Purification, Biochemical Characterization, and Structure of Recombinant Endo-1,4-β-xylanase XylE
T. V. Fedorova1, A. M. Chulkin1, E. A. Vavilova1, I. G. Maisuradze1, A. A. Trofimov1,2, I. N. Zorov1, V. P. Khotchenkov1, K. M. Polyakov1,2, S. V. Benevolensky1, O. V. Koroleva1*, and V. S. Lamzin3
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; fax: (495) 954-2732; E-mail: koroleva@inbi.ras.ru2Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; fax: (499) 135-1405; E-mail: isinfo@eimb.ru
3European Molecular Biology Laboratory (EMBL), c/o DESY, Build. 25A, Notkestrasse 85, 22603 Hamburg, Germany; fax: +49(0)4089-902149; E-mail: info@embl-hamburg.de
* To whom correspondence should be addressed.
Received June 20, 2012; Revision received July 7, 2012
The gene xylE encoding endo-1,4-β-xylanase from the 10th family of glycosyl hydrolases produced by the mycelial fungus Penicillium canescens has been expressed under the control of the strong promoter of the bgaS gene encoding β-galactosidase from P. canescens. As a result, a strain-producer of endoxylanase XylE was developed. The recombinant enzyme was isolated and purified to homogeneity with specific activity of 50 U/mg. The physicochemical and biochemical properties of the endoxylanase were studied. The maximal enzymatic activity was observed at pH 6.0 and 70°C. Endoxylanase XylE was shown to be a highly thermostable enzyme with half-inactivation period τ1/2 of 7 h at 60ºC. The kinetic parameters were 0.52 mg/ml (Km) and 75 µmol/min per mg (Vmax) using birch xylan as the substrate. Crystals of endoxylonase XylE were obtained, and the 3D structure was solved at 1.47 Å resolution. The 3D structure of an endo-1,4-β-xylanase from the 10th family containing carbohydrate and unique cyclic structure located at the C-terminus of the polypeptide chain was obtained for the first time.
KEY WORDS: Penicillium canescens, xylanase, homologous expression, XylE, 3D structure, biochemical and physicochemical properties, substrate specificityDOI: 10.1134/S0006297912100112
β-1,4-Xylan is a major cell wall polysaccharide of plants; it is second only to cellulose in prevalence of the carbohydrate of plants. In terms of chemical structure, xylan is a heteropolymer consisting of a linear chain of β-(1,4)-D-xylose that is partially acetylated, and it has a different degree of substitution in the side chains of D-glucuronosyl- and α-L-arabinosyl units. The complexity of its structure obstructs the degradation of the heteropolymer. In nature, this process is performed by a wide range of hydrolases. The key enzymes in the degradation of xylan are endo-β-(1,4)-xylanase (EC 3.2.1.8) cleaving the outer β-(1→4) bonds in the unmodified residues forming xylooligosaccharides of different lengths [1].
Interest in xylanase has increased dramatically in recent years due to the possibility of their application in biotechnology. The biotechnological application of xylanases includes the pulp and paper, food, fodder, and textile industries [1, 2]. They are also used in the production of biofuels [1, 2] and in the technology of conversion of plant residues to monosaccharides [1, 2]. Therefore, the research activity is focused on screening of different microorganisms including bacteria and fungi for xylanases with target properties required for industrial application. Fungi as a source of xylanases are of a greatest interest for biotechnology due to high production of extracellular enzymes [1]. Most endoxylanases of filamentous fungi belong to the 10th and 11th families of glycosyl hydrolases: xylanases with high molecular weight (>30 kDa) and low isoelectric point belong to the 10th family, and enzymes with low molecular weight (19-25 kDa) and high isoelectric point belong to the 11th family [3]. Xylanases from the 10th glycosyl hydrolase family depolymerize shorter unsubstituted xylopyranosyl residues compared to the xylanases from 11th family [4].
For some species of filamentous fungi various forms of xylanases have been found. One organism may produce several enzymes belonging both to the 10th and 11th families of glycosyl hydrolases. The genes xynB and xynC of P. funiculosum encode two xylanases from the 11th family, and the gene xylD encodes a xylanase from the 10th family [5-7]. Penicillium purpurogenum also produces two xylanases belonging to different families [8, 9]. In studying P. canescens, a natural producer of xylanases, a family of genes encoding endoxylanases was identified [10, 11], moreover, these endoxylanases have a low degree of homology and belong to different branches of the phylogenetic tree. The established regularity is observed for endoxylanases of both the 10th and 11th families. These data suggest a divergence of genes of this group at an early stage of evolution [11]. According to our preliminary studies, the level of production of most P. canescens endoxylanases when cultured in media of different composition is extremely low or absent. Thus, it is not possible to characterize new enzymes using a natural P. canescens strain. Therefore, the aim of this study was the homologous expression of this family gene, xylE, to produce a recombinant enzyme with subsequent characterization and determination of its 3D structure.
MATERIALS AND METHODS
Strains used and culture growth conditions. In this work we used P. canescens F178 (VKPM) strain and its mutant P. canescens PCE-7 deficient in the nitrate reductase gene (niaD–) and the endoxylanase gene (xylA–) [12]. The composition of the media for the cultivation of fungi and the cultivation conditions were as described in [13]. Methods for formation of protoplasts and co-transformation of the PCE-7 strain were described in [12, 13]. Escherichia coli XL1-Blue strain (Stratagene, USA) was used for plasmid transformation.
Methods for manipulation with DNA. Restriction, ligation, dephosphorylation, precipitation of nucleic acids, electrophoresis of DNA, plasmid transformation of E. coli, and isolation of plasmid DNA were performed by the methods given in [14]. Isolation of fungal chromosomal DNA was performed according to [13]. Purification of PCR fragments from agarose gels was performed using the set of reagents in the GFX PCR and Gel Band Purification Kit (GE Healthcare, USA) according to the manufacturer’s recommendations.
Determination of nucleotide sequences. The nucleotide sequences of the cloned DNA fragments were determined using ABI PRISM® BigDye™ Terminator v.3.1 with subsequent analysis of reaction products on an Applied Biosystems ABI PRISM 3730 automated DNA sequencer at the Center for Collective Use “Genome” of the Engelhardt Institute of Molecular Biology (Russian Academy of Sciences).
Sequence analysis. The sequence of the signal peptide was predicted using the SignalP program (http://www.cbs.dtu.dk/services/SignalP/). The molecular weight of the mature peptide was predicted using the Vector NTI 7.0 program. Homology search in GenBank was performed using the BLAST server (http://www.ncbi.nlm.nih.gov/BLAST). The ClustalW program (http://www.ebi.ac.uk/clustalW) was used for multiple alignments of amino acid sequences of the proteins.
Homologous expression of the xylE gene in P. canescens. Construction of a plasmid for xylE expression. For expression of the previously cloned P. canescens xylE gene of endoxylanase [11], we used a strong promoter of the bgaS gene of extracellular β-galactosidase of P. canescens [15].
A 1450-bp-long fragment of the xylE gene with the BglII restriction site introduced after the transcription terminator containing the complete encoding sequence and the terminator region was amplified using PfuI polymerase and a pair of primers XlBglD (5′-TTGCTCACCATGCGTCCATCCCTAGTGATCG-3′) and XlBglR (5′-CCAGATCTATACCCAGACCAAACGA-3′).
The resulting xylE gene fragment was docked with the 3′-end of the bgaS gene promoter using “nested” PCR. For this, the PCR fragment of the bgaS gene promoter was amplified using primers CR1337D (5′-CGTACACCAAGGCTGGGGATGAGGAGGACC-3′) and BgXlR (5′-TGGACGCATGGTGAGCAAAGTCGACGA-3′) and pPCG2.6 plasmid DNA [16] carrying a complete bgaS gene for β-galactosidase as a template. A mixture of DNA of the resulting fragment and DNA of PCR fragment of the xylE gene was amplified using primers CR1337D and XlBglR.
The resulting PCR fragment with a length of ~2670 bp containing an incomplete sequence of the bgaS gene promoter and coding and terminator regions of the xylE gene was treated with NcoI and BglII restriction enzymes and then cloned into the pPCG2.6 plasmid vector. The resulting construct was verified by sequencing for mutations.
Culture of the transformed P. canescens PCE-7/pBGXylE strain – producer of xylanase E. The P. canescens PCE-7/pBGXylE strain was cultivated in shaker flasks in medium of the following composition (fermentation medium, g/liter): beet pulp, 30; peptone, 50; KH2PO4, 25. The liquid fermentation medium was inoculated with conidial suspension (1·107 spores per 100 ml) and incubated on a shaker at 30°C and 200 rpm for 6 days.
Isolation and purification of the mutant enzyme. Extracellular proteins were precipitated by ammonium sulfate (80% saturation) and separated by centrifugation at 10,000g for 20 min at 4ºC. The pellet was resuspended in 30 ml of 35 mM Tris-HCl, pH 8.25 (buffer A). After repeated centrifugation to remove the undissolved fraction at 30,000g for 20 min at 4ºC, the supernatant was desalted on a Sephadex G-25M (2.6 × 40 cm) column equilibrated with buffer A.
Desalted protein solution (50 ml) was applied to a DEAE-Toyopearl 650M (2.6 × 34 cm; GE Healthcare) column equilibrated with buffer A. Proteins were eluted with a NaCl (0-0.2 M) gradient in the same buffer at a rate of 7.5 ml/min. Xylanase was eluted as a single peak at 0.125 M NaCl. The fractions with enzymatic activity were collected and precipitated with ammonium sulfate (27% saturation). The protein precipitate was separated by centrifugation at 30,000g for 20 min at 4ºC.
The enzyme was purified further by chromatography on a phenyl-Sepharose FF HS (1.6 × 36 cm; GE Healthcare) column equilibrated with 50 mM Na-phosphate buffer (pH 6.8) containing ammonium sulfate (30% saturation) (buffer B). Elution was carried out by a linear gradient of ammonium sulfate (30 to 0%) at a flow rate of 2 ml/min. The xylanase eluted as a single peak at 13% saturation of ammonium sulfate. The fractions with enzymatic activity were collected and transferred into 40 mM Na-acetate buffer, pH 4.8 (buffer C), using a Sephadex G-25M (2.6 × 40 cm) column.
The resulting enzyme solution was applied to a Source 15S (1.5 × 21 cm; GE Healthcare) column equilibrated with buffer C. Elution was performed using a linear gradient of NaCl (0-0.375 M) in the same buffer at 4 ml/min. The xylanase was eluted as a single peak at 0.25 M NaCl. Fractions containing the enzyme activity were collected and concentrated by ultrafiltration at 4000g for 20 min using a PP-10 VIVASPIN filter (Sartorius Stedim Biotech, Germany).
Protein concentration was determined using a BCA Protein kit (Pierce, USA). Bovine serum albumin was used as a standard.
Determination of enzymatic activity. To determine xylanase activity, birch xylan (Sigma, USA) was used as a substrate. The reaction medium contained 0.3 ml of 1% (w/v) suspension of the substrate in deionized water and 0.1 ml of enzyme solution in 0.1 M acetate buffer, pH 5.0. After 10 min incubation at 50°C, the amount of reducing sugar was determined by the Somogyi–Nelson method. The amount of the enzyme catalyzing the formation of 1 µmol of xylose equivalents from xylan in 1 min was taken as one unit (U) of xylanase activity.
Substrate specificity and catalytic parameters. The substrate specificity of XylE was studied using xylans from oat, birch, and larch and microcrystalline cellulose as substrates. The reaction mixture containing 0.3 ml of 1% substrate in 0.1 M acetate buffer, pH 5.0, and 0.1 ml of the enzyme solution was incubated for 10 min at 50ºC. The amount of reducing sugars was measured by Somogyi–Nelson.
Kinetic parameters, Km and apparent Vmax, were determined in the 1-10 mg/ml concentration range of birch xylan. The values of the catalytic parameters are means of three independent experiments with three parallel runs in each.
Identification of the enzyme and its characterization. The proteins were separated by SDS-PAGE with 4% concentrating gel and 12% separating gel using a Mini PROTEAN 3 device (Bio-Rad). The gels were stained with Coomassie Brilliant Blue-R250. Molecular weight was determined using the standard molecular weight PageRuler #SM0671 (Fermentas, Lithuania) kit. The isoelectric point of the enzyme was determined using ampholytes with pH 5.0-7.0 (Ampholyte; Bio-Rad) in a Mini IEF Cell system (Bio-Rad). A set of standards with range of isoelectric points of 2.5-6.5 (Amersham) was used. For detection of glycoproteins, gels after protein separation using denaturing 11% SDS-PAGE were incubated with concanavalin–peroxidase conjugate (Sigma, USA) with subsequent staining with 4-chloro-1-naphthol (Sigma). BSA (Sigma) was used as a negative control.
To identify the proteins, the bands under study were cut from the gel, subjected to trypsinolysis, and the resulting fragments were analyzed on a MALDI-TOF/TOF Ultraflex II BRUKER mass spectrometer (Germany) with UV laser (Nd).
Physicochemical properties of the XylE. The pH dependence of catalytic activity was investigated in the pH range 3.0-7.5 in 0.1 M acetate-phosphate buffer. Activity was determined as described above. The temperature dependence of the enzyme activity was determined in the range from 25 to 90ºC using 0.1 M acetate buffer, pH 5.0. The thermal stability of the enzyme was determined at 70 and 60ºC in 0.1 M acetate buffer, pH 5.0.
The calorimetric study was carried out using an adiabatic DASM-4M differential scanning microcalorimeter (Special Design Bureau, Russian Academy of Sciences) with 430-µl platinum capillary cells. To obtain heat absorption curves, xylanase in 0.1 M acetate buffer, pH 5.0, was placed in the cell and heated at a constant rate of 1 K/min from 20 to 80°C at a constant pressure of 2 atm. The control cell contained the same buffer solution. Calibrating power was 25 µW. The dependency of the excess molar heat capacity on temperature was plotted.
Determination of reaction products of xylan hydrolysis. The reaction medium containing 0.01 mg/ml of purified XylE and 2 mg/ml birch xylan in 0.1 M acetate buffer, pH 5.0, was incubated at 50ºC. The samples were taken after 15, 60, 120, 870, and 1440 min of incubation. The reaction products were analyzed by high-performance anion-exchange chromatography on an Agilent 1100 chromatograph (Agilent, USA) with an Agilent Chemstation ver. A09.01, 1312A binary pump, 1367A microplate autosampler, and an ESA Coulochem III electrochemical detector (ESA Inc, USA) with a 5040 cell containing a gold electrode in cyclic amperometry mode. Components of the reaction mixture were separated using an analytical Dionex CarboPak PA100 (4 × 250 mm) column with a PA100 pre-column (4 × 50 mm) at a flow rate of 1 ml/min. Xylose, xylobiose, xylotriose, xylotetrose, and xylopentose (Sigma, USA and Merck, Germany) were used as standards.
Enzyme crystallization. XylE was crystallized by the hanging drop vapor diffusion method using a 10 mg/ml protein solution in deionized water and a precipitant solution of the following composition: 0.1 M HEPES, pH 7.0, 22% (w/v) PEG 3350. Drops of 4 µl contained equal volumes of the protein solution and the precipitant solution. The crystals grew within two or three days at 18°C.
Data collection and structure refinement. Diffraction data were collected at the beam-line K4.4 of the Kurchatov Synchrotron Nano Centre (Moscow) using a CCD-detector with cooling to 100 K. Before data collection, crystals were soaked in a cryoprotectant solution containing the reservoir solution with 20% PEG 600. The diffraction data were processed using the XDS and XSCALE programs [17]. The structure was solved by the molecular replacement method with the use of the program BALBES [18]. The structure was refined using the REFMAC program [19] taking into account the contribution of hydrogen atoms in the scattering. The models were manually adjusted in accordance with the electron density maps using the COOT program [20]. The CCP4 software package was used for this work [21]. The structure was deposited in the PDB database with code 4F8X.
RESULTS
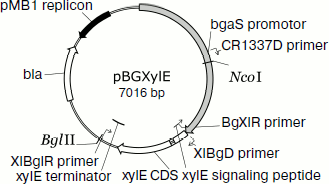
Homologous expression of the endoxylanase xylE gene in P. canescens. A DNA fragment containing the coding sequence including the signal peptide sequence and the transcription terminator of the XylE gene was cloned into plasmid vector pPCG2.6 [16] for efficient transcription of xylE under the control of the bgaS promoter. The final expression cassette pBGXylE (Fig. 1) was introduced into the P. canescens PCE-7 mutant strain deficient in the nitrate reductase gene (niaD–) and endoxylanase gene (xylA–) [12] by co-transformation with plasmid pSTA-10 [22] carrying a complementing niaD gene from Aspergillus niger. On selective medium containing sodium nitrate as a nitrogen source, the clone PCE-7/pBGXylE was selected having endoxylanase activity increased 24-fold compared to the recipient strain (the maximum was 120 U/ml). Protein electrophoresis of the culture liquid from the recombinant strains showed that the introduction of the plasmid did not lead to the emergence of novel proteins, but it increased the amount of protein with molecular weight of about 40 kDa (Fig. 2), which is close to the calculated molecular mass of the XylE protein (36 kDa) [11].
Fig. 1. An expression cassette pBGXylE.
Purification and characterization of recombinant XylE. Table 1 summarizes the stages of chromatographic purification of the extracellular endoxylanase from the 10th XylE family of the fungal strain P. canescens PCE-7. Purification by the anion-exchange (DEAE-Toyopearl), hydrophobic (P-Sepharose), and cation-exchange (Source 15S) chromatography yielded a homogeneous protein with 27.78-fold purification, and the XylE specific activity reached 50 U/mg. The purified protein had a molecular mass of 40 kDa according to SDS-PAGE (Fig. 2a, lane 4) and pI around 6.5 (Fig. 2b), which is typical for most filamentous fungal endoxylanases of the 10th family. The theoretically calculated molecular mass of XylE is 36 kDa [11], which is slightly less than value obtained by electrophoresis (40 kDa). This could be due to posttranslational glycosylation of xylanase E, which is a glycoprotein (staining for carbohydrates with 4-chloro-1-naphthol; Fig. 2c, lane 3), in contrast to P. canescens XylA [10]. Indeed, extracellular fungal xylanases can be glycoproteins. The amino acid sequence of xylanase XYL10C has six potential N-glycosylation sites [23], whereas the amino acid sequence of XylE [11] has only two such sites.
Table 1. Purification of recombinant
XylE
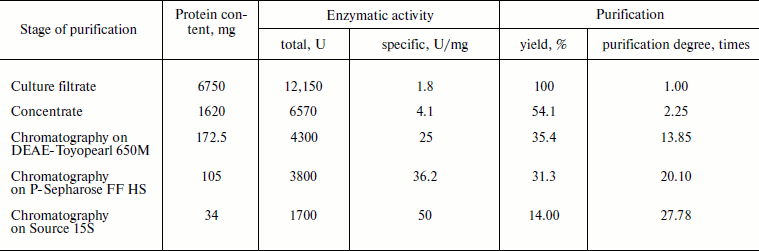
Fig. 2. Electrophoregrams of recombinant P. canescens XylE after denaturing PAGE. a) Lanes: 1) molecular weight markers; 2) culture liquid of transformant with pBGXylE; 3) culture liquid of P. canescens PCE-7; 4) XylE after purification. b) Isoelectrofocusing of purified XylE (lane 2). Molecular pI markers (lane 1). c) Carbohydrate staining of purified XylE (lane 3), molecular weight markers (lane 1) and BSA as a negative control (lane 2).
The protein band on electrophoresis corresponding to a molecular mass of 40 kDa was analyzed using MALDI-TOF/TOF mass spectrometry. Nine peptides were identified (TVQHFGR, STAVLQLVSNLR, ANLDVAVTELDVR, QNFGEITPANAMK, FSTVPYYTAAAQK, YAQEALAQIGANDVK, LYYNDYGIENPGTK, GKHWFGTAADIPGTAETTDAAYLK, and IDGVGLESHFIVGETPSLADQLATK), which were present in XylE amino acid sequence (GenBank ACP27611). Thus, the purified protein is indeed a recombinant xylanase E.
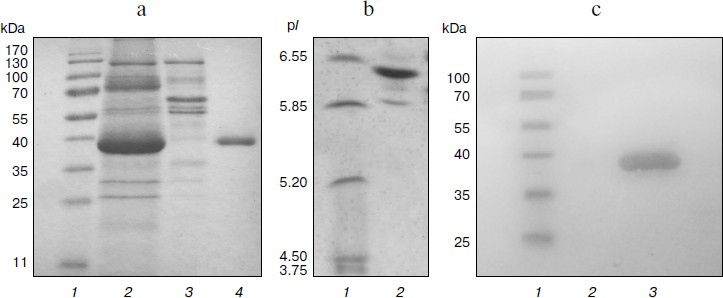
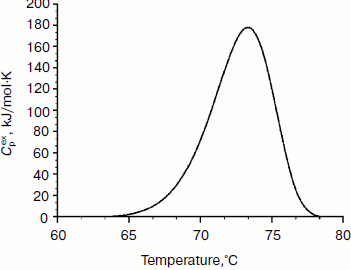
Physicochemical characterization of purified XylE and analysis of xylan hydrolysis products. Purified recombinant XylE is active over the pH range 4.0-8.0 (measurements were performed at 50ºC), with the peak of enzymatic activity at pH 5.5-6.0 (Fig. 3a). The optimum temperature for XylE enzymatic activity is about 70ºC (taken as 100%) (Fig. 3b). At 65 and 75ºC, the xylanase activity was 80% of the maximum value, and at 80 and 85ºC it was 60 and 20%, respectively. Indeed, according to differential scanning calorimetry data, the melting point of the XylE protein globule is about 73ºC (Fig. 4), which causes a sharp drop in enzyme activity when heated above 75ºC. The half-life time of XylE at 60ºC was about 7 h (Fig. 3c). After incubation of XylE at 70ºC for 15 min, the enzyme activity was 20% of the initial activity, and it was completely inactivated by preincubation for 30 min.
Fig. 3. Physicochemical properties of the XylE: a) pH dependence of enzymatic activity in 0.1 M citrate-phosphate buffer; b) dependence of enzymatic activity on temperature in 0.1 M citrate-phosphate buffer, pH 5.0; c) thermostability of XylE at 60ºC in 0.1 M acetate buffer, pH 5.0. Birch xylan was used as a substrate. Each data point is the average of three measurements ± SD (n = 3).
Fig. 4. Thermal denaturation of XylE. The dependence of the excess heat capacity (Cpex) obtained at the enzyme concentration of 1 mg/ml and scanning rate of 1 K/min.
XylE shows high activity towards larch xylan (78 U/mg) and slightly less activity with oat and birch xylans (55 and 50 U/mg, respectively). However, in contrast to other xylanases from the 10th family having rather broad substrate specificity, XylE is not active with cellulose substrates such as CMC-Na and β-glucan. The Km and Vmax values for hydrolysis of birch xylan were 0.52 mg/ml and 75 µmol/min per mg, respectively, which is in agreement with the literature data. Thus, for fungal endoxylanases Km values vary within 0.09-40.9 mg/ml and Vmax values within 0.106-6300 µmol/min per mg [2].
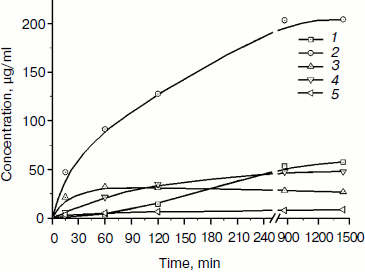
The products of birch xylan enzymatic hydrolysis by the purified xylanase XylE were analyzed. Xylan hydrolysis products were determined by the HPLC technique during 24-h incubation of the reaction mixture (Fig. 5). Xylobiose was the main product of the hydrolysis. Also, xylotriose accumulation was observed at the initial stage of the hydrolysis. Xylose accumulation starts after a rather long incubation of the reaction mixture.
Fig. 5. Accumulation of products of birch xylan hydrolysis by the purified xylanase XylE: 1) xylose; 2) xylobiose; 3) xylotriose; 4) xylotetraose; 5) xylopentose.
Structure of XylE. X-Ray data-collection statistics for the recombinant XylE are shown in Table 2. There is one molecule of xylanase per asymmetric unit. The structure of the enzyme shows 335 residues. Residues 1-25 are not seen in the electron density map (disordered).
Table 2. Data-collection and refinement
statistics. Values in parentheses are for the highest resolution
shell

The tertiary structure of XylE is manifested as an (α/β)8 barrel typical for the 10th family xylanases, and in general it differs slightly from other analogous xylanases (Fig. 6a; see color insert). The catalytic residues are Glu158 and Glu270. The distance between them in the structure is 4.98 Å. At a distance of 2.58 Å from Glu158, the residue Glu238 is located. In all known structures of xylanases of the 10th family this position is occupied by a glutamine residue. The observed substitution does not affect the structure of the enzyme active site.
Fig. 6. 3D structure of recombinant XylE. a) Cartoon representation of the XylE structure showing disulfide bridges and sugar chains. b) C-Terminal part of XylE. The electron density map 2Fo-Fc (1 σ) is shown. c) Superposition of the XylE (yellow; PDB code: 4F8X) and XylA (gray; PDB code: 1B3Z) chains on equivalent Cα atoms. Cyan marks differences near the substrate binding pockets of XylE and XylA. Linked sugars are assigned in accordance with the substrate binding subsites.
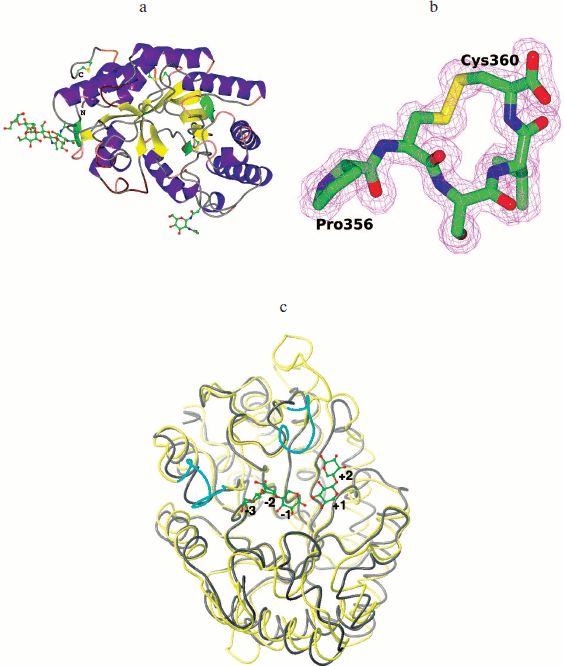
The XylE structure contains the maximum number (compared to analogous xylanases) of cysteine residues (seven), six of which are interconnected by three disulfide bonds. As a result of the Cys357–Cys360 bond formation, a cyclic structure is formed at the C-terminal of XylE (Fig. 6b; see color insert); this is unique among xylanases of the 10th family.
The structure of XylE is the only structure among xylanases of the 10th family that contains sugars covalently bound to the protein in the glycosylation sites. The XylE sequence contains two possible glycosylation sites – Asn88 and Asn336 residues. The Asn336 residue is linked to a carbohydrate chain consisting of three units, which is followed by branching. The first two units in the chain (residues of N-acetyl-D-glucosamine linked via 1,4-β-glycosidic bond) and the third unit (D-mannose residue) are characterized by full occupancy. The electron density near the Asn88 residue was interpreted by the N-acetyl-D-glucosamine residue with 1/2 occupancy.
DISCUSSION
XylE shows a low degree of homology (31%) with XylA (GenBank AAV65488) expressed by the fungus P. canescens under the normal culture growth conditions [11]. XylE and XylA have different biochemical properties. Thus, the Km value for birch xylan hydrolysis by XylA is 3.4 mg/ml [24], which is 6.5 times more than for XylE. XylA has broader substrate specificity and is able to hydrolyze cellulose substrates such as CMC-Na and β-glucan, whereas XylE shows narrower substrate specificity. The temperature maximum of XylE activity is 70ºC, and that of XylA is 55ºC [24]. The thermal stability of XylE is much higher than that of XylA. Half-life periods at 60ºC for XylE and XylA are 7 h and 5 min, respectively [24].
XylA from P. canescens is 100% identical with xylanase from P. simplicissimum having known structure [25]. The maximal structural differences between XylE and XylA are insertions of amino acid residues 192-196, 274-288, 323-329, and 353-360 (C terminus) in the XylE sequence but not in XylA (Fig. 6c; see color insert).
Most characterized xylanases from the 10th family have five or six substrate-binding subsites, although there are enzymes containing either four or seven subsites. Subsites –2, –1, and +1 are highly conserved, and amino acid residues within them play a key role in recognition and binding of the substrate. Amino acid substitutions in these subsites change the enzymatic properties of xylanases from the 10th family. Thus, it was shown that replacement of Tyr to Phe in binding subsite +1 greatly reduces the catalytic efficiency of xylanases towards glucuronoxylans (birch xylan) [26]. Indeed, subsites –1 in XylE and XylA are conserved. In XylA subsite –2 contains a side chain of the Glu47 residue, while XylE has in this position Ala72 incapable of forming a hydrogen bond with the substrate. Subsites +1 of XylE and XylA have identical structural organization; they contain a tyrosine residue (Tyr206 XylE), which explains their high catalytic efficiency towards birch xylan.
Subsites –3, +2, and +3 in xylanases of the 10th family are less conserved, and xylanase substrate specificity can vary greatly depending on the amino acid residues located there. Based on the analysis of catalytic and structural features of xylanase from Cellvibrio japonicas 10C, it was suggested that the presence of the additional tyrosine residue Tyr340 in subsite –3 provides high affinity of the –3 subsite to the substrate and increases the efficiency of catalysis [27]. Analysis of the XylE structure showed that there is a side chain of Tyr79 residue near subsite –3 and virtual subsite –4 that could determine the high affinity of these subsites. XylA has Ala54 instead of this residue. Obviously, the observed structural differences in subsite –3 affect the specificity of XylE and provide its high binding constant (Km) towards glucuronoxylan, since methylglucuronic acid, which is a component of glucuronoxylan, can bind only to subsites +1 and/or –3 [26, 28]. It should be noted that unusually high Km of XylE compared to other xylanases of the 10th family may be due to change in the course of the main chain for residues 46-52 near substrate-binding subsite –3 (Fig. 6c). Furthermore, the XylE structure contains near subsite +2 a side chain of the Phe326 residue from the loop 323-329, which can form hydrophobic contacts with the substrate and lead to a deepening of the gap where the substrate binds (Fig. 6c), which can also result in an increase in the enzyme affinity towards the substrate.
Conserved subsites –2 and –1 of xylanases of the 10th family can bind glucose molecules, but the catalytic efficiency of hydrolysis of glucose-containing oligosaccharides varies widely among the members of this family. Amino acids forming subsites –2 and –1 must be conformationally labile to bind additional hydroxymethyl group of glucose. A correlation between the lability of these amino acid residues and the catalytic efficiency of hydrolysis of glucose-containing oligosaccharides was established for xylanases of the 10th family [26, 28]. Thus, the Trp276 residue in the XylA structure (located near subsites –2 and –1) has more conformational freedom in comparison with the respective Trp321 residue in the XylE structure forming hydrophobic contacts with residues from the loop 323-329, which explains the lack of activity of XylE towards glucose-containing substrates in contrast to XylA.
The high thermal stability of XylE as compared to XylA is possibly due to the presence of glycosylation and three S–S-bonds in its structure. One of these S–S-bonds, i.e. that between residues Cys300 and Cys307, is conserved in a number of xylanases, while the Cys357–Cys360 bond in the C-terminus of XylE is unique for 10th family xylanases.
Today we know that the structure of the TIM-barrel of xylanases of the 10th family is stabilized via ionic and hydrophobic interactions as well as hydrogen bonds between the structural elements [29, 30]. In silico analysis of protein structures available in the PDB (protein structure database) revealed a direct correlation between the presence of a cluster of aromatic amino acids in the structure and thermal stability of these proteins [31]. And recent studies [32] have shown that bacterial xylanases of the 10th family have at their N- and C-termini an aromatic cluster, which includes amino acids Phe4, Trp6, and Tyr343 providing contact between the N- and C-termini in the structure. At the C-terminus of xylanase E we found a cyclic structure formed due to the Cys357–Cys360 bond. The presence of such clusters at the C-terminus may be important for protein folding and stability in different extreme conditions such as high temperature, high and low pH, high salt concentration, and the presence of protease activity.
A comparison of the amino acid sequence of P. canescens XylE with amino acid sequences of several fungal 1-4-β-endoxylanases of the 10th family (from Penicillium, Aspergillus, and other genera) available in the GenBank database [11] showed that there is a whole group of xylanases with a conserved KPCSVC sequence at the C-terminus. Thus, these xylanases with cysteine residues in positions 357 and 360 form a disulfide bond at the C-terminus similar to that found in the XylE structure. Perhaps this structure is identical in its functional role to the aromatic cluster of bacterial xylanases of the 10th family.
Thus, we suggest that the XylE plays an important role in hydrolysis of xylan of cell walls in extreme conditions, while it is not expressed by the fungus under the normal conditions. So, further structural and functional studies of XylE under different growth conditions are required to understand the biological role of this enzyme.
This work was financed by the Ministry of Education and Science of the Russian Federation (state contracts No. 16.512.11.2150 and 14.740.11.0005).
REFERENCES
1.Polizeli, M. L. T. M., Rizzatti, A. C. S., Monti,
R., Terenzi, H. F., Jorge, J. A., and Amorim, D. S. (2005) Appl.
Microbiol. Biotechnol., 67, 577-591.
2.Beg, Q. K., Kapoor, M., Mahajan, L., and Hoondal,
G. S. (2001) Appl. Microbiol. Biotechnol., 56,
326-338.
3.Collinsa, T., Gerdaya, C., and Fellera, G. (2005)
FEMS Microbiol. Rev., 29, 3-23.
4.Biely, P., Vrsanska, M., Tenkanen, M., and
Kluepfel, D. (1997) J. Biotechnol., 57, 151-166.
5.Alcocer, M. J. C., Furniss, C. S. M., and Kroon, P.
A. (2003) Appl. Microbiol. Biotechnol., 60, 726-732.
6.Furniss, C. S. M., Belshaw, N. J., and Alcocer, M.
J. C. (2002) Biochim. Biophys. Acta, 1598, 24-29.
7.Furniss, C. S. M., Williamson, G., and Kroon, P. A.
(2005) J. Sci. Food. Agric., 85, 574-582.
8.Chavez, R., Almarza, C., and Schachter, K. (2001)
Biol. Res., 34, 217-226.
9.Diaz, R., Sapag, A., and Peirano, A. (1997)
Gene, 187, 247-251.
10.Serebryany, V. A., Vavilova, E. A., and Chulkin,
A. M. (2002) Appl. Biochim. Microbiol., 38,
495-501.
11.Maisuradze, I. G., Chulkin, A. M., Vavilova, E.
A., and Benevolensky, S. V. (2011) Genetika, 47, 1-9.
12.Benevolensky, S. V., Vavilova, E. A., Chulkin, A.
M., Abyanova, A. R., Maisuradze, I. G., Zatsepin, S. S., Novozhilov, E.
V., and Benevolensky, M. S. (2011) RF Patent No. 2412246.
13.Nikolaev, I. V., Bekker, O. B., Serebryany, V.
A., Chulkin, A. M., and Vinetsky, Yu. P. (1999) Biotekhnologiya,
3, 3-13.
14.Maniatis, T., Fritsch, E., and Sambrook, J.
(1984) Molecular Cloning: A Laboratory Manual [Russian
translation], Mir, Moscow.
15.Nikolaev, I. V., and Vinetsky, Yu. P. (1998)
Biochemistry (Moscow), 63, 1294-1298.
16.Nikolaev, I. V., Vinetsky, Yu. P., Bekker, O. B.,
and Serebryany, V. A. (1999) RF Patent No. 2126049.
17.Kabsch, W. (1993) J. Appl. Cryst.,
26, 795-800.
18.Long, F., Vagin, A., Young, P., and Murshudov, G.
N. (2008) Acta Cryst., D64, 125-132.
19.Murshudov, G. N., Vagin, A. A., and Dodson, E. J.
(1997) Acta Cryst., D53, 240-255.
20.Emsley, P., and Cowtan, K. (2004) Acta
Cryst., D60, 2126-2132.
21.Winn, M. D., Ballard, C. C., Cowtan, K. D.,
Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E.
B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N.,
Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A.,
and Wilson, K. S. (2011) Acta Cryst., D53,
235-242.
22.Unkles, S. E., Campbell, E. I., Punt, P. J.,
Hawker, K. L., Contreras, R., Hawkins, A. R., Van den Hondel, C. A.,
and Kinghorn, J. R. (1992) Gene, 111, 149-155.
23.Luo, H., Li, J., Yang, J., Wang, H., Yang, Y.,
Huang, H., Shi, P., Yuan, T., Fan, Y., and Yao, B. (2009)
Extremophiles, 13, 849-857; DOI:
10.1007/s00792-009-0272-0.
24.Sinitsyna, O. A., Gusakov, A. V., Okunev, O. N.,
Serebryany, V. A., Vavilova, E. A., Vinetsky, Yu. P., and Sinitsyn, A.
P. (2003) Biochemistry (Moscow), 68, 1313-1319.
25.Schmidt, A., Schlacher, A., Steiner, W., Schwab,
H., and Kratky, C. (1998) Protein Sci., 7, 2081-2088.
26.Pollet, A., Delcour, J. A., and Courtin, C. M.
(2010) Crit. Rev. Biotechnol., 30, 176-191.
27.Pell, G., Szabo, L., Charnock, S. J., Xie, H.,
Gloster, T. M., Davies, G. J., and Gilbert, H. J. (2004) J. Biol.
Chem., 279, 11777-11788.
28.Pell, G., Taylor, E. J., Gloster, T. M.,
Turkenburg, J. P., Fontes, C. M., Ferreira, L. M., Nagy, T., Clark, S.
J., Davies, G. J., and Gilbert, H. J. (2004) J. Biol. Chem.,
279, 9597-9605.
29.Ihsanawati, T., Kumasaka, T., Morokuma, C.,
Yatsunami, R., Sato, T., Nakamura, S., and Tanaka, N. (2005)
PROTEINS: Structure, Function, and Bioinformatics,
61, 999-1009.
30.Mamoa, G., Thunnissen, M., Hatti-Kaul, R., and
Mattiasson, B. (2009) Biochimie, 91, 1187-1196.
31.Kannan, N., and Vishveshwara, S. (2000)
Protein Eng., 13, 753-761.
32.Bhardwaj, A., Leelavathi, S., Mazumdar-Leighton,
S., Ghosh, A., Ramakumar, S., and Reddy, V. S. (2010) PLoS ONE,
5, e11347.