formed only in response to increasing concentrations of nitrite. But for TvNiR and TvPaR it was shown that both proteins are constitutive, being present in the cells at all stages of growth with their content reaching 5-9% of soluble cell protein (A. V. Tikhonov, personal communication).
As already noted, sulfite reductase activity of ONR is comparable with the activity of natural siroheme-containing sulfite reductases [33, 43], suggesting that ONR can participate in the conversion of sulfur-containing compounds that play a key role in the metabolism of sulfur-oxidizing bacteria of the genus Thioalkalivibrio. In addition, the presence of ONR genes in all known genomes of bacteria of the genus Geobacter, characterized by high respiratory flexibility and the ability to use the oxides of various metals including iron and radioactive metals like uranium as an electron acceptor during anaerobic growth, suggests that ONR could be involved in respiratory processes with acceptors other than nitrite and sulfite.
ONR as an intermediate link in evolutionary chain of multiheme oxidoreductases involved in the nitrogen cycle. Comparative analysis of the properties and structure of multiheme oxidoreductases of nitrogen cycle has revealed significant similarity of these enzymes despite the low homology of amino acid sequences and the differences in oligomeric state, the structure of the active site, and the reactions catalyzed in cells. One of the most obvious general properties of all the above proteins was already mentioned – a conserved heme arrangement within the monomer. Axial ligands (His) of all hemes, except for the catalytic heme, are also conserved. Another important similarity is the presence of three long α-helices in the C-terminus of the NrfA, HAO, and ONR molecules, which are involved in the formation of intersubunit contacts in NrfA dimers [17], HAO trimers [28], and ONR hexamers [35]. A third similarity is that the reactions of conversion of nitrogen compounds catalyzed by multiheme oxidoreductases – oxidation of hydroxylamine to nitrite by HAO and reduction of nitrite, nitric oxide, and hydroxylamine to ammonium by NrfA and ONR, as well as the reduction of nitrite and hydroxylamine by HAO and OTR in vitro – appear to have similar mechanisms and proceed through the same intermediate states. All these signs point to the evolutionary relationship of the reviewed multiheme oxidoreductases.
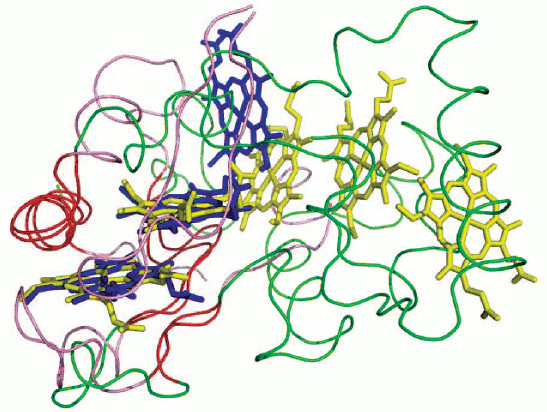
Based on analysis of amino acid sequence [31, 32], secondary structure elements, and available 3D structures of multiheme oxidoreductases of the nitrogen cycle, it has been postulated that the pentaheme dimeric nitrite NrfA reductases are evolutionary precursors of a variety of octaheme multisubunit proteins that reduce and oxidize nitrogen-containing compounds and possibly also sulfur-containing compounds. From NrfA two evolutionary groups are derived – the OTR and the HAO branches. In the second branch, ONR are regarded as an intermediate link. It is assumed that ONR were derived directly from NrfA by a single 5′-terminal insertion into a gene of a sequence that contains three heme-binding motifs [31], for example, a fragment of a gene (which is located immediately after the NrfA gene) of the electron donor (tetraheme cytochrome c NrfH for NrfA from δ-proteobacteria or pentaheme cytochrome c NrfB for NrfA from γ-proteobacteria E. coli). This assumption is confirmed by the superposition of the structure of TvNiR and the structure of NrfB taken from the structural model of the NrfB–NrfA complex from E. coli [49]. In the NrfB–NrfA model, hemes 1 and 2 of NrfB are exposed to the solution and, apparently, serve as a point of contact and electron transfer to the following member of the electron transport chain, NrfC. Hemes 1 and 2 of NrfB and solution-exposed hemes 1 and 2 of ONR superimpose (rmsd 0.8 Å) as well as the fragments of the protein structure surrounding hemes (rmsd 0.8 Å) (Fig. 6; see color insert) [35].
In addition to heme-binding motifs, ONR inherited from NrfA the residues forming the substrate channel and the residues involved in binding of calcium ions and the substrate. The newly acquired feature of ONR is cysteine located near the active site. In the active site of ONR, this cysteine forms a covalent bond with the catalytic tyrosine residue and cannot interact with the substrate. This bond is a distinguishing feature of ONR within multiheme oxidoreductases [31, 32].Fig. 6. Superimposition of hemes 1 and 2 from TvNiR (PDB 2OT4 [35]) and hemes 1 and 2 from NrfB [49]. Hemes from TvNiR are shown in blue, the elements of secondary structure are shown in pink, hemes from NrfA are shown in yellow, the elements of secondary structure are shown in green, and matching secondary structure elements are shown in red.
CONCLUSION
Octaheme oxidoreductases are widespread in different taxa of bacteria involved in the biogeochemical nitrogen cycle and possibly also the sulfur cycle [1, 31, 32]. Molecular genetic and structure-function analysis of genomes and proteins allowed to formulate a possible path of evolution of octaheme oxidoreductases of the nitrogen cycle from more ancient pentaheme nitrite reductases, with a change in their function from the reduction of nitrogen oxides to their oxidation under the influence of changing environmental conditions. Octaheme nitrite reductases are a transitional form combining structural and functional features of pentaheme nitrite reductases (NrfA) and octaheme oxidases (HAO). Packing of the eight hemes of ONR coincides with packing of hemes in HAO, while the structure of the catalytic domain, the presence of lysine-coordinated catalytic heme, and the structure of the active site correspond to those in NrfA. Despite the similarities, the structure of ONR demonstrates some distinctive features such as: covalent bond between the tyrosine and cysteine residues in the active site leading to a change in the properties of the catalytic tyrosine and providing more efficient nitrite reduction; hexameric organization of ONR in solution and, apparently, in the cell, which results in the release of the reaction product into the cavity inside the hexamer. Perhaps the formation of the hexameric structure in the two haloalkaliphilic ONR proteins studied is a consequence of adaptation to extreme environmental conditions and provides stability to the molecule at high salt concentrations and high pH values. Thus, it becomes clear that the formation of a stable hexamer containing 48 hemes does not lead to the formation of a common electron transport chain that unites the catalytic sites located on different subunits, as was shown for NrfA and HAO. The question of the possible role of oligomeric structure in catalysis and the question of the physiological function of ONR family proteins remains a subject of further research.
This work was financially supported by the Ministry of Education and Science (State contracts 14.740.11.0632 and P1197) and the Russian Foundation for Basic Research (grants 10-04-01695 and 11-04-01613).
REFERENCES
1.Simon, J., Kern, M., Hermann, B., Einsle, O., and
Butt, J. N. (2011) Biochem. Soc. Trans., 39,
1864-1870.
2.Simon, J. (2002) FEMS Microbiol. Rev.,
26, 285-309.
3.Kajie, S., and Anraku, Y. (1986) Eur. J.
Biochem., 154, 457-463.
4.Almeida, M. G., Macieira, S., Goncalves, L. L.,
Huber, R., Cunha, C. A., Romao, M. J., Costa, C., Lampreia, J., Moura,
J. J. G., and Moura, I. (2003) Eur. J. Biochem., 270,
3904-3915.
5.Pereira, I. A. C., LeGall, J., Xavier, A. V., and
Teixeira, M. (2000) Biochim. Biophys. Acta, 1481,
119-130.
6.Stach, P., Einsle, O., Schumacher, W., Kurun, E.,
and Kroneck, P. M. H. (2000) J. Inorg. Biochem., 79,
381-385.
7.Rudolf, M., Einsle, O., Neese, F., and Kroneck, P.
M. H. (2002) Biochem. Soc. Trans., 30, 649-653.
8.Angove, H. C., Cole, J. A., Richardson, D. J., and
Butt, J. N. (2002) J. Biol. Chem., 277, 23374-23381.
9.Clarke, T. A., Hemmings, A. M., Burlat, B., Butt,
J. N., Cole, J. A., and Richardson, D. J. (2006) Biochem. Soc.
Trans., 34, 143-145.
10.Van Wonderen, J. H., Burlat, B., Richardson, D.
J., Cheesman, M. R., and Butt, J. N. (2008) J. Biol. Chem.,
283, 9587-9594.
11.Poock, S. R., Leach, E. R., Moir, J. W. B., Cole,
J. A., and Richardson, D. J. (2002) J. Biol. Chem., 277,
23664-23669.
12.Liu, M.-C., and Peck, H. D., Jr. (1981) J.
Biol. Chem., 256, 13159-13164.
13.Pereira, I. C., Abreu, I. A., Xavier, A. V.,
LeGall, J., and Teixeira, M. (1996) Biochem. Biophys. Res.
Commun., 224, 611-618.
14.Lukat, P., Rudolf, M., Stach, P., Messerschmidt,
A., Kroneck, P. M. H., Simon, J., and Einsle, O. (2008)
Biochemistry, 47, 2080-2086.
15.Kemp, G. L., Clarke, T. A., Marritt, S. J.,
Lockwood, C., Poock, S. R., Hemmings, A. M., Richardson, D. J.,
Cheesman, M. R., and Butt, J. N. (2010) Biochem. J., 431,
73-80.
16.Kern, M., Volz, J., and Simon, J. (2011)
Environ. Microbiol., 13, 2478-2494.
17.Einsle, O., Messerschmidt, A., Stach, P.,
Bourenkov, G. P., Bartunik, H. D., Huber, R., and Kroneck, P. M. (1999)
Nature, 400, 476-480.
18.Einsle, O., Stach, P., Messerschmidt, A., Simon,
J., Kroger, A., Huber, R., and Kroneck, P. M. (2000) J. Biol.
Chem., 275, 39608-39616.
19.Bamford, V. A., Angove, H. C., Seward, H. E.,
Thomson, A. J., Cole, J. A., Butt, J. N., Hemmings, A. M., and
Richardson, D. J. (2002) Biochemistry, 41, 2921-2931.
20.Cunha, C. A., Macieira, S., Dias, J. M., Almeida,
G., Goncalves, L. L., Costa, C., Lampreia, J., Huber, R., Moura, J. J.,
Moura, I., and Romao, M. J. (2003) J. Biol. Chem., 278,
17455-17465.
21.Rodrigues, M. L., Oliveira, T. F., Pereira, I.
A., and Archer, M. (2006) EMBO J., 25, 5951-5960.
22.Lockwood, C. W. J., Clarke, T. A., Butt, J. N.,
Hemmings, A. M., and Richardson, D. J. (2011) Biochem. Soc.
Trans., 39, 1871-1875.
23.Einsle, O., Messerschmidt, A., Huber, R.,
Kroneck, P. M. H., and Neese, F. (2002) J. Am. Chem. Soc.,
124, 11737-11745.
24.Moser, C. C., Anderson, J. L., and Dutton, P. L.
(2010) Biochim. Biophys. Acta, 1797, 1573-1586.
25.Fonseca, B. M., Paquete, C. M., Salgueiro, C. A.,
and Louro, R. O. (2012) FEBS Lett., 586, 504-509.
26.Lockwood, C., Butt, J. N., Clarke, T. A., and
Richardson, D. J. (2011) Biochem. Soc. Trans., 39,
263-268.
27.Mowat, C. G., and Chapman, S. K. (2005) Dalton
Trans., 3381-3389.
28.Igarashi, N., Moriyama, H., Fujiwara, T.,
Fukumori, Y., and Tanaka, N. (1997) Nat. Struct. Biol.,
4, 276-284.
29.Mowat, C. G., Rothery, E., Miles, C. S., McIver,
L., Doherty, M. K., Drewette, K., Taylor, P., Walkinshaw, M. D.,
Chapman, S. K., and Reid, G. A. (2004) Nat. Struct. Mol. Biol.,
11, 1023-1024.
30.Atkinson, S. J., Mowat, C. G., Reid, G. A., and
Chapman, S. K. (2007) FEBS Lett., 581, 3805-3808.
31.Klotz, M. G., Schmid, M. C., Strous, M., Op den
Camp, H. J. M., Jetten, M. S. M., and Hooper, A. B. (2008)
Environ. Microbiol., 10, 3150-3163.
32.Bergmann, D. J., Hooper, A. B., and Klotz, M. G.
(2005) Appl. Environ. Microbiol., 71, 5371-5382.
33.Tikhonova, T., Tikhonov, A., Trofimov, A.,
Polyakov, K., Boyko, K., Cherkashin, E., Rakitina, T., Sorokin, D., and
Popov, V. (2012) FEBS J., in press.
34.Tikhonova, T. V., Slutsky, A., Antipov, A. N.,
Boyko, K. M., Polyakov, K. M., Sorokin, D. Y., Zvyagilskaya, R. A., and
Popov, V. O. (2006) Biochim. Biophys. Acta, 1764,
715-723.
35.Polyakov, K. M., Boyko, K. M., Tikhonova, T. V.,
Slutsky, A., Antipov, A. N., Zvyagilskaya, R. A., Popov, A. N.,
Bourenkov, G. P., Lamzin, V. S., and Popov, V. O. (2009) J. Mol.
Biol., 389, 846-862.
36.Foti, M., Ma, S., Sorokin, D. Y., Rademaker, J.
L. W., Kuenen, J. G., and Muyzer, G. (2006) FEMS Microbiol.
Ecol., 56, 95-101.
37.Sorokin, D. Yu., Tourova, T. P., Lysenko, A. M.,
Mityushina, L. L., and Kuenen, J. G. (2002) Int. J. Syst. Evol.
Microbiol., 52, 657-664.
38.Sorokin, D. Yu., Tourova, T. P., Sjollema, K. A.,
and Kuenen, J. G. (2003) Int. J. Syst. Evol. Microbiol.,
53, 1779-1783.
39.Hendrich, M. P., Upadhyay, A. K., Riga, J.,
Arciero, D. M., and Hooper, A. B. (2002) Biochemistry,
41, 4603-4611.
40.Fernandez, M. L., Estrin, D. A., and Bari, S. E.
(2008) J. Inorg. Biochem., 102, 1523-1530.
41.Kostera, J., Youngblut, M. D., Slosarczyk, J. M.,
and Pacheco, A. A. (2008) J. Biol. Inorg. Chem., 13,
1073-1083.
42.Kostera, J., McGarry, J., and Pacheco, A. A.
(2010) Biochemistry, 49, 8546-8553.
43.Trofimov, A. A., Polyakov, K. M., Boyko, K. M.,
Tikhonova, T. V., Safonova, T. N., Tikhonov, A. V., Popov, A. N., and
Popov, V. O. (2010) Acta Crystallogr. D Biol. Crystallogr.,
66, 1043-1047.
44.Tikhonova, T. V., Slutskaya, E. S., Filimonenkov,
A. A., Boyko, K. M., Kleimenov, S. Y., Konarev, P. V., Polyakov, K. M.,
Svergun, D. I., Trofimov, A. A., Khomenkov, V. G., Zvyagilskaya, R. A.,
and Popov, V. O. (2008) Biochemistry (Moscow), 73,
164-170.
45.Trofimov, A. A., Polyakov, K. M., Tikhonova, T.
V., Tikhonov, A. V., Safonova, T. N., Boyko, K. M., Dorovatovsky, P.
V., and Popov, V. O. (2012) Acta Crystallogr. D Biol.
Crystallogr., 68, 144-153.
46.Clarke, T. A., Kemp, G. L., van Wonderen, J. H.,
Doyle, R. M., Cole, J. A., Tovell, N., Cheesman, M. R., Butt, J.
N., Richardson, D. J., and Hemmings, A. M. (2008) Biochemistry,
47, 3789-3799.
47.Trofimov, A. A., Polyakov, K. M., Boyko, K. M.,
Filimonenkov, A. A., Dorovatovskii, P. V., Tikhonova, T. V., Popov, V.
O., and Kovalchuk, M. V. (2010) Crystallogr. Rep., 55,
58-64.
48.Sorokin, D. Y., Antipov, A. N., and Kuenen, J. G.
(2003) Arch. Microbiol., 180, 127-133.
49.Clarke, T. A., Cole, J. A., Richardson, D. J.,
and Hemmings, A. M. (2007) Biochem. J., 406, 19-30.