Role of cis- and trans-Interactions in Manifestations of Amyloidogenic Properties of Variable Domains of Bence-Jones Proteins TIM and LUS
V. M. Tishchenko
Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: tischen@vega.protres.ru
Received October 1, 2012; Revision received December 10, 2012
Intact Bence-Jones proteins TIM and LUS under simulated physiological conditions (10 mM phosphate buffer, pH 7.0, 100 mM NaCl, 37°C) did not display amyloidogenic properties. However, their isolated variable domains exhibit these qualities in full measure. Therefore, both intact proteins and their variable domains were studied using a complex of physical methods (scanning microcalorimetry, analytical centrifugation, optics) that allowed us to assess the stability of their tertiary and quaternary structures. The experimentally obtained thermodynamic functions indicated that the stability of isolated variable domains of TIM and LUS was comparable to the stability of similar domains in amyloidogenic proteins described earlier. However, inside the whole protein their stability was comparable to the stability of VL domains of ordinary Bence-Jones proteins. The decreased stability of the isolated variable domains of TIM and LUS was shown to be due both to weak interactions between a pair of variable domains (trans-interaction) and to a natural lack of interaction with the constant domains (cis-interaction).
KEY WORDS: Bence-Jones proteins, amyloid fibrils, variable domains, stabilization free energy, interdomain interactionsDOI: 10.1134/S0006297913040056
Abbreviations: CL, light chain constant domains; DSS, bis(N-hydroxysuccinimide suberate) ester; DSS-FV, a fragment with variable domains of the light chain cross-linked with the corresponding reagent; Fb-subunit (fragment), a structure formed by a pair of constant domains CL-CL; FITC, fluorescein isothiocyanate; Fv-subunit (fragment), a structure formed by a pair of variable domains VL-VL; FITC-V(C)L, FITC-labeled variable (constant) domain; IgG, immunoglobulins of G class; VL, light chain variable domains.
Under certain conditions, many even well-soluble proteins are prone to
produce either unordered or, by contrast, highly ordered aggregations.
In particular, production of highly ordered aggregations also
reproducible in vitro under physiologic-like conditions results
in formation of insoluble fibrils and underlies some hereditary and
acquired human diseases [1]. Such diseases include
type-II diabetes, some hereditary neuropathies, Alzheimer’s
disease, and primary systemic amyloidosis. All these diseases have in
common a molecular mechanism characterized by deposition in the tissues
of regular cross-β-fibrils (amyloid deposits) with structure not
significantly depending on the precursor protein [2]. Renal amyloidosis in multiple myeloma is also such
a disease [3]. This complication develops in 10-15%
of patients with multiple myeloma, is progressing rapidly, and is the
main cause of the patients’ death within 1.5-2 years after
beginning of the disease. Multiple myeloma is a malignant B-cell
dyscrasia, i.e. disorders in the normal division and functioning of
B-lymphocytes associated with an uncontrolled synthesis of monoclonal
immunoglobulins. Production of these proteins, in particular, manifests
itself by appearance in the patients’ blood of large amounts of
free light chains of immunoglobulins. Some of them are not retained in
the organism and are filtered by the kidneys into the urine, but
another part loses its thermodynamic stability and forms in the kidneys
insoluble aggregations.
Until recently, studies on the molecular basis of formation of amyloid fibrils by physicochemical approaches were focused on structural features and stability of variable domains of Bence-Jones proteins. Therefore, the main results were associated with detection of differences between physicochemical characteristics of variable domains of amyloidogenic and ordinary Bence-Jones proteins.
Once it seemed that the correlation between the stability of variable domains and their tendency to form amyloid fibrils was the most important finding [4-7], i.e. that the free energy of stabilization of the native structure ΔG of variable parts of the amyloidogenic protein was lower than such energy of variable parts of the protein not producing such aggregations. However, the weak point of these works was, first, the use of indirect approaches for determination of thermodynamic parameters and, second, inadequate control or even the absence of data about the form (monomers or dimers) of the variable domains under study. These considerations mean that it is necessary to use more direct experimental approaches (scanning and isothermal calorimetry) for determination of thermodynamic parameters and to take into account the interdomain interactions.
In fact, we have shown that in Bence-Jones proteins where the formation of amyloid fibrils is associated with constant and not with variable domains, not only the internal stability of the domains must be taken into account [8]. Interdomain interactions, which can level the whole amyloidogenic potential recorded in the isolated domains, must be also considered [9, 10].
A priori, it is clear that the interdomain interactions must be taken into account also in studies on Bence-Jones proteins when the variable parts are responsible for the ability of the whole protein or its fragments to form amyloid fibrils. This is reasonable because in the Bence-Jones proteins the variable domains interact through a greater number of contacts [11, 12] and more intensively than the constant domains [13, 14].
The Bence-Jones proteins TIM and LUS (light chain dimers) do not produce amyloid structures in vitro. Numerous analyses (including those on amyloid deposits in tissues) revealed in these patients renal amyloidosis; therefore, the proteins themselves and their fragments were studied using various physicochemical approaches. In this case, the purpose of the present study was not only to show important interdomain interactions influencing the properties of these proteins but to study them in detail, because in Bence-Jones proteins there are both contacts between the domains within the same chain and interactions between two light chains forming these proteins (respectively, cis- and trans-interactions).
MATERIALS AND METHODS
Preparation of proteins and fragments, control and testing, chemical modifications. All Bence-Jones proteins under study were prepared using the routine method from 0.7 liter of the daily urine of patients with multiple myeloma [15]. Upon fractionation in 70% ammonium sulfate, the proteins were separated by ion-exchange chromatography on DEAE-Sephacel (10 mM Na2HPO4, pH 8.1, the NaCl linear gradient was 0-300 mM). The immunoglobulin light chain fraction was additionally purified on sorbents Blue-Sepharose 4B and Protein A-Sepharose 4B. The final stage of the protein isolation was gel filtration on Sephacryl S100 HR. The resulting preparation of the purified native protein was stored in a buffer solution consisting of 10 mM Na2HPO4, 100 mM NaCl, 1 mM NaN3 (pH 7.4) at 4°C in a sterile glass vessel for no more than 24 h. Immediately before any experiment, gel filtration was performed to remove aggregations and to transfer the protein solution into the required buffer.
Homogeneity of the intact proteins and their fragments was monitored by electrophoresis under native conditions (pH 7.5), by standard SDS-electrophoresis in polyacrylamide gel [16], and also by two-dimensional electrophoresis according to [17] with commercial poly- and monospecific antisera. The light chain isotype was determined by immunoelectrophoresis in 1% agarose gel with a polyspecific antiserum to human blood plasma proteins (ICN, USA) and with monospecific antisera to light chain isotypes of human immunoglobulin (Sigma, USA).
The limited proteolysis procedures leading to cleavage of the peptide bond between the constant and variable domains and the subsequent separation of constant (Fb-fragment) and variable (FV-fragment) parts of the protein were performed in general as described in work [18]. But because the yield of variable domains in the case of protein LUS was decreased, the method was modified as follows: not the isolated protein LUS was subjected to limited proteolysis, but its complex with the Fab-fragment [9]. This fragment was prepared by the standard method from antibodies (rabbit IgG) against the LUS variable domains. This approach was earlier shown to significantly increase the yield of those constant domains, the stability of which significantly decreased on limited proteolysis of the Bence-Jones proteins VAD and PER [9, 10]. This approach was also productive for preparing variable domains of the protein LUS and resulted in more than twofold increase in the yield of intact domains upon the proteolysis.
Dimers of variable domains connected by covalent bonds were obtained using the homobifunctional reagent bis(N-hydroxysuccinimide suberate) ester (DSS) as described earlier in works [15, 19]. The cross-linked dimers and residual monomers were separated by gel filtration on a column with Sephacryl S100 HR at low protein concentration (0.5 mg/ml), when the equilibrium of the fraction of residual free domains was sharply shifted to monomers.
Both intact proteins and their fragments were labeled with fluorescein isothiocyanate (FITC) as described in works [20, 21]. Whole Bence-Jones proteins with the fluorescent label located either in the constant or in the variable part were obtained by affinity chromatography with immobilized antibodies against the FITC-Fb-fragment (FITC-VL-domain) [20].
Monitoring of formation of amyloid fibrils. The ability to produce amyloid fibrils was studied in the initial solutions of whole proteins TIM and LUS (their variable or constant fragments), and kinetics of fibril production were monitored based on the general approach described in [15, 22, 23].
The specimens (1.5 mg/ml) in buffer solution were passed through a 0.22 µm filter (Millipore, USA) and incubated at 37°C for 7 days. Every 24 h (6 h at the first stages), several samples of 50-100 µl were taken to control the self-association of the proteins or of their fragments using approaches based on binding dyes.
The binding of thioflavin T was recorded after mixing of 40 µl of the protein (or its fragment) solution and 2 ml of solution consisting of 100 mM phosphate buffer (pH 7.5), 100 mM NaCl, and 18 µM thioflavin T. The fluorescence intensity of the solution was measured with an MPF-4B fluorimeter (Japan) at 520 nm with excitation wavelength of 450 nm.
The binding of Congo Red was monitored 1 h after mixing and incubation of two solutions (100 µl of the protein solution and 200 µl of 100 mM phosphate buffer (pH 7.5) supplemented with 100 mM NaCl, and 20 µM Congo Red). The differential spectrum in the range of 400-700 nm was recorded with a Hitachi 2000M spectrophotometer (Japan). To obtain kinetic curves of formation of amyloid structures, each point was measured at least five times.
Electron microscopy. Solutions of free variable domains of TIM and of free variable domains of LUS cross-linked with DSS were incubated for 4 days and then mixed at the ratio of 1 : 1. Later the specimens were prepared and subjected to measurements performed with a JEM-100C electron microscope (Japan) as described in our previous work [15].
Determination of molecular weights. Molecular weights of the proteins and of their fragments were determined by analytical ultracentrifugation with a Beckman Spinco model E centrifuge (USA). Using high-speed and low-speed equilibrium centrifugation (according to [24] and [25], respectively), the average weight molecular mass Mw and the z-average molecular mass Mz were determined by the formulas:
Mw = 2RT/(1 – νρsol)ω2 × dlny/dr2, (1)
Mz = RT/ω2(1 – νρsol) × d(1/r dc/dr)/dc, (2)
where y is the displacement of the interference band (or of the optical density) along the y axis (proportional to the protein concentration), r is the rotation radius, ω is the angular speed in rad/s, R is the universal gas constant, T is absolute temperature, ν is the specific partial volume, ρsol is the solvent density.
The protein distribution in the cell was recorded using the Rayleigh’s interference system (at the protein concentration of 0.3 mg/ml) or the absorption two-beam ultraviolet system at the wavelengths of 280 and 230 nm (the protein concentrations, respectively, 1 and 0.1 mg/ml). To decrease the time of the experiment, it was performed in partially filled cells because the time of equilibrium establishment in the cell is proportional to thickness of the solution layer [26].
Studies on temperature-dependent conformational changes in proteins by fluorescence and scanning thermometry and calculation of thermodynamic parameters of stability. Conformational changes in the intact Bence-Jones proteins and in their fragments were studied by measuring the fluorescence with a Shimadzu RF-5301 PC spectrofluorimeter (Japan) using a thermostatted cell. The resulting temperature dependences of fluorescence were analyzed as described in works [20, 21, 27].
Scanning thermometry was performed with a DASM-4A microcalorimeter equipped with a gold 1-ml capillary cell and a DASM-4 microcalorimeter equipped with a 0.43-ml platinum cell. The protein concentration in the solution was varied in the range of 1.2-6.0 mg/ml. The measurements were performed with the rate of temperature change of 0.5-2°C/min in 10 mM phosphate buffer (pH 7.0-8.0) and 10 mM glycine buffer (pH 3.2). The calorimetric data were analyzed as described in work [28]. Value of the free energy of stabilization of the native structure of the cooperative block ΔG(T) in dependence on temperature was calculated using the equation:
ΔG(T) = ΔHm (1 – T/Tm) – ΔCp [(Tm – T) + T ln(T/Tm)], (3)
where ΔHm is enthalpy of the mentioned cooperative structure, Tm is the temperature of its melting, ΔCp is the change in heat capacity during this process. The first member of Eq. (3) corresponds to changes in the enthalpy, and the second member corresponds to changes in the entropy.
RESULTS AND DISCUSSION
Formation in solution by variable domains of proteins TIM and LUS of amyloid fibrils and their structure. Figure 1 shows kinetics of binding of Congo Red (a fluorescent probe) and thioflavin T (an absorption probe) by proteins TIM and LUS and their fragments during long-term incubation. Earlier, it was established that changes in the optical parameters of these probes were caused by their binding with elongated β-folded layers produced by monomers of amyloidogenic proteins on placement lengthwise the long axis of fibrils [23, 29]. The Bence-Jones proteins TIM and LUS did not display the tendency to bind the standard dyes (Congo Red and thioflavin T), which indicated their inability to form amyloid structures. The constant domains (Fb-fragments) also lack this property. However, isolated variable domains bound with both dyes that indicated that VL (FITC-VL) domains of TIM and LUS could produce amyloid structures, but they could not produce such structures when they were inside the intact proteins.
Fig. 1. a) Kinetics of formation of VL and FITC-VL fibrils by the LUS domains (light and dark symbols, respectively). The processes were followed by binding of thioflavin T (circles) and of Congo Red (triangles). Similar data are presented for VL and FITC-VL domains of TIM. The fluorescence intensity is decreased by an order of magnitude. The first method did not revealed fibril production by the intact proteins TIM and LUS (squares), and the other method gave similar results. Experimental errors are presented only for one curve, for other curves their values are similar. b) Formation of fibrils after binding the dyes for DSS-FITC-FV of TIM (circles) and of LUS (triangles). The Fb fragments of these proteins do not form fibrils (squares).
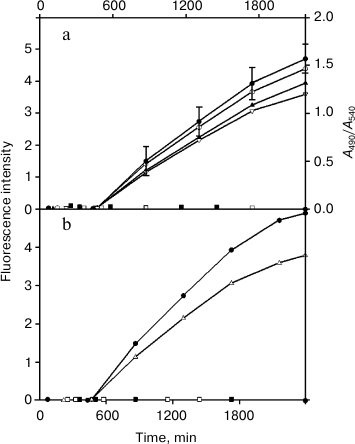
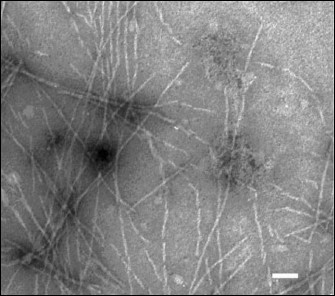
Figure 2 shows that the produced amyloid structures are non-branched fibrils of different length with ~60 Å in diameter.
Fig. 2. Electron microphotograph of fibrils of the TIM and LUS proteins. The label value on the photograph is ~450 Å.
Determination of molecular weights of the proteins and their fragments. Molecular weights determined by centrifugation were Mw = 50.9 ± 1.5 and 50.7 ± 1.5 kDa and Mz = 51.2 ± 2.5 and 51.1 ± 2.5 kDa for TIM and LUS, respectively. Because the two molecular weights are differently sensitive to aggregations, their equality means that the two proteins exist in solution as monomers. And they are unchanged at least for two days.
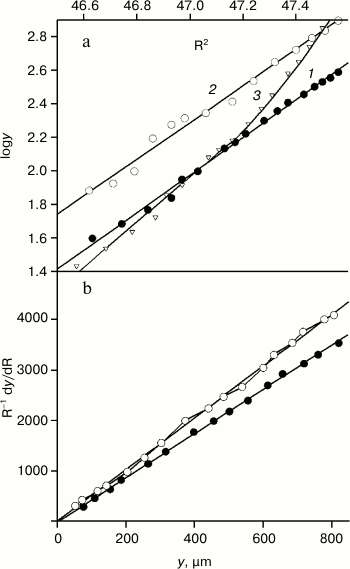
On the contrary, molecular weights of their VL domains display the equilibrium between monomers and dimers, which is indicated by the nonlinear dependence of logy on R2 (Fig. 3a, curve 3). And the equilibrium is not sharply shifted to either side at the concentrations of 1.2-3.5 mg/ml. In work [30], the dissociation constant of the reaction VL + VL ↔ FV was shown to be high and equal to 2.1·10–4 M at 20°C. Under such conditions, it is impossible to obtain thermodynamic curves characterizing only the internal stability of the VL domains. Therefore, the FITC-VL domains were studied with Mw = 12.5 ± 0.4 and 12.7 ± 0.4 kDa and Mz = 12.6 ± 0.6 and 12.5 ± 0.6 kDa for TIM and LUS, respectively (Fig. 3, a and b). These domains exist as monomers at least for 6 h (the time required for experiments with the equilibrium centrifugation).
Fig. 3. Determination of molecular weights of the FITC-VL domains by equilibrium centrifugation. a) Data for FITC-VL of TIM (dark circles, straight line 1 slope is 1.121), for FITC-VL of LUS (light circles, straight line 2 slope is 1.092), for VL of TIM (light triangles, curve 3) were obtained using high-speed centrifugation. The rotor rotation rates were 56410, 54360, and 57890 rpm. b) Similar data for FITC-TIM and FITC-LUS were obtained using low-speed centrifugation. The rotor rotation rates were 52450 and 51180 rpm, and the straight line slopes are 5.1 and 4.4, respectively.
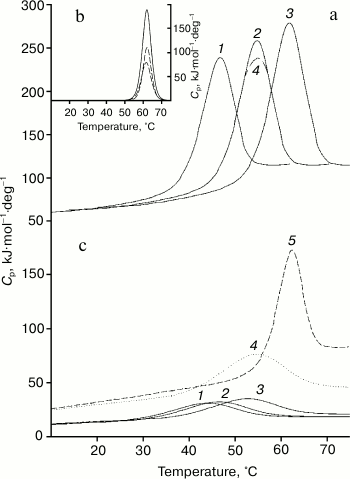
Results of studies on temperature-induced conformational changes. Thermodynamic functions characterizing stability of the isolated domains and of the same domains inside the whole Bence-Jones proteins were obtained by determination of their temperature-induced extreme conformational changes (thermal denaturation). Figure 4 (a and c) presents calorimetric data for the TIM protein obtained at different pH values. It should be especially emphasized that all melting curves of the proteins and their fragments characterize an equilibrium process, because the experiments revealed neither dependence on the concentration of the preparation studied [31] nor dependence on the scanning rate [32, 33]. Similar data were also obtained for the LUS protein and its fragments. The data suggest a block structure of the protein because the calorimetric enthalpy is much higher than the effective one (calculated also from optical curves of the melting, see table) that is specific for the Bence-Jones proteins [9, 10, 18]. For interpretation of the results, it was necessary to establish just what domains could form the blocks and also to determine thermodynamic parameters of these cooperative structures.
Fig. 4. Results of calorimetric measurements of protein TIM. Very similar data were also obtained for protein LUS. a) Temperature dependences of the molar thermal capacities of the intact protein at pH values of 2.1, 3.05, 7.0 (curves 1-3; curve 4 shows the melting reversibility at pH 3.05). b) Expansion of the excess heat absorption (solid line) of the protein TIM melting curve at pH 7.0 into elementary transitions (dashed lines). c) Temperature dependences of molar thermal capacities of the FITC-VL domain at pH values of 2.1, 3.05, 7.0 (curves 1-3, respectively), DSS-FITC-FV fragment at pH 7.0 (curve 4); for comparison, the melting curve (5) of the FV-subunit is presented at pH 7.0 obtained by deconvolution (Fig. 4b).
Thermodynamic parameters of melting of Bence-Jones proteins and their
fragments
Note: On determination of enthalpy, the error is no more than 7%; on
calculations of the free energy, the error is no more than 15%.
1 Data were obtained for the intact protein.
2,3 Data were obtained either as a result of deconvolution of
the calorimetric curve of the intact protein for the first and second
elementary transitions, by the fluorescence for melting of blocks of
variable and constant domains inside the intact protein, or for
isolated domains by calorimetry (2) or optics (3).
4 Data were obtained for the intact protein by
fluorescence.
First of all, expansion of the excess heat absorption curves into elementary transitions each of which corresponding to the model of two states [28] reveals the presence of two such transitions corresponding to melting of two cooperative blocks (Fig. 4b and the table). Based on earlier data obtained for both Bence-Jones proteins [9, 10, 18] and for the Fab fragments [34, 35], it was supposed that they could be formed by a pair of the constant and a pair of the variable domains.
However, these data do not reveal just what elementary transition corresponds to the melting of one or the other protein. The standard approach to this question, i.e. the comparison of thermodynamic parameters obtained for the intact protein and its isolated variable and constant parts, is not promising for the situation. This is, in particular, due to similar values of the melting temperatures of the two elementary transitions. Moreover, it is difficult to directly compare thermodynamic parameters of melting of the isolated V domains and of the same domains inside the whole Bence-Jones protein because in the first case they are in a heterogeneous mixture (mainly as monomers at the operating concentrations of ~10–4 M), whereas in the other case they are components of the cooperative block1.
1 Using high concentrations at calorimetric measurements does not simplify the situation, although VL domains are already present as dimers. First, there are problems because the meltings of the FV fragment and FV subunit are, respectively, first and second order reactions. Second, for high concentration of the protein, the heat denaturation at some pH values is associated with aggregations. Therefore, the problem was solved with an approach that we have used before in studies on some multidomain proteins including the Bence-Jones proteins [9, 10, 19-21, 36]. This approach presents a modification of a protein under study with a fluorescent label, and then preparation of the intact protein fractions with selectively labeled either variable or constant domains [9, 10, 20].
Data obtained on the melting of such proteins are presented in Fig. 5, and results of their calculation are given in the table. The comparison with the results of the calorimetric curves expansion shows, first of all, that the temperatures and effective enthalpies of melting of both variable and constant domains calculated from the optical curves by the “two states” model virtually coincide with similar calorimetric parameters for `the cooperative proteins. Thus, we conclude that the two pairs of mentioned domains form inside the whole protein cooperative blocks coinciding, respectively, with the FV and FB subunits.
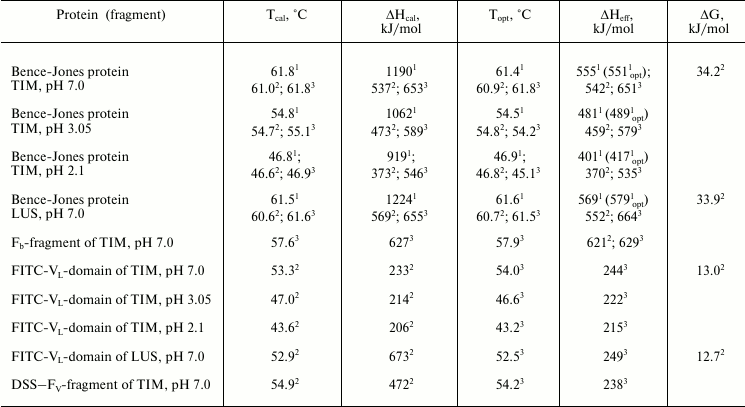
Fig. 5. Change in the fraction of the denatured state of the Fv (dark circles) and Fb subunits (light circles) at pH values of 7.0 and 2.1 calculated from changes in the fluorescence on melting of the Bence-Jones protein TIM. Effective enthalpies calculated from these curves coincide with calorimetric enthalpies for the first and second elementary transitions on the melting of the intact protein TIM (Fig. 4b and table).
The obtained thermodynamic parameters for elementary transitions with the melting of the definite cooperative structure are compared as follows. Both the optical data (Fig. 5) and the calorimetrical data (Fig. 4b) show the closeness of temperatures of the two transitions. Moreover, the experimental error on determination of the transition temperature is only ±0.25°C for the differential calorimetric curve, whereas for the integral optical curve it is significantly higher (no less than ±0.75°C). Therefore, the above-formulated problem cannot be solved based only on the data on thermostability obtained by the two methods.
However, comparison of the melting enthalpies calculated based on calorimetric and optical curves (Figs. 4 and 5 and the table) results in a quite unambiguous conclusion. Thus, the transition with the higher enthalpy corresponds to melting of the Fb subunit (the cooperative block of CL domains), whereas the lower enthalpy corresponds to melting of the FV subunit (the cooperative block of VL domains) within the whole range of pH values studied. This was especially demonstrative at low pH values (Fig. 5, melting curves at pH 2.1).
And because within the range of concentrations of (1.5-3.0)·10–4 M (typical for calorimetry) and lower (typical for fluorescence) the FITC-VL domains exist as monomers, and the cooperative block size changes. A preliminary conclusion based not even on the qualitative but on quantitative analysis can be immediately made. Because the cooperative block size is decreased twofold, the free energy of stabilization of this structure also has to decrease strongly. Based on these considerations, the ability to produce amyloid structures by isolated VL domains and their full loss of amyloidogenicity inside the intact protein seem quite reasonable and natural.
These considerations based on the qualitative analysis are to be confirmed and developed using quantitative data. First of all, we have in mind the determination of the melting enthalpies of the VL domains in two states, the values of which were obtained by both calorimetrical and optical approaches in the wide range of pH values. Note that the comparison of data on the calorimetric and effective enthalpy of melting of the isolated VL domains (table) shows that in this state the domain itself is a cooperative block. Certainly, this could be expected based on studies on small globular proteins, in particular, non-interacting IgG domains (CH2 domains) [20, 37, 38].
The comparison of melting enthalpies of the FV subunit and of the isolated VL domain also shows that the first is more than twofold greater than the second. This is not surprising because the joining of variable domains in the united cooperative block inside the intact molecule implies the presence of sufficiently intense VL–VL interactions. Moreover, the disintegration of the tertiary structure of the FV subunit is accompanied by a break of its contacts with the Fb subunit, which has to additionally increase the melting enthalpy.
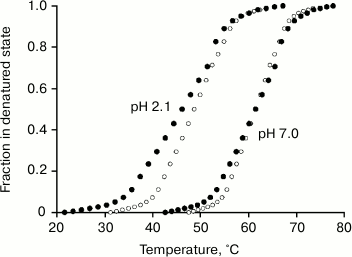
Figures 4 and 5 and the table show that decrease in pH leads to destabilization of both FV subunit and VL domains, which allows us to obtain the dependence of their melting enthalpies on the denaturation temperature. And this, in turn, allows us to determine changes in the partial heat capacities ΔCp and to calculate the free energy value ΔG of their native structural stabilization (Fig. 6) [28, 37]2. It follows from Eq. (3) and data of the table and of Figs. 4 and 5 that at 25°C molar values of ΔG25° for the FITC-VL domain and the FV subunit are, respectively:
ΔGV = 233(1 – 298.2/320.0) – 4.1[(320.0 – 298.2) + 298.2ln(298.2/320.0)] = 15.9 – 3.1 = 12.8 kJ/mol (4)
and
ΔGFV = 537(1 – 298.2/334.2) – 11.6[(334.2 – 298.2) + 298.2ln(298.2/334.2)] = 57.8 – 23.6 = 34.2 kJ/mol, (5)
that quite corresponds to values of amyloidogenic and usual structure of the IgG domains [4-7, 9, 10, 35]. The specific values of ΔG are:
ΔGV = 1.3 – 0.3 = 1.0 J/g,
ΔGFV = 2.3 – 0.9 = 1.4 J/g.
2 We assume the ΔCp value to be independent of temperature (despite different viewpoints on this question [28, 37, 39]) because the protein stability changes in a small temperature range of relatively low temperatures, where ΔCp is more reasonable to be considered as a constant parameter.
Fig. 6. Temperature dependence of the molar enthalpy of melting of the FV-subunit (dark circles) and of the FITC-FV-fragment (light circles).
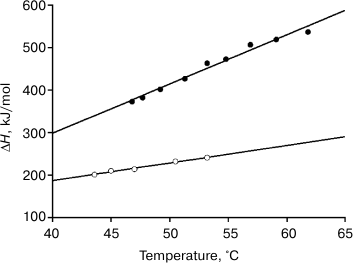
Thus, the above-mentioned hypothesis about causes of appearance in the VL domains of the amyloidogenic potential based on purely qualitative considerations is confirmed by quantitative calculations. Differences in the molar values of ΔG for the FITC-VL domain and the FV subunit are very great and not so significant in specific values. This finding emphasizes once more that the main difference between the same protein structures capable and incapable of producing amyloid fibrils is associated with the twofold greater size of the cooperative unite in the first case. Just this is responsible for the great difference in the free energies of stabilization of the two structures.
Moreover, analysis of the data reveals that the free energy decreases because of decreases in both the enthalpic and entropic members of Eqs. (4) and (5), and the contribution of the entropic member to these changes is more significant. In this connection, it is necessary to emphasize once more the decisive role of the Fb subunits in the elimination of the amyloidogenic potential of the intact Bence-Jones proteins TIM and LUS. Even the appearance of a covalent bond between isolated variable domains did not lead to a noticeable change in their thermodynamic parameters (the melting curve of the DSS-FITC-FV fragment in Fig. 4c). Therefore, it was supposed that the constant domains not only could act as a kind of platform promoting orientation of the VL domains relative to one another in the definite position as we mentioned earlier in work [35]. The Fb subunit also induces in the V domains the corresponding conformational changes, resulting in adaptation of their structure to strong interdomain interactions and leading to formation of a cooperative block of two variable domains [40, 41].
Thus, on one hand the present work has confirmed rather multiple data on the correlation between the amyloidogenic potential of the protein structure and its decreased stability. On the other hand, the findings have shown a very important role of interdomain interactions in maintaining (leveling) of this potential. Both these findings appear especially topical from the standpoint of new data indicating disorders in this regularity. According to work [42], the V domain of the amyloidogenic Bence-Jones protein 6aJL2 has higher free energy of stabilization of the native ΔG25°C structure than many of the variable domains studied before. This energy is 21.8 kJ/mol, and this either coincides with the stability of common domains [43] or is even higher [44].
However, a careful analysis of data presented in work [42] reveals that the ΔG25°C value of the V domain of 6aJL2 has been calculated with some admissions, which principally need experimental confirmation. Thus, on determination of this parameter from the melting and titration curves of GuHCl, the domains were suggested to be monomers. It seems that the authors believed this suggestion quite reasonable because the transition curves were obtained by fluorescence at relatively low concentrations of the protein (~5·10–6 M). But it should be noted that if dimerization of the VL domains is described by the dissociation constant of 10–6 M (and this value is quite reasonable and admissible), the V domains of 6aJL2 will exist mainly as dimers.
Then, on calculations of the free energy after Eq. (3) in work [42] the value of the heat capacity jump ΔCp at the denaturation was assumed to be 5.9 kJ/mol·deg. This value was not determined experimentally but calculated theoretically, as described in work [45]. But it follows from the findings of our work (table and Fig. 5) that the heat capacity jump ΔCp is lower (4.1 kJ/mol·deg). Note that in their later work [46], the authors used scanning calorimetry to study the melting of the V domains of 6aJL2 at different concentrations of urea. During that work, the ΔCp value of 7.3 kJ/mol·deg was obtained from the temperature dependence of enthalpy. And it should be taken into account that on melting of globular proteins in the presence of various denaturing agents, the slope of the temperature dependence of enthalpy is much steeper, i.e. the ΔCp value increases because the experimentally determined enthalpy of the melting includes not only decomposition of the tertiary structure of the protein, but also solvation of the denaturing agent molecules on the newly opened surface of the protein molecules [28, 38].
Thus, the analysis of the data presented in work [42] describing abnormal high value of free energy ΔG25°C causes us to doubt both the experimentally results obtained for an insufficiently characterized preparation and the reasonability of using indirect approaches for obtaining thermodynamic parameters of its stability. On the whole, the totality of original experimental findings described in the present work and the critical analysis of the previously published data convincingly indicate that the published results should be revised at least to correct the quantitative data and to show the urgent necessity to use more direct experimental approaches and to take into account the interdomain interactions.
The author is grateful to O. P. Bliznyukov (Institute of Immunology, Ministry of Health of Russian Federation) for help in measurements with electron microscope and to S. A. Potekhin and V. V. Filimonov (Institute of Protein Research, Russian Academy of Sciences) for fruitful discussion.
This work was supported by the Russian Foundation for Basic Research (projects 07-04-12199-ofi and 11-04-00064-a).
REFERENCES
1.Falk, R. H., Comenzo, R. L., and Skinner, M. (1997)
N. Engl. J. Med., 337, 898-909.
2.Huff, M. E., Balch, W. E., and Kelly, J. W. (2003)
Curr. Opin. Struct. Biol., 13, 674-682.
3.Pozzi, C., and Locatelli, F. (2002) Semin.
Nephrol., 22, 319-330.
4.Helms, L. R., and Wetzel, R. (1995) Protein
Sci., 4, 2073-2081.
5.Wall, J., Schell, M., Murphy, C., Hrncic, R.,
Stevens, F. J., and Solomon, A. (1999) Biochemistry, 38,
14101-14108.
6.Raffen, R., Dieckman, L. J., Szpunar, M., Wanschl,
C., Rokkuluri, P. R., Dave, R., Wilkins, Stevens, P., Cai, X.,
Schiffer, M., and Stevens, F. J. (1999) Protein Sci., 8,
509-517.
7.Kim, Y., Wall, J. S., Meyer, J., Murphy, C.,
Randolph, T. W., Manning, M., Solomon, A., and Carpenter, J. F. (2000)
J. Biol. Chem., 275, 1570-1574.
8.Solomon, A., Weiss, D. T., Murphy, C. L., Hrncic,
R., Wall, J. S., and Schell, M. (1998) Proc. Natl. Acad. Sci.
USA, 95, 9547-9551.
9.Tishchenko, V. M., Khristoforov, V. S., and
Bliznyukov, O. P. (2009) Mol. Biol., 43,
148-156.
10.Tishchenko, V. M. (2011) Mol. Biol.,
45, 1055-1064.
11.Epp, O., Lattman, E. E., Schiffer, M., Huber, R.,
and Palm, W. (1975) Biochemistry, 14, 943-952.
12.Chang, C. H., Short, M. T., Westholm, F. A.,
Stevens, F. J., Wang, B. C., Furey, W., Jr., Solomon, A., and Schiffer,
M. (1985) Biochemistry, 24, 4890-4897.
13.Schiffer, M., Chang, C.-H., Naik, V. M., and
Stevens, F. J. (1988) J. Mol. Biol., 203,
799-802.
14.Miller, S. (1990) J. Mol. Biol.,
216, 965-973.
15.Bliznyukov, O. P., Kozmin, L. D., Vysotskaya, L.
L., Golenkov, A. K., Tishchenko, V. M., Samoilovich, M. P., and
Klimovich, V. P. (2005) Biochemistry (Moscow), 70,
458-466.
16.Laemmli, U. K. (1970) Nature, 227,
680-685.
17.Laurell, C. B. (1965) Analyt. Biochem.,
10, 358-361.
18.Zav’yalov, V. P., Troitsky, G. V.,
Khechinashvili, N. N., and Privalov, P. L. (1977) Biochim. Biophys.
Acta, 492, 102-111.
19.Tishchenko, V. M. (2001) Biochemistry
(Moscow), 66, 1352-1355.
20.Tischenko, V. M., Abramov, V. M., and
Zav’yalov, V. P. (1998) Biochemistry, 37,
5576-5581.
21.Tischenko, V. M., and Zav’yalov, V. P.
(2002) Immunol. Lett., 84, 241-245.
22.Eulitz, M., Chang, L.-Y., Zirkel, C., Schell, M.,
Weiss, D. T., and Solomon, A. (1995) J. Immunol., 154,
3256-3265.
23.Wall, J., Murphy, C. L., and Solomon, A. (1999)
Methods Enzymol., 309, 204-216.
24.Yphantis, D. A. (1964) Biochemistry,
3, 297-317.
25.Van Holde, K. E., and Baldwin, R. L. (1958) J.
Phys. Chem., 62, 734-743.
26.Bowen, T. (1971) in An Introduction to
Ultracentrifugation (Degly, S., ed.) Wiley-Interscience,
London-N.-Y.-Sydney-Toronto, pp. 107-108.
27.Tishchenko, V. M. (2000) Mol. Biol.,
34, 116-122.
28.Privalov, P. L., and Potekhin, S. A. (1986)
Methods Enzymol., 131, 4-51.
29.Wetzel, R. (1997) Adv. Prot. Chem.,
50, 183-242.
30.Tishchenko, V. M. (2012) Biofizika, in
press.
31.Filimonov, V. V., and Rogov, V. V. (1996) J.
Mol. Biol., 255, 767-777.
32.Potekhin, S. A., and Kovrigin, E. L. (1998)
Biofizika, 43, 223-232.
33.Potekhin, S. A., Loseva, O. I., Tiktopulo, E. I.,
and Dobritsa, A. P. (1999) Biochemistry, 38,
4121-4127.
34.Tischenko, V. M., Zav’yalov, V. P.,
Medgyesi, G. A., Potekhin, S. A., and Privalov, P. L. (1982) Eur. J.
Biochem., 126, 517-521.
35.Zav’yalov, V. P., and Tishchenko, V. M.
(1991) Scand. J. Immunol., 33, 755-762.
36.Tischenko, V. M., and Zav’yalov, V. P.
(2003) Immunol. Lett., 86, 281-285.
37.Privalov, P. L. (1979) Adv. Prot. Chem.,
33, 167-241.
38.Privalov, P. L. (1982) Adv. Prot. Chem.,
35, 1-104.
39.Privalov, P. L., and Makhatadze, G. I. (1992)
J. Mol. Biol., 224, 715-723.
40.Tischenko, V. M. (1999) in Abst. XIth Int.
Conf. on Small-Angle Scattering, Brookhaven National Laboratory, N.
Y., p. 93.
41.Tishchenko, V. M. (2013) Mol. Biol., in
press.
42.Del Pozo Yauner, L., Ortiz, E., Sanchez, R.,
Sanchez-Lopez, R., Guereca, L., Murphy, C. L., Allen, A., Wall, J. S.,
Fernandez-Velasco, D. A., Solomon, A., and Becerril, B. (2008)
Proteins, 72, 684-692.
43.Rowe, E. S., and Tanford, C. (1970)
Biochemistry, 24, 4822-4827.
44.Goto, Y., Ichimura, N., and Hamaguchi, K. (1988)
Biochemistry, 27, 1670-1677.
45.Pace, C. N., Shirley, B. A., and Thomson, J. A.
(1989) in Protein Structure: A Practical Approach (Creighton, T.
E., ed.) IRL Press, Oxford, pp. 311-330.
46.Blancas-Mejia, L. M., Tellez, L. A., del
Pozo-Yauner, L., Becerril, B., Sanchez-Ruiz, J. M., and
Fernandez-Velasco, D. A. (2009) J. Mol. Biol., 386,
1153-1166.