REVIEW: Multifunctionality of PIWI Proteins in Control of Germline Stem Cell Fate
E. Y. Yakushev, O. A. Sokolova, V. A. Gvozdev, and M. S. Klenov*
Institute of Molecular Genetics, Russian Academy of Sciences, Kurchatov sq. 2, 123182 Moscow, Russia; E-mail: klenov@img.ras.ru* To whom correspondence should be addressed.
Received March 26, 2013
PIWI proteins interacting with specific type of small RNAs (piRNAs) repress transposable elements in animals. Besides, they have been shown to participate in various cellular processes: in the regulation of heterochromatin formation including telomere structures, in the control of translation and the cell cycle, and in DNA rearrangements. PIWI proteins were first identified by their roles in the self-renewal of germline stem cells. PIWI protein functions are not limited to gonadogenesis, but the role in determining the fate of stem cells is their specific feature conserved throughout the evolution of animals. Molecular mechanisms underlying these processes are far from being understood. This review focuses on the role of PIWI proteins in the control of maintenance and proliferation of germinal stem cells and its relation to the known function of PIWI in transposon repression.
KEY WORDS: small RNA, RNA silencing, piRNA, heterochromatin, stem cells, PIWIDOI: 10.1134/S0006297913060047
PIWI AND piRNA SILENCING
PIWI proteins belong to a subfamily of the protein family Argonaute, which bind small RNAs and are responsible for sequence-specific gene silencing via the RNA-interference mechanism [1, 2]. The term PIWI is derived from the drosophila protein Piwi, which was the first member characterized [3]. The PIWI subfamily proteins interact with a specific class of endogenous small RNAs, piRNAs (PIWI-interacting RNAs), which usually have length of 23-31 nucleotides and are mechanistically different in the biogenesis and functions from other well-studied classes of small RNAs, such as microRNAs and siRNAs. The central function of piRNAs is silencing of transposable elements and, thus, maintaining genome stability. Both piRNA-mediated silencing and RNA-interference are based on recognizing the RNA target using a complementary molecule of small RNA [1, 2].
PIWI proteins and piRNAs are present mainly in animal gonads (testes and ovaries) and function during different development stages of germinal cells, but they are found also in somatic tissues [4]. The preferential gonad-specific expression of PIWI/piRNAs can be possibly due to the strongest influence of transposons on the germinal cells, which are gamete precursors. The transposable elements have a tendency to multiply in the germinal cells because new insertions are transmitted through the germline to subsequent generations of the host. Transpositions of the mobile elements can be associated with different defects, such as damage of protein-coding genes, chromosome breaks, and genome rearrangements [1, 5]. Absence of PIWI proteins or other components of the piRNA-silencing system usually result in sterility. On the other hand, the preferential expression of PIWI in gonads can be related to the role of these proteins in the maintenance and development of germline stem cells. Both functions of PIWI are conserved during the evolution of multicellular animals from sponges to mammals, but their interrelation is still unclear [4].
In drosophila ovaries used as a model for investigation of piRNA silencing mechanisms, the majority of piRNAs is produced from transcripts of certain genomic regions, called piRNA clusters, predominantly located in pericentromeric and subtelomeric regions of chromosomes (concerning piRNA clusters, see the review [6]). piRNA clusters mainly consist of broken copies of transposons incapable of transposing and of their fragments [7]. Clusters are transcribed into long single-stranded RNAs (predominantly in the antisense polarity to mRNA of transposable elements), which are cut during so-called primary processing into mature piRNAs. As differentiated from two other classes of small RNAs, microRNAs (miRNAs) and small interfering RNAs (siRNAs), piRNAs are produced without involvement of Dicer nucleases [2, 8] that cleave double-stranded RNA structures. With the help of auxiliary proteins, piRNAs are loaded into RNP complexes with PIWI proteins, which suppress the expression of transposons complementary to piRNAs. piRNA clusters are a kind of database exemplifying sequences that are harmful for the cell. Inside a piRNA cluster, any DNA fragment will be a source of piRNAs, which will suppress all homologous sequences [9-11]. Mechanisms of transcription and processing of piRNA clusters remain unclear. It is unknown what features of single-stranded RNAs produced by piRNA clusters discriminate them from other cellular RNAs, which do not undergo processing into piRNAs. Chromatin of piRNA clusters is thought to be marked with specific proteins [12, 13].
piRNAs can also be produced with involvement of an amplification loop, called ping-pong. During this amplification the PIWI complex with an “antisense” piRNA cuts a complementary transcript of a transposon RNA producing the 5′-end of the future “sense” piRNA. Then an unknown nuclease forms the 3′-end of the piRNA. The resulting secondary piRNAs can initiate the formation of new “antisense” piRNAs from transcripts of piRNA clusters [7, 14]. Ping-pong amplification has been found in various organisms [1, 2].
Drosophila melanogaster has three piRNA-binding proteins of the PIWI subfamily: the nuclear protein Piwi and cytoplasmic proteins Aub and Ago3. The ovaries of drosophila consist of somatic and germinal cells. In the somatic cells piRNAs are generated only by primary processing, whereas in the germinal cells the majority of piRNAs are produced through ping-pong amplification [15]. The Piwi protein is loaded mainly with primary piRNAs and suppresses transposable elements in both germinal and somatic cells of the ovaries, whereas Aub and Ago3 proteins are involved in the ping-pong amplification of piRNAs and suppress transposons in the cytoplasm of germinal cells [15-17].
In addition to evidences of the involvement of piRNAs in post-transcriptional regulation (slicing of target transcripts) [7, 14, 18], some PIWI proteins in complex with piRNAs suppress the expression of their targets at the transcriptional level, as has been shown also for other types of Argonaute proteins, which bind siRNAs or other classes of small RNAs [19]. In mice the PIWI proteins Mili and Miwi2 are involved in de novo DNA methylation of transposons [20-22]. The piRNA-dependent methylation of DNA is shown to contribute to parental genomic imprinting [23]. In the nematode Caenorhabditis elegans the PIWI protein Prg-1 together with chromatin factors is involved in initiation of transcriptional silencing of transgenes in germinal cells [24, 25]. Nuclear protein Piwi in drosophila is responsible for suppression of transposon transcription in ovaries. This suppression is probably realized through interaction of Piwi–piRNA complexes with complementary transcripts, which are close to the chromosome sites that have produced them. It is suggested that Piwi can recruit to the corresponding genomic locus histone methyltransferases responsible for di- and trimethylation of histone H3 residue K9 that results in binding of the heterochromatic protein HP1 and suppression of transcription [26-29].
PIWI and other proteins of the piRNA system of drosophila have also been shown to participate in formation of heterochromatin in somatic tissues outside the gonads and also in the silencing by Polycomb group proteins [30-34]. However, mutations in drosophila piRNA pathway genes lead to sterility and defects in the germinal cells but do not cause pronounced defects in development of the somatic tissues.
piRNAs are found in somatic stem cells and in various somatic tissues of different animals including drosophila and mammals [35-38]. Several cases of involvement of piRNAs in processes of gene regulation in somatic cells have been described, in particular, in serotonin-dependent methylation of promoter DNA of the gene CREB2 responsible for long-term memory formation in neurons of the mollusk Aplysia [39]. piRNAs were shown to contribute to methylation of DNA of genes encoding receptors in human immune killer cells, providing their variability [40].
PIWI proteins are also involved in the regulation of chromosome structures, in particular, the telomere cap, which plays a critical role during meiotic divisions. In drosophila, Piwi is thought to participate in the formation of the cap structure due to interaction with a specific class of small RNAs with length different from that of piRNAs suppressing transposons [41]. Zili, one of two PIWI proteins in Danio rerio, is required for meiosis that is not related to its function in transposon silencing [42]. In ciliated protozoa PIWI proteins are involved in developmental genome rearrangements. In these organisms the mechanism of PIWI-dependent chromatin repression is adapted for the elimination of “unnecessary” DNA sequences during the development of somatic nuclei [43].
Mutations in PIWI proteins in drosophila are associated with a number of abnormalities in germinal tissue development and result in sterility of females and in a partial or complete sterility of males [3, 16, 44, 45]. As follows from the above description of the multiplicity of PIWI protein functions, causes of these abnormalities can be different. Some developmental defects can be explained by activation of transposable element expression or by the role of these proteins during the formation of chromatin and chromosome structures. Besides, PIWI proteins can also have specific functions associated with stem cells.
PIWI AND SELF-RENEWAL OF GERMLINE STEM CELLS
Germline stem cells (GSC) are characterized by asymmetric divisions resulting in a new stem cell (self-renewal) and in a differentiating germ cell, which is a precursor of gametes [46, 47]. Oogenesis of drosophila has been studied for several decades as a model of GSC differentiation. The ovaries of drosophila consist of ovarioles, which are chains of egg chambers linked with the germarium – a zone containing germinal and somatic cells where oogenesis starts. GSCs are located in a special microenvironment, a niche formed by somatic supporting cells. Studies on interactions of these two cell types in drosophila became the basis for concepts of signaling pathways used by the niche cells to mountain the adjacent stem cells [47-49]. These fundamental principles are the same in drosophila and in mammals [50]. The niche cells maintain the stem cell phenotype and prevent their differentiation and also program asymmetry of the subsequent divisions of GSCs, providing the place inside the niche only for one of two daughter cells. The daughter cell adjacent to the niche cells obtains their signaling molecules, which inhibit the differentiation, and retains itself as a stem cell. The other daughter cell lacking the contact with the niche begins to differentiate to a cystoblast undergoing four mitoses, after which the resulting 16-cellular cyst forms an egg chamber and leaves the germarium [47-49].
Signaling ligands secreted by the niche cells for maintaining GSCs include Dpp (decapentaplegic) and Gbb (glass bottom boat) proteins. The ligands interact with receptors (in particular, with Thick Veins (TKV)) on the GSC surface that results in phosphorylation of a protein Mad (Smad) of the signaling pathway responsible for the transcriptional repression of the gene bag-of-marbles (bam) [51, 52]. In the cells that get outside the niche (precystoblasts) and receive less Dpp and Gbb ligands, active transcription of the bam gene begins (figure). The protein Bam produced in the cystoblasts promotes translation of mRNAs activating the differentiation and inhibits translation of mRNAs specific for undifferentiated GSCs [48]. The regulation of the bam gene through Dpp exemplifies the signaling pathway TGF-β (transforming growth factor beta), which is realized in various types of cells in both adult organisms and during embryogenesis.
Suggested functions of Piwi during the maintenance and division of germline stem cells (GSCs) in the ovaries of drosophila. Somatic cells of the niche prevent the differentiation of GSCs due to secretion of the dpp peptides, which activate the signaling pathway suppressing transcription of the bam gene. When a GSC divides, one of the daughter cells (the precystoblast) loses contact with the niche, stops to receive dpp, and differentiates into a cystoblast. The proteins Piwi and Yb located in the cytoplasm of the niche cells are also necessary for maintenance of GSCs, but not due to influence on dpp secretion. Piwi and Yb interact in Yb-bodies, where Piwi forms a complex with piRNA. Piwi–piRNA complexes cause silencing of transposable elements (TE) in nuclei of the somatic and germinal cells. The nuclear location of Piwi in GSCs is necessary for their divisions. Derepression of TE in the germline or somatic cells blocks differentiation of precystoblasts due to activation of the check-point system
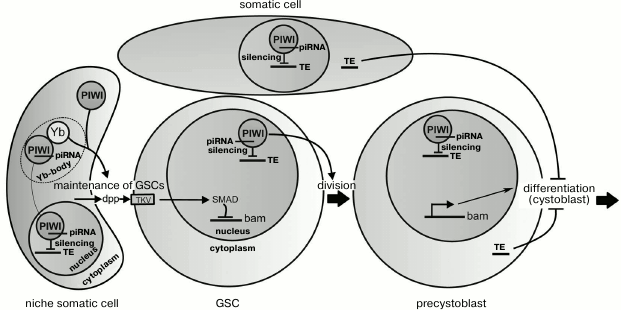
The piwi gene was first described to play a role in maintenance of GSCs in the testes and ovaries of Drosophila melanogaster [3]. Both female and male flies with mutations in the piwi gene lose GSCs because of their differentiation without self-renewal divisions [3, 45]. In ovaries, Piwi is present in both germinal and somatic cells [7, 53], whereas the maintenance of GSCs depends on its presence only in niche cells [53, 54], and its overexpression in these cells increases GSC number [53, 55]. Therefore, it was suggested that the Piwi protein is involved in the regulation of signals from the niche aimed to maintain the stem cells (figure). The underlying mechanism is not clear, but Piwi was shown to downregulate Bam expression in GSCs, similar to the ligand Dpp secreted by the niche [56]. It seems that Piwi does not influence the production of Dpp but is involved in formation of a signal of unknown nature also secreted by the niche cells [56]. It is suggested that this Piwi-dependent signal can suppress the ubiquitin ligase Smurf, which inhibits Dpp-signaling through acceleration of TKV receptor degradation [56, 57]. Inactivation of smurf results in an increase in GSC number [58], similarly to the effect of Piwi overexpression.
Two other Drosophila Piwi-family proteins, Aub and Ago3, have been found only in the germinal cells of ovaries [7] and are not involved in GSC maintenance in females [16, 44]. Interestingly, the Ago3 protein is required for GSC maintenance in males [16].
It seems unlikely that the loss of stem cells observed in the absence of Piwi is caused by derepression of transposons. Mutations affecting some components of the piRNA silencing system in the ovarian somatic cells fail to markedly influence GSC maintenance [27, 59, 60]. Moreover, the nuclear location of Piwi has been found to be necessary for the transposon silencing but not for GSC maintenance, which can involve cytoplasmic Piwi (figure) [27]. Thus, in the niche cells Piwi seems to have a specific molecular function in supporting the stem cells that is not directly associated with the piRNA-dependent regulation of chromatin.
The ability of cytoplasmic Piwi to maintain GSCs was an unexpected finding because Piwi is a mostly nuclear protein. However, by immunoprecipitation approaches the wild type Piwi has been found not only in the nucleus but also in cytoplasmic Yb-bodies, where the Piwi–piRNA complex becomes loaded that is suggested to be required for its delivery into the nucleus [61-64]. Yb-bodies are found in somatic cells of the ovaries, including the niche cells [64, 65]. The protein Yb, which is a key component of these bodies, is also needed for the maintenance of GSCs [55, 65, 66]. Mutations in the yb gene result in the same loss of GSCs as null-mutations of piwi, and Yb overexpression in somatic cells, similarly to Piwi, increases the number of GSCs. It can be suggested that formation of a signal aimed at the maintenance of GSCs depends on the interaction of Piwi and Yb in the cytoplasm of the niche cells (figure). Whether the integrity of Yb-bodies is important for GSC maintenance is still unknown.
Loss of the nuclease Zucchini (Zuc) and the protein Vreteno (Vret) involved in primary processing of piRNAs [63, 67, 68] abolishes the transport of unloaded Piwi into the nuclei of somatic cells, leading to its accumulation in Yb-bodies [61, 62]. Mutations in zuc and vret do not lead to defects in GSC maintenance [27, 60, 69] therefore indicating that this process does not depend on piRNAs. However, damage of the PAZ domain responsible for Piwi binding with piRNAs leads to the loss of GSCs [70], but this can be caused by inability of the Piwi mutant to interact with other proteins.
What can be the mechanism behind piRNA-independent functioning of Piwi in GSC maintenance? Several recent works describe examples of the involvement of PIWI subfamily proteins in signaling pathways through protein–protein interactions, which likely do not depend on small RNAs. The nuclear protein Zili (a PIWI ortholog in Danio rerio) suppresses the TGF-β pathway specifically binding protein Smad4 and preventing formation of its complex with other proteins of the Smad group. This function of Zili is required for mesoderm formation during embryogenesis [71]. In human embryonic kidney cells, a PIWI subfamily protein Hili also suppresses the TGF-β signaling pathway, but in this case Hili physically interacts with the chaperone Hsp90 preventing its association with a receptor that is ubiquitinated and degraded [72]. Note that in drosophila embryos Piwi protein has been also detected in complex with Hsp90 [73]. An example of the RNA silencing-independent functioning of the drosophila Piwi and Aub proteins is their role in formation of the pole plasm, a specialized cytoplasmic region of embryo where germ cells are formed. It is suggested that during this process Piwi and Aub interact in the oocyte cytoplasm with other components of the pole plasm, in particular with the Vasa protein [44, 74].
ROLE OF PIWI PROTEINS IN PROLIFERATION AND DIFFERENTIATION OF
STEM CELLS
In addition to GSC maintenance, the drosophila Piwi protein is also required for normal rate of stem cell divisions that depends on Piwi located within GSCs and not within niche cells [53, 54]. Interestingly, cytoplasmic Piwi is unable to provide the normal level of GSC proliferation (Yakushev and Klenov, unpublished data). This finding suggests that Piwi-dependent nuclear silencing of transposons can be necessary for cell divisions, as discriminated from the role of cytoplasmic Piwi in GSC maintenance (figure). Stem cell divisions have been shown to be blocked in the absence of some proteins of the piRNA-silencing system [75] and also during P-M hybrid dysgenesis – transposon mobilization in the offspring of crosses between certain drosophila strains [10]. Transpositions of mobile elements in the stem cells probably induce DNA breaks. Damage in DNA can be recognized by the check-point system of ATR and Chk2 kinases, which trigger a signaling pathway arresting the cell cycle that is necessary to provide the time required for DNA repair [76].
The proteins Vasa and Aub of the piRNA system are also suggested to associate directly with mitotic chromosomes and promote their condensation due to recruiting of condensin components during GSC divisions [75].
Accumulation of undifferentiated GSCs in the germaria is another phenotypic manifestation of mutations in some drosophila piRNA pathway genes [59, 60]. This developmental arrest can also be attributed to the activation of transposons and the check-point system (figure), because the GSC differentiation is delayed during hybrid dysgenesis, and this defect is suppressed by mutation of the check-point kinase chk2 [59]. It is interesting that mutations leading to absence of piRNAs in somatic cells of the ovaries are also accompanied by the block of germinal cell differentiation [59, 60]. These results can be explained by the ability of transposon transcripts to be transported from the ovarian somatic cells to the germinal ones, where they can insert into the genome inducing DNA breaks and activating check-point signaling pathway. This model needs to be tested experimentally.
Active expression of PIWI proteins has been shown in various cancer cells, and the level of their expression correlates with the proliferation rate of tumors, including those in humans [77-80]. This is consistent with the idea that cancer cells for their survival can express proteins characteristic for normal stem cells. Drosophila brain tumors caused by the l(3)mbt gene mutation express several proteins specific for the germinal cells including Piwi and Aub, and inactivation of piwi and aub genes suppressed the tumor growth [81]. It is possible that PIWI functioning in the tumor proliferation could be associated with transposon repression, as it occurs during the division of germinal cells. The protein Hiwi (a human PIWI protein) has recently been shown to promote the proliferation of sarcoma cells by directing DNA methylation [82]. Thus, sarcoma cells use the gene silencing mechanism, similar to that used by PIWI—piRNA complexes in the germinal cells of mammals for repression of transposon transcription [20, 21]. It is known that an activity of transposable elements can promote malignization by inducing certain mutations [83]. However, unrestrained transposon activation may reduce tumor cell viability, in particular, the proliferation of cancer stem cells. Possibly, some tumors can use an increased level of PIWI proteins as a defense mechanism against hyperexpression of transposable elements.
In this review we have focused mainly on studies performed on drosophila because they reveal complicated interrelations of different functions of PIWI proteins and the piRNA system with processes responsible for the maintenance, division, and differentiation of germline stem cells. Mechanisms of PIWI protein functioning that are not related to transposon silencing, as well as elucidation of the role of these proteins in cell proliferation, seem to be especially interesting for further studies.
This work was supported by the Russian Foundation for Basic Research (project No. 13-04-02156A) and by the Russian Academy of Sciences Presidium programs “Molecular and Cellular Biology” and “Basic Sciences – to Medicine”.
REFERENCES
1.Malone, C. D., and Hannon, G. J. (2009)
Cell, 136, 656-668.
2.Siomi, M. C., Sato, K., Pezic, D., and Aravin, A.
A. (2011) Nat. Rev. Mol. Cell. Biol., 12, 246-258.
3.Lin, H., and Spradling, A. C. (1997)
Development, 124, 2463-2476.
4.Juliano, C., Wang, J., and Lin, H. (2011) Annu.
Rev. Genet., 45, 447-469.
5.Goodier, J. L., and Kazazian, H. H., Jr. (2008)
Cell, 135, 23-35.
6.Olovnikov, I. A., and Kalmykova, A. I. (2013)
Biochemistry (Moscow), 78, 572-584.
7.Brennecke, J., Aravin, A. A., Stark, A., Dus, M.,
Kellis, M., Sachidanandam, R., and Hannon, G. J. (2007) Cell,
128, 1089-1103.
8.Vagin, V. V., Sigova, A., Li, C., Seitz, H.,
Gvozdev, V., and Zamore, P. D. (2006) Science, 313,
320-324.
9.Muerdter, F., Olovnikov, I., Molaro, A., Rozhkov,
N. V., Czech, B., Gordon, A., Hannon, G. J., and Aravin, A. A. (2012)
RNA, 18, 42-52.
10.Khurana, J. S., Wang, J., Xu, J., Koppetsch, B.
S., Thomson, T. C., Nowosielska, A., Li, C., Zamore, P. D., Weng, Z.,
and Theurkauf, W. E. (2011) Cell, 147, 1551-1563.
11.De Vanssay, A., Bouge, A. L., Boivin, A.,
Hermant, C., Teysset, L., Delmarre, V., Antoniewski, C., and Ronsseray,
S. (2012) Nature, 490, 112-115.
12.Klattenhoff, C., Xi, H., Li, C., Lee, S., Xu, J.,
Khurana, J. S., Zhang, F., Schultz, N., Koppetsch, B. S., Nowosielska,
A., Seitz, H., Zamore, P. D., Weng, Z., and Theurkauf, W. E. (2009)
Cell, 138, 1137-1149.
13.Pane, A., Jiang, P., Zhao, D. Y., Singh, M., and
Schupbach, T. (2011) EMBO J., 30, 4601-4615.
14.Gunawardane, L. S., Saito, K., Nishida, K. M.,
Miyoshi, K., Kawamura, Y., Nagami, T., Siomi, H., and Siomi, M. C.
(2007) Science, 315, 1587-1590.
15.Malone, C. D., Brennecke, J., Dus, M., Stark, A.,
McCombie, W. R., Sachidanandam, R., and Hannon, G. J. (2009)
Cell, 137, 522-535.
16.Li, C., Vagin, V. V., Lee, S., Xu, J., Ma, S.,
Xi, H., Seitz, H., Horwich, M. D., Syrzycha, M., Honda, B. M., Kittler,
E. L., Zapp, M. L., Klattenhoff, C., Schulz, N., Theurkauf, W. E.,
Weng, Z., and Zamore, P. D. (2009) Cell, 137,
509-521.
17.Guzzardo, P. M., Muerdter, F., and Hannon, G. J.
(2013) Curr. Opin. Genet. Dev., Epub ahead of print.
18.Lim, A. K., Tao, L., and Kai, T. (2009) J.
Cell. Biol., 186, 333-342.
19.Castel, S. E., and Martienssen, R. A. (2013)
Nat. Rev. Genet., 14, 100-112.
20.Aravin, A. A., Sachidanandam, R.,
Bourc’his, D., Schaefer, C., Pezic, D., Toth, K. F., Bestor, T.,
and Hannon, G. J. (2008) Mol. Cell, 31, 785-799.
21.Kuramochi-Miyagawa, S., Watanabe, T., Gotoh, K.,
Totoki, Y., Toyoda, A., Ikawa, M., Asada, N., Kojima, K., Yamaguchi,
Y., Ijiri, T. W., Hata, K., Li, E., Matsuda, Y., Kimura, T., Okabe, M.,
Sakaki, Y., Sasaki, H., and Nakano, T. (2008) Genes Dev.,
22, 908-917.
22.Bortvin, A. (2013) Biochemistry
(Moscow), 78, 592-602.
23.Watanabe, T., Tomizawa, S., Mitsuya, K., Totoki,
Y., Yamamoto, Y., Kuramochi-Miyagawa, S., Iida, N., Hoki, Y., Murphy,
P. J., Toyoda, A., Gotoh, K., Hiura, H., Arima, T., Fujiyama, A., Sado,
T., Shibata, T., Nakano, T., Lin, H., Ichiyanagi, K., Soloway, P. D.,
and Sasaki, H. (2011) Science, 332, 848-852.
24.Lee, H. C., Gu, W., Shirayama, M., Youngman, E.,
Conte, D., Jr., and Mello, C. C. (2012) Cell, 150,
78-87.
25.Shirayama, M., Seth, M., Lee, H. C., Gu, W.,
Ishidate, T., Conte, D., Jr., and Mello, C. C. (2012) Cell,
150, 65-77.
26.Klenov, M. S., Lavrov, S. A., Stolyarenko, A. D.,
Ryazansky, S. S., Aravin, A. A., Tuschl, T., and Gvozdev, V. A. (2007)
Nucleic Acids Res., 35, 5430-5438.
27.Klenov, M. S., Sokolova, O. A., Yakushev, E. Y.,
Stolyarenko, A. D., Mikhaleva, E. A., Lavrov, S. A., and Gvozdev, V. A.
(2011) Proc. Natl. Acad. Sci. USA, 108, 18760-18765.
28.Shpiz, S., Olovnikov, I., Sergeeva, A., Lavrov,
S., Abramov, Y., Savitsky, M., and Kalmykova, A. (2011) Nucleic
Acids Res., 39, 8703-8711.
29.Wang, S. H., and Elgin, S. C. (2011) Proc.
Natl. Acad. Sci. USA, 108, 21164-21169.
30.Pal-Bhadra, M., Leibovitch, B. A., Gandhi, S. G.,
Rao, M., Bhadra, U., Birchler, J. A., and Elgin, S. C. (2004)
Science, 303, 669-672.
31.Brower-Toland, B., Findley, S. D., Jiang, L.,
Liu, L., Yin, H., Dus, M., Zhou, P., Elgin, S. C., and Lin, H. (2007)
Genes Dev., 21, 2300-2311.
32.Yin, H., and Lin, H. (2007) Nature,
450, 304-308.
33.Grimaud, C., Bantignies, F., Pal-Bhadra, M.,
Ghana, P., Bhadra, U., and Cavalli, G. (2006) Cell, 124,
957-971.
34.Kavi, H. H., and Birchler, J. A. (2009)
Epigenetics Chromatin, 2, 15.
35.Ro, S., Park, C., Song, R., Nguyen, D., Jin, J.,
Sanders, K. M., McCarrey, J. R., and Yan, W. (2007) RNA,
13, 1693-1702.
36.Ghildiyal, M., Seitz, H., Horwich, M. D., Li, C.,
Du, T., Lee, S., Xu, J., Kittler, E. L., Zapp, M. L., Weng, Z., and
Zamore, P. D. (2008) Science, 320, 1077-1081.
37.Lee, E. J., Banerjee, S., Zhou, H.,
Jammalamadaka, A., Arcila, M., Manjunath, B. S., and Kosik, K. S.
(2011) RNA, 17, 1090-1099.
38.Yan, Z., Hu, H. Y., Jiang, X., Maierhofer, V.,
Neb, E., He, L., Hu, Y., Hu, H., Li, N., Chen, W., and Khaitovich, P.
(2011) Nucleic Acids Res., 39, 6596-6607.
39.Rajasethupathy, P., Antonov, I., Sheridan, R.,
Frey, S., Sander, C., Tuschl, T., and Kandel, E. R. (2012) Cell,
149, 693-707.
40.Cichocki, F., Lenvik, T., Sharma, N., Yun, G.,
Anderson, S. K., and Miller, J. S. (2010) J. Immunol.,
185, 2009-2012.
41.Khurana, J. S., Xu, J., Weng, Z., and Theurkauf,
W. E. (2010) PLoS Genet., 6, e1001246.
42.Houwing, S., Berezikov, E., and Ketting, R. F.
(2008) EMBO J., 27, 2702-2711.
43.Chalker, D. L., and Yao, M. (2011) Annu. Rev.
Genet., 45, 227-246.
44.Harris, A. N., and Macdonald, P. M. (2001)
Development, 128, 2823-2832.
45.Cox, D. N., Chao, A., Baker, J., Chang, L., Qiao,
D., and Lin, H. (1998) Genes Dev., 12, 3715-3727.
46.Kimble, J. (2011) Cold Spring Harb. Perspect.
Biol., 3, a002683.
47.Spradling, A. C., Fuller, M. T., Braun, R. E.,
and Yoshida, S. (2011) Cold Spring Harb. Perspect. Biol.,
3, a002642.
48.Kirilly, D., and Xie, T. (2007) Cell Res.,
17, 15-25.
49.Xie, T., Song, X., Jin, Z., Pan, L., Weng, C.,
Chen, S., and Zhang, N. (2008) Cold Spring Harb. Perspect.
Biol., 73, 39-47.
50.Morrison, S. J., and Spradling, A. S. (2008)
Cell, 132, 598-611.
51.Xie, T., and Spradling, A. C. (1998) Cell,
94, 251-260.
52.Chen, D., and McKearin, D. (2003) Curr.
Biol., 13, 1786-1791.
53.Cox, D. N., Chao, A., and Lin, H. (2000)
Development, 127, 503-514.
54.Szakmary, A., Cox, D. N., Wang, Z., and Lin, H.
(2005) Curr. Biol., 15, 171-178.
55.King, F. J., Szakmary, A., Cox, D. N., and Lin,
H. (2001) Mol. Cell., 7, 497-508.
56.Chen, D., and McKearin, D. (2005) Curr.
Biol., 15, 179-184.
57.Xia, L., Jia, S., Huang, S., Wang, H., Zhu, Y.,
Mu, Y., Kan, L., Zheng, W., Wu, D., Li, X., Sun, Q., Meng, A., and
Chen, D. (2010) Cell, 143, 978-990.
58.Casanueva, M. O., and Ferguson, E. L. (2004)
Development, 131, 1881-1890.
59.Rangan, P., Malone, C. D., Navarro, C., Newbold,
S. P., Hayes, P. S., Sachidanandam, R., Hannon, G. J., and Lehmann, R.
(2011) Curr. Biol., 21, 1373-1379.
60.Zamparini, A. L., Davis, M. Y., Malone, C. D,
Vieira, E., Zavadil, J., Sachidanandam, R., Hannon, G. J., and Lehmann,
R. (2011) Development, 138, 4039-4050.
61.Olivieri, D., Sykora, M. M., Sachidanandam, R.,
Mechtler, K., and Brennecke, J. (2010) EMBO J., 29,
3301-3317.
62.Saito, K., Ishizu, H., Komai, M., Kotani, H.,
Kawamura, Y., Nishida, K. M., Siomi, H., and Siomi, M. C. (2010)
Genes Dev., 24, 2493-2498.
63.Handler, D., Olivieri, D., Novatchkova, M.,
Gruber, F. S., Meixner, K., Mechtler, K., Stark, A., Sachidanandam, R.,
and Brennecke, J. (2011) EMBO J., 30, 3977-3993.
64.Qi, H., Watanabe, T., Ku, H. Y., Liu, N., Zhong,
M., and Lin, H. (2011) J. Biol. Chem., 286,
3789-3797.
65.Szakmary, A., Reedy, M., Qi, H., and Lin, H.
(2009) J. Cell Biol., 185, 613-627.
66.King, F. J., and Lin, H. (1999)
Development, 126, 1833-1844.
67.Ipsaro, J. J., Haase, A. D., Knott, S. R.,
Joshua-Tor, L., and Hannon, G. J. (2012) Nature, 491,
279-283.
68.Nishimasu, H., Ishizu, H., Saito, K., Fukuhara,
S., Kamatani, M. K., Bonnefond, L., Matsumoto, N., Nishizawa, T.,
Nakanaga, K., Aoki, J., Ishitani, R., Siomi, H., Siomi, M. C., and
Nureki, O. (2012) Nature, 491, 284-287.
69.Pane, A., Wehr, K., and Schupbach, T. (2007)
Dev. Cell, 12, 851-862.
70.Le Thomas, A., Rogers, A. K., Webster, A.,
Marinov, G. K., Liao, S. E., Perkins, E. M., Hur, J. K., Aravin, A. A.,
and Toth, K. F. (2013) Genes Dev., 27, 390-399.
71.Sun, H., Li, D., Chen, S., Liu, Y., Liao, X.,
Deng, W., Li, N., Zeng, M., Tao, D., and Ma, Y. (2010) J. Biol.
Chem., 285, 4243-4250.
72.Zhang, K., Lu, Y., Yang, P., Li, C., Sun, H.,
Tao, D., Liu, Y., Zhang, S., and Ma, Y. (2012) PLoS One,
7, e41973.
73.Gangaraju, V. K., Yin, H., Weiner, M. M., Wang,
J., Huang, X. A., and Lin, H. (2011) Nat. Genet., 43,
153-158.
74.Megosh, H. B., Cox, D. N., Campbell, C., and Lin,
H. (2006) Curr. Biol., 16, 1884-1894.
75.Pek, J. W., and Kai, T. (2011) Curr.
Biol., 21, 39-44.
76.Lazzaro, F., Giannattasio, M., Puddu, F.,
Granata, M., Pellicioli, A., Plevani, P., and Muzi-Falconi, M. (2009)
DNA Repair (Amst.), 8, 1055-1067.
77.Chen, L., Shen, R., Ye, Y., Pu, X. A., Liu, X.,
Duan, W., Wen, J., Zimmerer, J., Wang, Y., Liu, Y., Lasky, L. C.,
Heerema, N. A., Perrotti, D., Ozato, K., Kuramochi-Miyagawa, S.,
Nakano, T., Yates, A. J., Carson, W. E., 3rd, Lin, H., Barsky, S. H.,
and Gao, J. X. (2007) PLoS ONE, 2, e293.
78.Lee, J. H., Schutte, D., Wulf, G., Fuzesi, L.,
Radzun, H. J., Schweyer, S., Engel, W., and Nayernia, K. (2006) Hum.
Mol. Genet., 15, 201-211.
79.Liu, X., Sun, Y., Guo, J., Ma, H., Li, J., Dong,
B., Jin, G., Zhang, J., Wu, J., Meng, L., and Shou, C. (2006) Int.
J. Cancer, 118, 1922-1929.
80.Suzuki, R., Honda, S., and Kirino, Y. (2012)
Front. Genet., 3, 204.
81.Janic, A., Mendizabal, L., Llamazares, S.,
Rossell, D., and Gonzalez, C. (2010) Science, 330,
1824-1827.
82.Siddiqi, S., Terry, M., and Matushansky, I.
(2012) PloS ONE, 7, e33711.
83.Chenais, B. (2013) Biochim. Biophys. Acta,
1835, 28-35.