REVIEW: Diversity of Mitochondrial Genome Organization
A. A. Kolesnikov* and E. S. Gerasimov
Biological Faculty, Lomonosov Moscow State University, 119234 Moscow, Russia; E-mail: aak330@yandex.ru* To whom correspondence should be addressed.
Received August 8, 2012; Revision received August 27, 2012
In this review, we discuss types of mitochondrial genome structural organization (architecture), which includes the following characteristic features: size and the shape of DNA molecule, number of encoded genes, presence of cryptogenes, and editing of primary transcripts.
KEY WORDS: mitochondrial genome, structural organization, mt-encoded genes, editingDOI: 10.1134/S0006297912130020
Abbreviations: ED, edited domain, a fragment of the primary transcript that needs to be edited; mRNA, messenger RNA; mt genome, mitochondrial genome; pan-editing, editing of the whole molecule of primary transcript.
The apparent simplicity of the organization of the mitochondrial genome
is very deceptive. Since the description of first examples of mtDNA
organization [1-3], the number
of questions “how” and “why” has been growing
exponentially. For a long time, it was believed that the mt genome is
represented generally by a circular DNA molecule virtually devoid of
proteins associated with it, and linear variants found in some Ciliata
(Tetrahymena and Paramecium) are exceptions from the
general rule. However, researchers now are faced with multiple types of
mt genome organization, and their number continues to increase,
particularly in the last decade. The differences concern the form of
organization of the molecule and its overall size, as well as the mode
of organization of the “nucleoid”, which differs in the set
of proteins encoded by the mitochondrial genome, and different
mechanisms used for expression of genetic information. Development of
whole genome sequencing techniques opened the possibility for analysis
of numerous genomes of organisms from protozoa to mammals. In the
beginning of 2012, GenBank contained about 3000 sequences of whole
mitochondrial genomes, including 2683 Metazoa, 109 fungal species,
69 Viridiplantae, and 90 Protista (www.ncbi.nlm.nih.gov/genomes). In this review, we
will discuss types of mitochondrial genome structural organization
(architecture), which includes the following characteristic features:
size and the shape of DNA molecule, number of encoded genes, presence
of cryptogenes, and editing of primary transcripts.
TYPES OF MITOCHONDRIAL GENOME ORGANIZATION. SIZE AND FORM OF DNA
MOLECULES
Through more than 50 years of study of the mitochondrial genome, it has become evident that its structure takes a wide variety of forms. Six main types of organization can be distinguished.
1) Circular molecule with size from 11-12 up to 28 kbp.
2) Circular molecule with size from 22 up to 1000 kbp.
3) Circular molecule of more than 22 kbp with simultaneous presence of plasmid-like molecules.
4) Heterogeneous population of circular molecules.
5) Homogenous population of linear molecules.
6) Population of heterogeneous linear molecules.
Further on, we will discuss different types of organization in more detail. All forms of molecules found in mitochondria will be discussed.
1) The classical view of the mitochondrial genome suggests that mitochondrial genomes of most animal cells are similar. Generally, it is a circular molecule of 14-20 kbp. Such a genome usually codes for two ribosomal RNAs, 13 specific polypeptides, and up to 25 tRNAs (the number of tRNAs encoded can vary from 2 to 24 in different species). Animal genomes contain a very small percentage of non-coding DNA, represented by a single control region (however, some exceptions are known when the control region is quite long [4-11]). The protein-coding genes do not contain introns. The tRNA genes are situated generally between protein-coding genes and play, besides their main function – transfer of amino acids during protein biosynthesis, the role of signals (“commas”), where polycistronic pre-mRNA is processed. Sometimes multimer circular molecules can be found (most often dimers of “head-to-tail” type). The mt genome of animal cells is an example of this type of organization (Fig. 1a).
Fig. 1. Examples of types 1, 2, 4, and 5 organization of the mt genome: a) Felis catus; b) Synedra acus; c) Trypanosoma brucei; d) Paramecium aurelia. The schemes were made in Vector NTI software with annotations from GenBank (NC_001700, GU002153, M94286, NC_001324). Genes and direction of transcription are designated by arrows.
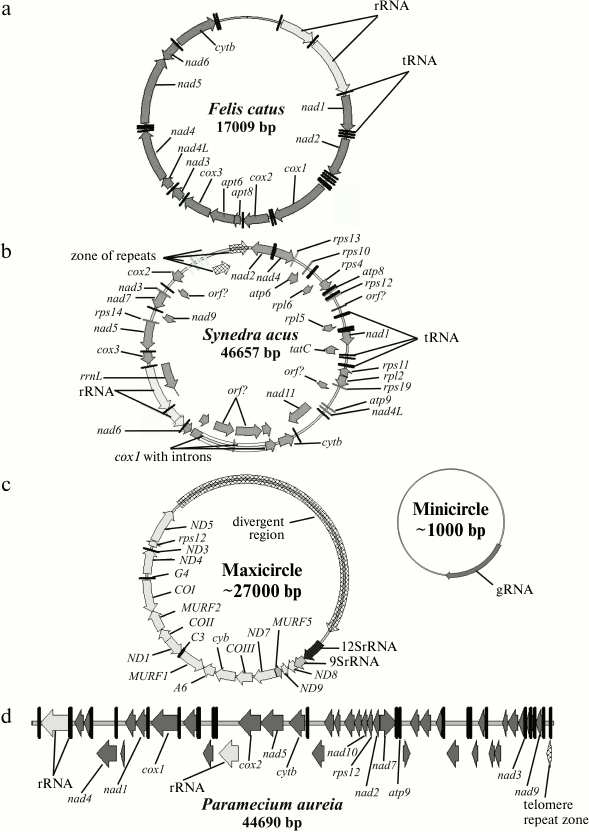
2) The second type of organization is also a circular molecule of size between 19 and 1000 kbp. The length of the mt genome of higher plants is about 100-1000 kbp. The main difference from the first type of organization is mostly the presence of additional genes and intergenic spacer sequences of different length. The number of identified genes varies between 40 and 156. In some species, structural genes contain type I and type II introns. The genomes of the diatomic alga Synedra acus [12] (Fig. 1b), of green and red algae [13], and the flagellate species Monosiga brevicollis [14] can be considered as examples of this type of organization.
3) The third type of organization is characterized by the presence of plasmid-like circular or linear molecules whose number and size vary largely, in addition to a circular molecule of 20-1000 kbp (size is similar to that of the second type). This type of organization is found in many fungi and higher plants.
4) The forth type of genome consists of several different circular molecules in one mitochondrion. For example, the mt genome of the primitive multicellular (about 20-30 cells) parasitic organism Dicyemida misakiense (or Rhombozoa) includes a set of minicircular DNA molecules with size from 1 to 2 kbp. One molecule generally contains one open reading frame. The number of molecules per cell varies from 100 to 1000 [15]. In the diplonemid Rhynchopus euleeides, two types of circular molecules have been identified with size of 7.0 and 7.7 kbp [16].
This variant was also found in 6 out of 10 species of lice (Insecta: Phthiraptera). Their genome has several variants: type 1 – full-length chromosome with 37 genes and a set of heterogeneous minicircular molecules; type 2 – a set of minicircles, smaller in size than a full chromosome, with one short control region; type 3 – a set of minicircles bearing 1-3 genes with long complex control region [17].
Unique organization of the mt genome was found in kinetoplastids. The class Kinetoplastida can be subdivided into two subclasses, Prokinetoplastina (accommodating bodonids Ichthyobodo and Perkinsiella) and Metakinetoplastina (containing other bodonids and trypanosomatids). Within the Metakinetoplastina group, four orders are distinguished: Neobodonida, Parabodonida, Eubodonida (Bodo saltans), and Trypanosomatida [18]. The single cell mitochondrion of Kinetoplastida contains a complex associate represented by two populations of circular DNA molecules: minicircular (105 molecules of size from 0.5 to 12 kbp) and maxicircular (20-50 molecules of size from 20 to 40 kbp) (Fig. 1c). The molecules are linked with each other as catenanes and form a complex structure referred to as a network, basket, or an associate. In this type of organization, a true analog of the mt genome (coding for standard mitochondrial genes) is maxicircular molecules. It is believed that the function of minicircular molecules is restricted to coding for small guide RNAs, which are essential for the editing of uridyl of multiple primary maxicircular transcripts. But we cannot exclude some other functions for minicircles. In bodonids, mtDNA associate is less compact, and the minicircular component includes longer molecules.
Surprisingly, in cells of several species of bivalves (in particular, in the genus Mytilis) there are two types of mt genome: “male, M” and “female, F”, which are inherited via two pathways: via ovule (F) or via sperm (M). This system of mtDNA transmission was called Doubly Uniparental Inheritance of the mt genome (DUI) compared to usual Strictly Maternal Inheritance (SMI) [19]. Sequences of the M and F genomes of Mytilus galloprovincialis differ by about 20%, although the sets of genes and their distribution in the genome are identical [20].
5) The fifth type comprises linear single-component mitochondrial genomes that are found in several independent phylogenetic branches: in some Ciliata (Tetrahymena pyriformis [21] and Paramecium aurelia [22] (Fig. 1d)), in Apicomplexa (Plasmodium and closely related species) [23], in fungi, green algae (Chlamydomonas and closely related species), and in some Cnidaria [24-26]. One of the largest characterized linear mt genome within Ciliata is that of Oxytricha trifallax [29]. In this organism, the mt genome includes one linear chromosome of about 70 kbp and a linear plasmid of about 5 kbp.
Linear mtDNA molecules contain various clusters of tandem repeats [27] that play the role of telomeres. Nosek and Tomaska [28] divided telomeric sequences of linear mtDNA into six variants. Five of them are formed by the DNA sequence itself: i) covalently linked hairpins on both ends (for example, in the infusorian Paramecium aurelia, in the yeast Pichia pijperii, and in Williopsis (previously Hansenula) mrakii); ii) a covalently linked hairpin on one end and a cluster of short repeats on the other end of the molecule (plasmids in mitochondria of Fusarium (Ascomycota)); iii) clusters of tandem repeats (for example, in the infusorian Tetrahymena thermophila) sometimes organized in telomeric loops (t-loops) (in the green alga Chlamydomonas parapsilosis); iv) inverted repeats and long single-stranded sequences on the 3′-ends (for example, in Chlamydomonas reinhardtii); v) complex clusters of repeats of different types (for example, in Theileria parva (Apicomplexa), Amoebidium parasiticum (Ichthyosporea), and in linear plasmids of the slime mold Physarum polycephalum (Myxogastrids). Another way to designate the end of a chromosome is defined by a specific protein that is covalently bound to the 5′-end of each DNA strand (for example, in linear plasmids of fungal mitochondria).
6) The sixth type of mitochondrial genome includes a heterogeneous population of linear molecules. For example, the genome of Amoebidium parasiticum consists of several hundred linear molecules (total length of the genome is more than 200 kbp) [30]. The same type of genome organization was found in Euglenozoa (E. gracilis). The overall genetic complexity of the genome is estimated at 60 kbp, although it includes a set of short linear fragments [31, 32]. Probably the same type of genome organization can be found in other Euglenozoa – Diplonemid and Euglenids [16]. It seems that the mt genome of dinoflagellates is also includes a complex set of linear molecules [33, 34]. A unique of structure of the mitochondrial genome was found in Alatina moseri (Cubozoa). Eighteen genes are located on eight linear chromosomes with size from 2.9 to 4.6 kbp; moreover all of the chromosomes have identical telomere sequences [26].
Many studies of mitochondrial genome structure have been done using electron microscopy. Some authors observed either a ring-type structure with a long linear tail, or branched. Such structures are, typically, the intermediates of the rolling circle DNA replication process [23]. In this regard, it should be noted that for isolation of mtDNA, cells in stationary phase should be used. The mtDNA samples obtained from actively dividing cells may contain a large number of replication intermediates. In particular, this fact raises a hot discussion about the structure of the mt genome of Saccharomyces cerevisiae [35].
Phenomenon of heteroplasmy. Heteroplasmy means the presence of different mitochondrial DNA molecules in a cell. In most cases, there are molecules of the same type with small differences. There are two types of heteroplasmy: by size and by sites. The first generally results from the presence in the control region of the molecule of different number of relatively short repeated sequences (10-100 nucleotides). Increase in the number of repeats is believed to be associated with the activation of the mtDNA transcription process. Size heteroplasmy has been found in many organisms, including fish, insects, bats, and others [36, 37]. Site heteroplasmy is generally associated with the accumulation of mutations [37]. In this review, we will not discuss this phenomenon.
Is there any specificity of mt genome organization type in certain groups of organisms? Is there any specificity in type of mt-genome organization in related groups of organisms? In vertebrates, there is only type 1 of mitochondrial genome – a compact circular molecule. On the other hand, in invertebrate we found examples of type 1, 2, and 5. However, in this group of animals the type 1 is predominant, the other types being rather an exception to the rule. In plants and algae the situation is less conserved. We should rather talk about trends. Typically, these organisms contain a genome of type 2, rather compact (100-500 kbp in higher plants, 40-90 kbp in green algae, except those from chlorophyte algae with size from 15 to 40 kbp) and possessing specific features. For example, the mt genome of higher plants contains predominantly group II introns, while in other organisms these introns are rather exceptional. In the average plant, the mt genome codes for 50-70 proteins (not taking into account unidentified open reading frames and reading frames inside the introns). The mt genome of higher plants codes for 5S rRNA – a gene that is not found in other organisms. As for the other groups of organisms, similarity can be observed on the level of families. This applies particularly to Protista. It should be noted that in most cases significant differences in mt genome structure are observed in parasitic organisms.
In the last decade, a new systematic of eukaryotes was proposed suggesting the division of all organisms in 5-8 large “supergroups” based on huge bioinformatic data sets (including data on morphology, biochemical characteristics, and genome structure) [38-40]. It is interesting to analyze the distribution of mt genome organization types on the phylogenetic tree of eukaryotes. Figure 2 presents schematically the phylogenetic tree of eukaryotes [37] with numbers corresponding to the type of mt genome organization as described above. We excluded a few examples that differ from the major type characteristic for the supergroup. It should be mentioned that the database containing data about mt genome structure is extremely uneven. For animals there are thousands examples, while for the Plantae supergroup there are hundreds, and for other groups only tens or single cases. For the Rhizaria supergroup, there is no information concerning full-sized mtDNA genomes. Therefore, this analysis of mt genome organization type distribution is preliminary. For more accurate assessment, the database should be significantly expanded. As can be seen from Fig. 2, the most frequent type is number 2, followed by types 3 and 5. Types 1 and 4 are highly specialized.
Fig. 2. Schematic representation of phylogenetic tree of eukaryotes [37]. Supergroups are printed in bold. Circled numbers correspond to the type of mt genome organization.
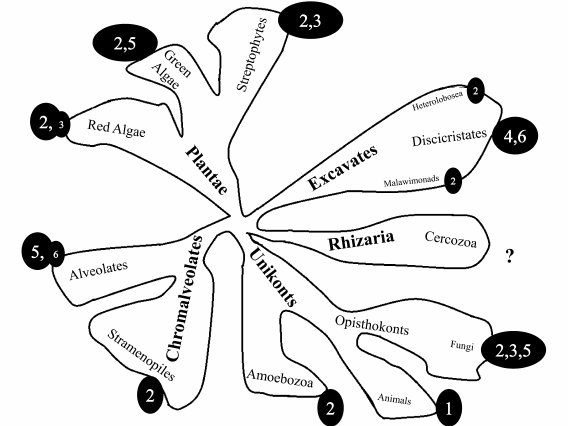
Figure 3 depicts the distribution of mt genome organization types among unicellular and multicellular organisms. As can be seen from the picture, unicellular eukaryotes (Protists) show the most important diversity.
Fig. 3. Frequency of different types of mt genome organization in unicellular and multicellular organisms.
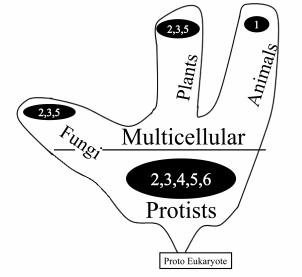
A similar conclusion can be drawn from the analysis of gene sets encoded by mt genome.
Mitochondrial nucleoid. The structure of mtDNA inside the mitochondria (nucleoid) can also be a source of mt genome diversity. Unfortunately, there are only a few experimental studies of nucleoid structure. In the last decade, proteins that bind specifically to mtDNA have been actively investigated. To date, about 30-35 proteins that can specifically participate in nucleoid organization have been identified [42]. Studies have been done on a limited number of objects, including yeast, frog oocytes, and human cell cultures [43-46]. According to the model of nucleoid organization suggested [45], it has a central part, where processes of replication and transcription occur, and a peripheral part, where translation takes place. However, there is a distinct lack of data for comparative analysis of nucleoid organization in various groups of organisms.
Sets of genes encoded by mitochondrial genome. Large variations in size of the mt genome suggest that there might be a correlation between the size of the genome and the number of genes encoded in it. A standard set of mitochondrial genes includes genes coding for complex I (NADH:ubiquinone oxidoreductase) subunits nad 1-9, complex III (ubiquinone:cytochrome c oxidoreductase) subunit cob, complex IV (cytochrome c oxidase) subunits cox 1-3, and complex V (ATP synthase) subunits atp 1,6,8. In addition, there are genes of ribosomal RNAs from large and small subunits and varying numbers of tRNA genes. This set of genes, with rare exceptions, is found in most mt genomes, and particularly in the genomes of the first group. According to the available database, in many organisms with other types of mt genome organization the number of genes can largely exceed this list. Among the additional genes, we should mention 3 to 27 genes coding for ribosomal proteins (mainly from the small subunit), genes coding for complex II (succinate:ubiquinone oxidoreductase) subunits sdh 2-4, and others coding mainly for unique reading frames with unknown functions [47]. The number of additional genes may differ even in closely related species. The set of genes encoded in the mt genome of Reclinomonas americana (97 genes) is often used for comparison [48]. This genome is believed to be the closest to the genome of the pro-mitochondrial predecessor. However, the number of genes encoded in the mt genome of higher plants can reach 156 in Beta macrocarpa and Nicotiana tabacum [49]. The presence of additional genes is likely to reflect the particularities of the organism’s development. For example, the conservation of ribosomal protein genes in certain genomes during evolution can be explained by the fact that these proteins might be multifunctional and play an additional role apart from participating in the formation of ribosomes [50]. Indeed, at least for 25 ribosomal proteins in different organisms (E. coli, Saccharomyces cerevisiae, Drosophila melanogaster, Xenopus laevis, Ascaris lumbricoides, Arabidopsis thaliana, Strongilocentrotus purpuratus, mouse, rat, human), additional cellular functions have been demonstrated.
We are not going to discuss the processes of replication and transcription of the mt genome in the present review. We will focus instead on the process of editing of the primary transcripts.
EDITING OF PRIMARY TRANSCRIPTS OF mtDNA
Analysis of presence or absence of cryptogenes contributes significantly to the assessment of the diversity of mt genome organization. An open reading frame whose primary transcript should undergo some modifications to become a fully functional template for protein synthesis (editing) is referred to as a cryptogene. Sometimes, when analyzing a set of genes encoded by the mt genome, it is difficult to identify a cryptogene. For example, editing of a 438-nucleotide-long cryptogene COIII in Trypanosoma brucei results in an mRNA of 866 nucleotides [41].
RNA editing is a broad term that covers a wide variety of modifications in almost all types of RNA – messenger, ribosomal, transport, and small non-coding RNAs [51, 52]. The very first example of editing was described for the COII mitochondrial gene in two trypanosomatid species: Trypanosoma brucei and Crithidia fasciculata [53]. This gene is highly conserved among trypanosomatids, indicating its functionality, but its reading frame is disrupted. Benne and colleagues [53] showed that insertion of four uridines in the pre-mRNA occurs posttranscriptionally and restores the normal reading frame. Later, other types of editing were described in other species.
It soon became clear that RNA editing is a widespread process and occurs in various species from viruses [54] to mammals and higher plants. There are three major types of RNA editing: base modification, posttranscriptional base insertion/deletion, and co-transcriptional base insertion/removal [55].
Further on we will use two abbreviations: ED and pan-editing. ED – from Edited Domain – is a fragment of pre-mRNA sequence that is subjected to editing. Pan-editing is the process when editing occurs though all the length of the pre-mRNA, in contrast to partially edited cryptogenes.
Editing in the kingdom of animals. The first and only known example of editing in multicellular animals was demonstrated in the trematode Teratocephalus lirellus. Insertion of 6 U into the cytochrome b mRNA was found [84]. Authors have also analyzed mRNA in 12 other nematodes, but no other examples of editing were found (analysis was done by comparison of cyt b gene sequence with the sequence of corresponding mRNA).
Editing in mitochondria of plants. RNA editing in the mitochondria of flowering plants consists in conversion of C to U by deamination [56-58]. In the mitochondrial genomes studied so far, there are about 400-500 deamination sites within open reading frames [59-61]. Deamination is catalyzed by a specific enzyme, probably a deaminase that was evolutionarily adapted to use polynucleotides as substrates [62, 63]. The C to U conversion is strictly site-specific, and moreover cis-acting factors responsible for specificity have been identified. First of all, there are 20-40 nucleotide sequences downstream to cytosine being modified. Changing of these sequences altered the number of edited transcripts. In some cases, approximately 10 nucleotides upstream from the edited site were also shown to be important [64, 65]. Bioinformatic analysis of cis-acting elements in A. thaliana did not reveal any significant similarity between them; they could not be even organized into groups. It was found that sequences surrounding different edited sites are absolutely different [59]. The cis-proximal elements described above did not form any consensus and rarely had any common features. Attempts to identify trans-acting factors responsible for specificity of editing failed. Initially, there was an idea to find RNA molecules analogous to gRNAs of trypanosomatids, which could play a role of trans-acting factors determining the sites of deamination. However, none of these studies succeeded.
In vitro studies showed that editing sites are recognized independently. A number of partially edited intermediates can be isolated, which are further used for editing of the remaining sites with equal probability. No evidence for polarity of the editing process (i.e. moving from the 3′ to 5′ end of the molecule or vice versa) was revealed.
U-to-C editing occurs rarely in mitochondria of flowering plants. In most studied species – Arabidopsis thaliana, Brassica napus, and Oryza sativa – no cases were reported [59-61]. To the contrary, this type of editing is common for hornwort mitochondria, where it is as frequent as C-to-U editing. In other bryophytes, as well as in all orders of liverworts except for Marchantiales, both types of editing (C-to-U, U-to-C) were found.
The only land plant in which editing was not found is a liverwort from the order Marchantiales, for example, Marchantia polymorpha [66]. Moreover, to date no editing has been found in green algae – the closest relatives of land plants [67].
Editing in slime molds. Editing in slime molds has been known for a long time. Large-scale editing of COI transcript of the first subunit of cytochrome c oxidase of Physarum polycephalum was shown by Gott et al. [68]. In this transcript, 66 base insertions and at least four conversions occur. Editing occurs via four different mechanisms: insertion of UU dinucleotide (and, possibly, CU), C-to-U conversion, and insertions of mononucleotides C and U. Insertions are made co-transcriptionally [69]. Analysis of possible evolutionary pathways for different types of COI editing led to the conclusion that these four types of editing have their unique histories [70, 71].
Editing by insertions of U is found in all slime molds, which suggests that this type of editing emerged in their common ancestor. Inserts of C are not found in the early diverged Clastoderma debaryanum, and a unique insertion of C is observed in Arcyria cinerea. In groups that diverged later, 30-40 C insertions are found. These data suggest that the mechanism of C insertion emerged within the slime mold taxon somewhere near the point of divergence of A. cinerea, and it apparently developed in groups that diverged later [72]. Dinucleotide insertion editing occurs only within one phylogenetic branch of slime molds, and it was not found in other branches. It should be mentioned that the mechanism of dinucleotide insertion is more complicated than sequential insertion of two single U bases [73, 74].
The C-to-U conversion, according to the phylogenetic data, either emerged several times, or it was lost by different groups of slime molds [75]. Such a variety of evolutionary pathways of different editing mechanisms aimed at providing functionality for a single gene points to an unusual plasticity of editing [72].
More than 500 sites of editing were found and about 500 sites were predicted in the mitochondrial genome of Physarum polycephalum [76]. In the case of dinucleotide insertion, the following variants were detected: AA, UU, GU, UA, GC, and CU inserts. In the transcript of the nad2 gene, a unique type of editing found only in slime molds was discovered: removal of four adenine residues [77]. Genes of mitochondrial ribosomal proteins in Didymium iridis (rpS12, rpS7, rpL2, rpS19, rpS3, and rpL16) are subjected to intense editing by the same mechanisms as used for the transcript of the COI gene [78].
Editing of two types of mitochondrial tRNAs has been described for Physarum polycephalum. Two methionine tRNAs (tRNAmet1, tRNAmet2) have unpaired bases in the acceptor stem, which are repaired posttranscriptionally by insertion of one G. This is the first and still unique known case of posttranscriptional insertion of G [79]. Thus, the editing system of mitochondrial transcripts of slime molds is extremely complicated. It includes several mechanisms and combines both co-transcriptional and posttranscriptional editing. Different types of RNA molecules are edited: mRNAs of respiratory chain proteins, mRNAs of ribosomal proteins, and tRNAs.
We have described taxa of wildlife where C-to-U editing has been demonstrated. It should be stressed that this form of editing is the most labile. It appears independently and in various forms in trypanosomatids (tRNA Trp editing [80]), in mitochondria and chloroplasts of higher plants [81], in apolipoprotein B (nuclear mammalian genome), and in the cellular slime mold Dictyostelium discoideum [82].
Editing in dinoflagellates. Extensive editing in genes cox1 and cob was found in three representatives of dinoflagellates. Editing by the mechanism of nucleotide substitutions (A-to-G, U-to-C, C-to-U) affects about 2% of gene coding sequences [83]. Editing results in changing of nucleotides in the first and second positions of the codon, leading to change in the encoded amino acid.
Mechanism of uridyl editing in trypanosomatids. The editing process in trypanosomatids, which was the first to be discovered, is one of the best studied to date. We will describe below in more detail how the genetic information from the kinetoplast genome (mitochondrial genome) of trypanosomatids is expressed. The Trypanosomatids are a group of unicellular organisms, most of which are parasites. We will discuss the close relationship between editing and structural organization of DNA molecules and will trace some evolutionary pathways of several cryptogenes.
A distinctive feature of editing in mitochondria of trypanosomatids is the presence of specialized RNA molecules – guide RNAs (gRNAs). The editing process can be briefly described as follows. A gRNA molecule complementary to the 5′ and 3′ terminal regions of a pre-mRNA molecule binds to it. Then a multienzyme complex catalyzes sequential digestion of the pre-mRNA, addition/removal of uridine bases (to achieve total complementation with the central part of the gRNA), and ligation of the pre-mRNA [85, 86].
The gRNAs encoded in the intergenic regions or adjacent to maxicircle divergent region were identified first [87, 88], but then it became clear that most gRNAs are encoded by minicircles [89, 90]. For the moment, it is the only proven genetic function of minicircles [91]. Minicircles can contain 1-4 gRNA genes, each of them flanked by short inverted repeats. The gRNA genes are transcribed polycistronically, and the transcripts are further processed by the 19S RNA processing complex to form individual gRNAs [92]. The gRNA gMURF2-II encoded in a maxicircle is unique, its gene being located within the coding region of the ND4 gene [93]. This is the only case known so far of intragenic localization of a functional gRNA gene. Despite the fact that transcription of maxicircle genes is polycistronic, the gRNA genes located in a maxicircle form individual transcription units [93]. In L. tarentolae, alternative editing was found for several genes, such as COIII, RPS12, and ND3 [94]. An example of alternative editing was also described in T. brucei: one gRNA leads to formation of an alternative form of COIII mRNA, which is translated into an alternative form of the protein [95]. Thus, alternative editing can be common, although still little studied, mechanism of generating protein diversity in the kinetoplast.
Let’s have a more detailed look at the structure of gRNA. The gRNAs are short (60 bp) RNA molecules. We can designate three functional regions within it [88]:
– the 5′-terminal region, also called in a number of papers the “anchor”, which is involved in anchoring of the gRNA on pre-mRNA upstream the site of editing. This part is usually about 10 bp and is fully complementary to the pre-mRNA of the edited cryptogene;
– the information region of the gRNA, starting from the first non-complementary to pre-mRNA nucleotide. This region contains information about the number of uridines to be inserted or deleted. Insertion or deletion of uridines in pre-mRNA results in restoration of complementarity with the information region of the gRNA. This region is called the editing block. In duplex formation between mRNA and gRNA within the editing unit, both canonical Watson–Crick pairs and noncanonical G:U pairs are involved [96];
– the 3′-terminal region consisting of uridine bases, which are added to gRNA posttranslationally, don’t have a fixed length. Addition of uridines is catalyzed by the special enzyme KRET1, a terminal transferase of uridyl nucleotides, that differs from an analogous enzyme involved in uridyl addition to pre-mRNA [97, 98]. It is involved in stabilization of the pre-mRNA duplex with gRNA during the editing process by interacting with the purine-rich (usually G-rich) sequence preceding the editing block. In cells where expression of KRET1 was suppressed by RNA interference, editing was not detected [97].
The gRNAs have no catalytic functions in the editing process, they provide only information. The reaction is catalyzed by a protein complex with multiple activities [85]. Great progress in studying of the editing process was achieved when the first in vitro system of editing was developed. Let’s discuss the main steps of the editing mechanism.
During the first step, gRNA molecules “recognize” the pre-mRNA to be edited through its complementary interaction with the 5′-end of the gRNA and form a duplex. Recent studies suggest that the key event is the formation of so-called “triple-helix” structure of gRNA:pre-mRNA (formation of three double-stranded regions) [99]. The first helix is formed by the anchor region of gRNA and pre-mRNA, the second helix is formed by bases of the gRNA information region, and the third results from the interaction between the poly-U region of the gRNA with pre-mRNA. Such structure of gRNA–pre-mRNA duplex, stabilized by proteins, serves as a basis for editing complex formation. The pre-mRNA digestion site is determined by the first unpaired nucleotide of the gRNA information region and results in formation of 5′ and 3′ fragments of pre-mRNA. The gRNA holds them together by interaction of the anchor region with the 3′ fragment and the poly-U region with the 5′ fragment. Then, depending on the structure of the information region of the gRNA, insertions or deletions of uridines in the 3′- or the 5′-fragments of the pre-mRNA occur [100]. It should be noted that no gRNA leading exclusively to deletions of uridines has been found. Such gRNA should have been shorter than the pre-mRNA, making impossible the triple-helix structure formation, where one helix should be formed by the information region of the gRNA, which is not complementary to the pre-mRNA before insertion of uridines. Generally, deletion of uridines is provided by a fragment of the gRNA information region, while the remaining part forms a triple-helical structure and is responsible for the insertion of uridines. Due to this fact, all gRNAs always form a triple-helix structure necessary for the assembly of the editing complex.
Free UTP is used as a source of uridines to be inserted [101]. The number of uridines is determined by the number of A or G in the information region of the gRNA, as these bases will form complementary base pairs. Insertions stop when the gRNA becomes fully complementary to the edited pre-mRNA [102]. Deletion of uridines occurs when the 3′-terminal uridines of the 5′-fragment pop up from the duplex. Deletion also results in the restoration of full complementarity between the gRNA and pre-mRNA [96]. Thus, in both cases of insertion or deletion of uridines, the gRNA brings genetic information into the mRNA. Finally, the 5′- and 3′-fragments of the pre-mRNA are ligated [100].
RECC proteins, 20S complex, and enzymatic cascade model. The enzymatic cascade model reflects the common view of the reactions of the editing process [103]. Digestion of pre-mRNA is catalyzed by a specific endonuclease. In the case of uridine deletions, popping up bases are removed from the 3′-end of the 5′-fragment by a specific exonuclease, and insertions of uridine are catalyzed by the terminal uridyl transferase KRET2. Further ligation of the pre-mRNA fragments is catalyzed by the ligase KREL1-2. All these activities are found in a large protein complex – the editosome [104].
Initially, in several studies so-called 20S complex was isolated and characterized. Some authors have designated such a set of proteins as the L-complex [83, 84, 90]. We will further use the name “20S complex”. This complex contains all enzymatic activities listed above, which co-sediment in a glycerol gradient with a 20S peak in Trypanosoma brucei [105] and a 25S peak in Leishmania tarentolae [106]. This complex can catalyze only one round of editing in vitro, i.e. to insert or remove uridines only in the first editing region of one gRNA molecule. Therefore, some authors suggest that the 20S complex represents only a part of the real active editosome, or vice versa, the 20S peak contains enzymes that do not belong to the in vivo editing apparatus [107]. The question about which protein plays the role of exonuclease is still open, while a number of proteins from the 20S complex possess such activity.
Three major types of core complex editing have been described [108, 109]: insertion subcomplex (complex called RECC2 – RNA Editing Core Complex 2), deletion subcomplex (complex RECC1), and a subcomplex performing editing with cis-gRNA (RECC3). A three-dimensional reconstruction of these complexes was made that enables us to determine the mode of interaction of protein molecules within these complexes. Presumably, all types of subcomplexes are present in the 20S editosome. An alternative hypothesis suggests that there are three types of editosomes in a cell, corresponding to the three types of subcomplexes.
The functional roles of some proteins from the editosome have recently been elucidated using the gene knockout technique and by inhibition of expression using RNA interference.
Evolution of edited domains of cryptogenes in trypanosomatids. It should be noted that the presence of a gene in the form of a cryptogene has to be examined for each individual organism, even within one taxon. The next two sections illustrate this situation.
The structure of cryptogenes in bodonids (the closest relatives of trypanosomatids) is still not very well studied. In Trypanoplasma borreli, the cyb and COI genes undergo editing. Both cryptogenes have two edited domains, one at the 5′- and another at the 3′-end; both are edited independently. This pattern is similar to the editing of the ND7 cryptogene in trypanosomatids. Gene COII, that in trypanosomatids undergoes insertion of 4 U, in bodonids T. borreli and Criptobia helicis is not edited [110]; moreover, there is no cis-gRNA downstream of COII [111]. In the bodonid Bodo saltnas, a species that is closer to trypanosomatids, COII and MURF2 are edited in positions similar to those of the trypanosomatids. In the COII gene, two out of four U inserted in trypanosomatids are encoded in the DNA. The cis-gRNA of the COII gene is found immediately downstream in a position similar to that in trypanosomatids. Unexpectedly, in B. saltans the transcript of ND5 is edited throughout its whole length. It should be mentioned that in trypanosomatids this gene was never present as a cryptogene [110]. These data indicate that editing has emerged before trypanosomes turned to a parasitic way of life, and the evolution of cryptogenes apparently occurred simultaneously either by loss of editing in some genes (ND5) or by switching to pan-editing in others.
Retroposition model of size reduction of an edited domain. As described above, editing within the ED is a polar process and occurs in the direction from the 3′-to-5′ end of the pre-mRNA molecule. The above-described ED structure comparison of homologous cryptogenes of different trypanosomatid species shows that the length of the ED of a cryptogene can be progressively reduced towards the 5′ end, up to the ED that consists of a single edited block (edited by one gRNA, for example, both domains of ND7 in L. tarentolae). As mentioned above, the A6 cryptogene in T. brucei is pan-edited by 8-10 gRNAs [112], and in L. tarentolae and other leishmaniae it is edited at the 5′ end by 4-6 gRNAs [113]. The same gene is edited by four gRNAs in Crithidia fasciculata and one gRNA in Angomonas culicis [114]. These facts allowed creating a model explaining the reduction of ED length by a retroposition of partially edited templates in a maxicircle [114-116]. Thus, the ancient pan-edited cryptogenes could have been replaced by partially edited ones [114, 116]. The fact that the genome of trypanosomatid contains a gene of reverse transcriptase and cell extracts possess corresponding activity [118, 119] defines the theoretical possibility of this process. The similarity of the 5′-edited form of the gene with a partially edited mRNA of the pan-edited homolog from other species is consistent with this model. At the same time, there are about 50 copies of maxicircle genome in a kinetoplast associate, whereas retroposition is very rare and occurs only in one maxicircle of a single cell. It is assumed that preservation of retroposition in one maxicircle may be the result of partial loss of certain types of minicircles during their stochastic segregation during cell division [114]. Thus, cultures that lose some types of minicircles, and consequently some gRNAs, lose also the ability to edit the corresponding transcript. In this case, cells containing a maxicircle with retroposition of a partially edited cryptogene are getting an advantage. Thus, retroposition can be considered as a mutation that compensates the loss of certain types of minicircles.
Dixenous trypanosomatids at one stage of their life cycle are obligate anaerobes with an inactive mitochondrial respiratory chain. Under these conditions, the absence of certain classes of minicircles will not be noticed until the cells are transferred to aerobic conditions. It can be suggested that among monoxenous trypanosomatids there should be a tendency to reduce the length of the ED in a number of genes.
In summary, we should say that variants and mechanisms of primary transcript editing differ dramatically. It can be suggested that editing types emerged and developed independently for each variant. The phenomenon of editing is probably not limited to the described processes. This is due to the fact that, to be able to say that a certain matrix is edited, detailed information about the primary structure of the gene and of the edited template is required, and the accumulation of information on gene structures is much ahead of the database on the structures of their transcripts.
CONCLUSION
The contemporary experimental data show that the mitochondrial genome can be organized in cells in different ways. First of all, this concerns the form of mitochondrial DNA itself (i.e. its architecture). According to the form and size of the molecule, at least six basic variants can be distinguished. The greatest diversity of mt genome was demonstrated for organisms from the Protista kingdom, which reflects the diversity of growing conditions of these organisms. With increasing complexity of the organism, which happened simultaneously with functional simplification of each cell type (specialization), the diversity of mt genome organization types decreases. One gets the impression that circular form is preferable to linear form. Mitochondrial genomes of the vast majority of multicellular organisms are circular.
The second important factor is the size of the molecule. It is logical that increase in size enables the cell to encode more proteins. However, no correlation was detected between the increase in DNA size and the number of genes, with a large part of the DNA occupied by intergenic spacers. Perhaps it is determined by some other tasks these sequences are playing. Perhaps these regions have some regulatory functions and interact with proteins that form mitochondrial nucleoid or have other functions. There are some hundreds of mtDNA molecules in a cell. Obviously, they have to be structured in the mitochondria. Unfortunately, the question of mt nucleoid organization remains poorly understood. The model of “multi-layer” structure [45] with different distribution of functional areas within the nucleoid was suggested. More and more proteins able to interact with mtDNA are identified, but whether they participate in the formation of nucleoid and functioning of the genome in vivo in different organisms is not yet very clear. The large number of DNA molecules in mitochondria raises the question whether these molecules are identical. The phenomenon of heteroplasmy is known. Moreover, it is known that during ontogenesis complex structural changes can occur, resulting in the presence of different mt genomes in the same organism.
The mt genome is characterized by great diversity of mechanisms used for realization of genetic information and gene expression, as we discussed in the second part of this review. Editing of transcripts is frequently used way of regulation and the generation of diversity in the mitochondrial genome. Mechanisms of editing vary in different groups of organisms, as well as the set of genes (cryptogenes) subjected to this process. Sometimes cryptogene encodes only half of the genetic information or even less, while the missing part is added from other loci of the molecule or other molecules; thus the information in the genome is shared between different compartments (e.g. mitochondrial maxi- and minicircles of trypanosomatids). Such complex organization is probably due to the fact that in addition to canonical function of energy production, mitochondria play a role in species-specific metabolic and regulatory pathways during a complex life cycle, division, and reorganization of the cell.
In conclusion, it should be noted that much experimental data has been obtained on model organisms. Therefore, one should be very careful in extrapolating the large variety of known types of mt genome organization to all organisms. For example, in recent years active use of transcriptome sequencing techniques made it more obvious that editing of transcripts is a much more widely spread phenomenon, and rather a rule than an exception in the genomes of organelles.
REFERENCES
1.Steinert, G., Firket, H., and Steinert, M. (1958)
Exp. Cell Res., 15, 632-635.
2.Ris, H., and Plaut, W. (1962) J. Cell Biol.,
13, 383-391.
3.Nass, S., and Nass, M. M. K. (1963) J.
Cell Biol., 19, 593-611.
4.Boyce, T. M., Zwick, M. E., and Aquadro, C. F.
(1989) Genetics, 123, 825-836.
5.Okimoto, R., Macfarlane, J. L., Clary, D. O., and
Wolstenholme, D. R. (1992) Genetics, 130, 471-498.
6.Azevedo, J., and Hyman, B. (1993)
Genetics, 133, 933-942.
7.Fuller, K. M., and Zouros, E. (1993) Curr.
Genet., 23, 365-369.
8.Boore, J. L. (1999) Nucleic Acids Res.,
27, 1767-1780.
9.Clayton, D. A. (2000) Exp. Cell Res.,
255, 4-9.
10.Delarbre, C., Rasmussen, A.-S., Amason, U., and Gachelin,
G. (2001) J. Mol. Evol., 53, 634-641.
11.Hu, M., and Gasser, R. B. (2006) Trends
Parasitol., 22, 78-84.
12.Ravin, N. V., Galachyants, Y. P., Mardanov, A.
V., Beletsky, A. V., Petrova, D. P., Sherbakova, T. A., Zakharova, Y.
R., Likhoshway, Y. V., Skryabin, K. G., and Grachev, M. A. (2010)
Curr. Genet., 56, 215-223.
13.Odintsova, M. S., and Yurina, N. P. (2002)
Genetika, 38, 773-788.
14.Lang, B. F., O’Kelly, C., Nerad, T., Gray, M. W., and
Burger, G. (2002) Curr. Biol., 12, 1773-1778.
15.Watanabe, K. I., Bessho, Y., Kawasaki, M., and
Hori, H. (1999) J. Mol. Biol., 286, 645-650.
16.Roy, J., Faktorova, D., Lukes, J., and Burger, G.
(2007) Protist, 158, 385-396.
17.Cameron, S. L., Yoshikawa, K., Mizukoshi, A.,
Whiting, M. F., and Lohnson, K. P. (2011) BMC Genomics,
12, 394.
18.Moreira, D., Lopez-Garcia, P., and Vickerman, K. (2004)
Int. J. Syst. Evol. Microbiol., 54, 1861-1875.
19.Theologidis, I., Fodelianakis, S., Gaspar, M. B.,
and Zouros, E. (2008) Evolution, 62, 959-970.
20.Mizi, A., Zouros, E., Moschonas, N., and Rodakis,
G. C. (2005) Mol. Biol. Evol., 22, 952-967.
21.Suyama, Y., and Miura, K. (1968) Proc. Natl.
Acad. Sci. USA, 60, 235-242.
22.Goddard, J. M., and Cummings, D. J. (1977) J.
Mol. Biol., 109, 327-344.
23.Wilson, R. J. M., and Williamson, D. H. (1997)
Microbiol. Mol. Biol. Rev., 61, 1-16.
24.Bridge, D., Cunningham, C. W., Schierwater, B.,
DeSalle, R., and Buss, L. W. (1992) Proc. Natl. Acad. Sci. USA,
89, 8750-8753.
25.Kayal, E., Bentlage, B., Collins, A. G., Kayal,
M., Pirro, S., and Lavrov, D. V. (2012) Genome Biol. Evol.,
4, 1-12.
26.Smith, D. R., Kayal, E., Yanaqihara, A. A.,
Collins, A. G., Pirro, S., and Keeling, P. J. (2012) Genome Biol.
Evol., 4, 52-58.
27.Morin, G. B., and Cech, T. R. (1988) Cell,
52, 367-374.
28.Nosek, J., and Tomaska, L. (2003) Curr. Genet.,
44, 73-84.
29.Swart, E. C., Nowacki, M., Shum, J., Stiles, H., Higgins, B.
P., Doak, T. G., Schotanus, K., Magrini, V. J., Minx, P., Mardis, E.
R., and Landweber, L. F. (2012) Genome. Biol. Evol., 4,
136-154.
30.Burger, G., Forget, L., Zhu Yun, Gray, M. W., and
Lang, B. F. (2003) Proc. Natl. Acad. Sci. USA, 100,
892-897.
31.Yasuhira, S., and Simpson, L. (1997) J. Mol.
Evol., 44, 341-347.
32.Spencer, D. F., and Gray, M. W. (2010) Mol.
Gen. Genom., 285, 19-31.
33.Nash, E. A., Nisbet, R. E. R., Barbrook, A. C.,
and Howe, C. J. (2008) Trends Genet., 24, 328-335.
34.Jackson, C. J., Norman, J. T., Schnare, M. N.,
Gray, M. W., Keeling, P. J., and Waller, R. F. (2007) BMC
Biology, 5:41 doi: 10.1186/1741-7007-5-41.
35.Bendich, A. J. (2010) Mol. Cell,
39, 831-832.
36.Arnason, E., and Rand, D. M. (1992)
Genetics, 132, 211-220.
37.Sato, A., Endo, H., Umetsu, K., Sone, H.,
Yanagisawa, Y., Saigusa, A., Aita, S., and Kagawa, Y. (2003) Biosci.
Rep., 23, 313-337.
38.Cavalier-Smith, T. (2003) Eur. J.
Protistol., 39, 338-348.
39.Baldauf, S. L. (2003) Science, 300,
1703-1706.
40.Keeling, P. J., Burger, G., Durnford, D. G.,
Lang, B. F., et al. (2005) Trends Ecol. Evol., 20,
670-676.
41.Feagin, J. E., Abraham, J. M., and Stuart, K.
(1988) Cell, 53, 413-422.
42.Wang, Y., and Bogenhagen, D. F. (2006) J.
Biol. Chem., 281, 25791-25802.
43.Bogenhagen, D. F., Wang, Y., Shen, E. L., and
Kobayashi, R. (2003) Mol. Cell Proteom., 2,
1205-1216.
44.Holt, I. J., He, J., Mao, C.-C., Boyd-Kirkup, J.
D., et al. (2007) Mitochondrion, 7, 311-321.
45.Bogenhagen, D. F., Rousseau, D., and Burke, S.
(2008) J. Biol. Chem., 283, 3665-3675.
46.Miyakawa, I., Okamuro, A., Kinski, S., Visacka,
K., Tomashka, L., and Nosek, J. (2009) Microbiology, 155,
1558-1568.
47.Gray, M. W. (1999) Curr. Opin. Genet.
Develop., 9, 678-687.
48.Lang, B. F., Burger, G., O’Kelly, C. J.,
Cedergren, R., Golding, G. B., Lemieux, C., Sankoff, D., Turmel, M.,
and Gray, M. W. (1997) Nature, 387, 493-497.
49.Sugiyama, Y., Watase, Y., Nagase, M., Makita, N.,
Yagura, S., Hirai, A., and Sugiura, M. (2005) Mol. Genet.
Genom., 272, 603-615.
50.Warner, J. R., and McIntosh, K. B. (2009) Mol.
Cell, 34, 3-11.
51.Gott, J. M., and Emeson, R. B. (2000) Annu
Rev. Genet., 34, 499-531.
52.Ohman, M. (2007) Biochimie, 89,
1171-1176.
53.Benne, R., Van den Burg, J., Brakenhoff, J.,
Sloof, P., Van Boom, J., and Tromp, M. (1986) Cell, 46,
819-826.
54.Vidal, S., Curran, J., and Kolakofsky, D. (1990)
EMBO J., 9, 2017-2022.
55.Chateigner-Boutin, A.-L., and Small, I. (2011)
WIREs RNA, 2, 493-506.
56.Covello, P. S., and Gray, M. W. (1989)
Nature, 341, 662-666.
57.Gualberto, J. M., Lamattina, L., Bonnard, G.,
Weil, J.-H., and Grienenberger, J. M. (1989) Nature, 341,
660-662.
58.Hiesel, R., Wissinger, B., Schuster, W., and
Brennicke, A. (1989) Science, 246, 1632-1634.
59.Giege, P., and Brennicke, A. (1999)
Proc. Natl. Acad. Sci. USA, 96, 15324-15329.
60.Handa, H. (2003) Nucleic Acids Res.,
31, 5907-5916.
61.Notsu, Y., Masood, S., Nishikawa, T., Kubo, N.,
Akiduki, G., Nakazono, M., Hirai, A., and Kadowaki, K. (2002)
Mol. Genet. Genom., 268, 434-445.
62.Faivre-Nitschke, S. E., Grienenberger, J. M., and
Gualberto, J. M. (1999) Eur. J. Biochem., 263,
896-903.
63.Takenaka, M., and Brennicke, A. (2003)
J. Biol. Chem., 278, 47526-47533.
64.Lippok, B., Wissinger, B., and Brennicke, A.
(1994) Mol. Gen. Genet., 243, 39-46.
65.Kubo, N., and Kadowaki, K. (1997) FEBS Lett.,
413, 40-44.
66.Oda, K., Yamato, K., Ohta, E., Nakamura, Y., Takemura, M.,
Nozato, N., Akashi, K., Kanegae, T., Ogura, Y., Kohchi, T., and Ohyama,
K. (1992) J. Mol. Biol., 223, 1-7.
67.Turmel, M., Otis, C., and Lemieux, C. (2003)
Plant Cell, 15, 1888-1903.
68.Gott, J. M., Visomirski, L. M., and Hunter, J. L.
(1993) J. Biol. Chem., 268, 25483-25486.
69.Gott, J. M., and Rhee, A. C. (2008) in
RNA Editing (Gott, J. M., and Rhee, A. C., eds.) Springer,
Berlin, pp. 85-104.
70.Visomirski-Robic, L. M., and Gott, J. M. (1995)
RNA, 1, 681-691.
71.Wang, S. S., Mahendran, R., and Miller, D. L.
(1999) J. Biol. Chem., 274, 2725-2731.
72.Horton, T., and Landweber, L. (2000)
RNA, 6, 1339-1346.
73.Visomirski-Robic, L. M., and Gott, J. M. (1997)
RNA, 3, 821-837.
74.Visomirski-Robic, L. M., and Gott, J. M. (1997) Proc. Natl.
Acad. Sci. USA, 94, 4324-4329.
75.Traphagen, S. J., Dimarco, M. J., and Silliker,
M. E. (2010) RNA, 16, 828-838.
76.Beargie, C., Liu, T., Corriveau, M., Lee, H. Y.,
Gott, J., and Bundschuh, R. (2008) Bioinformatics, 24,
2571-2578.
77.Gott, J. M., Parimi, N., and Bundschuh, R. (2005)
Nucleic Acids Res., 33, 5063-5072.
78.Hendrickson, P. G., and Silliker, M. E. (2010)
Curr. Genet., 56, 203-213.
79.Gott, J. M., Somerlot, B. H., and Gray, M. W.
(2010) RNA, 16, 482-488.
80.Alfonzo, J. D., Thiemann, O. H., and Simpson,
L. (1998) J. Biol. Chem., 273, 30003-30011.
81.Pring, D., Brennicke, A., and Schuster, W.
(1993) Plant Mol. Biol., 21, 1163-1170.
82.Barth, C., Greferath, U., Kotsifas, M., and
Fisher, P. R. (1999) Curr. Genet., 36, 55-61.
83.Lin, S., Zhang, H., Spencer, D. F., Norman, J.
E., and Gray, M. W. (2002) J. Mol. Biol., 320,
727-739.
84.Vanfleteren, J. R., and Vierstraete, A. R.
(1999) RNA, 5, 622-624.
85.Lukes, J., Hashimi, H., and Zikova, A. (2005)
Curr. Genet., 48, 277-299.
86.Simpson, L., Sbicego, S., and Aphasizhev, R.
(2003) RNA, 9, 265-276.
87.Blum, B., Bakalara, N., and Simpson, L. (1990)
Cell, 60, 189-198.
88.Blum, B., and Simpson, L. (1990) Cell,
62, 391-397.
89.Bhat, G. J., Koslowsky, D. J., Feagin, J. E.,
Smiley, B. L., and Stuart, K. (1990) Cell, 61,
885-894.
90.Koslowsky, D. J., Bhat, G. J., Perrolaz, A. L.,
Feagin, J. E., and Stuart, K. (1990) Cell, 62,
901-911.
91.Sturm, N. R., and Simpson, L. (1990) Cell,
61, 879-884.
92.Grams, J., McManus, M. T., and Hajduk, S. L.
(2000) EMBO J., 19, 5525-5532.
93.Clement, S. L., Mingler, M. K., and Koslowsky, D.
J. (2004) Eukaryote Cell, 3, 862-869.
94.Maslov, D. A., and Simpson, L. (1992)
Cell, 70, 459-467.
95.Ochsenreiter, T., and Hajduk, S. L. (2006)
EMBO Rep., 7, 1128-1133.
96.Igo, R. P., Jr., Lawson, S. D., and Stuart, K.
(2002) Mol. Cell Biol., 22, 1567-1576.
97.Aphasizhev, R., Aphasizheva, I., Nelson, R. E.,
and Simpson, L. (2003) RNA, 9, 62-76.
98.Aphasizhev, R., Aphasizheva, I., and Simpson, L.
(2003) Proc. Natl. Acad. Sci. USA, 100, 10617-10622.
99.Reifur, L., and Koslowsky, D. J. (2008)
RNA, 14, 2195-2211.
100.Seiwert, S. D., Heidmann, S., and Stuart, K.
(1996) Cell, 84, 831-841.
101.Kable, M. L., Seiwert, S. D., Heidmann, S., and
Stuart, K. (1996) Science, 273, 1189-1195.
102.Seiwert, S. D., and Stuart, K. (1994)
Science, 266, 114-117.
103.Stuart, K. D., Feagin, J. E., and Abraham, J.
M. (1989) Gene, 15, 155-160.
104.Aphasizhev, R., Aphasizheva, I., Nelson, R. E.,
Gao, G. H., Simpson, A. M., Kang, X. D., Falick, A. M., Sbicego, S.,
and Simpson, L. (2003) EMBO J., 22, 913-924.
105.Falick, A. M., Sbicego, S., and Simpson, L.
(2003) EMBO J., 22, 913-924.
106.Peris, M., Frech, G. C., Simpson, A. M.,
Bringaud, F., Byrne, E., Bakker, A., and Simpson, L. (1994) EMBO
J., 13, 1664-1672.
107.Corell, R. A., Read, L. K., Riley, G. R.,
Nellissery, J. K., Allen, T. E., Kable, M. L., Wachal, M. D., Seiwert,
S. D., Myler, P. J., and Stuart, K. D. (1996) Mol Cell Biol.,
16, 1410-1418.
108.Panigrahi, A. K., Ernst, N. L., Domingo, G. J.,
Fleck, M., Salavati, R., and Stuart, K. D. (2006) RNA,
12, 1038-1049.
109.Simpson, L., Aphasizhev, R., Gao, G., and Kang, X.
(2004) RNA, 10, 159-170.
110.Schnaufer, A., Wu, M., Park, Y. J., Nakai, T.,
Deng, J., Proff, R., Hol, W. G., and Stuart, K. D. (2010) J.
Biol. Chem., 285, 5282-5295.
111.Panigrahi, A. K., Schnaufer, A., Carmean, N.,
Igo, R. P., Gygi, S. P., Ernst, N. L., Palazzo, S. S., Weston, D. S.,
Abersold, R., Salavati, R., and Stuart, K. D. (2001) Mol. Cell
Biol., 21, 6833-6840.
112.Weston, D. S., Abersold, R., Salavati, R., and
Stuart, K. D. (2001) Mol. Cell Biol., 21, 6833-6840.
113.Blom, D., de Haan, A., van den Berg, M., Sloof,
P., Jirku, M., Lukes, J., and Benne, R. (1998) Nucleic Acids
Res., 26, 1205-1213.
114.Lukes, J., Arts, G. J., van den Burg, J., de
Haan, A., Opperdoes, F., Sloof, P., and Benne, R. (1994) EMBO
J., 13, 5086-5098.
115.Ochs, D. E., Ots, K., Teixeira, S. M. R.,
Moser, D. R., and Kirchhoff, L. V. (1996) Mol. Biochem.
Parasitol., 76, 267-278.
116.Kolesnikov, A. A., Schonian, G., and Presber,
V. (2000) Mol. Biol. (Moscow), 34, 183-185.
117.Simpson, L., and Maslov, D. A. (1999)
Ann. NY Acad. Sci., 870, 190-205.
118.Kolesnikov, A. A., Merzlyak, E. M.,
Bessolitsina, E. A., Fediakov, A. V., and Schonian, G. (2003) Mol.
Biol. (Moscow), 37, 539-543.
119.Landweber, L. F., and Gilbert, W. (1994)
Proc. Natl. Acad. Sci. USA, 91, 918-921.