Assay of Insulator Enhancer-Blocking Activity with the Use of Transient Transfection
N. A. Smirnov, D. A. Didych, S. B. Akopov*, L. G. Nikolaev, and E. D. Sverdlov
Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: (495) 330-6538; E-mail: akser@ibch.ru* To whom correspondence should be addressed.
Received March 25, 2013
We used a transient transfection of cultured cells with linearized plasmids to analyze the enhancer-blocking activity of potential insulators including the standard cHS4 chicken beta-globin insulator and several DNA fragments selected from the human genome sequence. About 60-80% of the potential insulators do reveal the enhancer-blocking activity when probed by the transient transfection assay. The activity of different sequences is characterized by certain tissue specificity and by dependence on the orientation of the fragments relative to the promoter. Thus, the transfection model may be used for quantitative analysis of the enhancer-blocking activity of the potential insulators.
KEY WORDS: human genome, gene expression regulation, insulator, enhancerDOI: 10.1134/S0006297913080051
Insulators are DNA sequences that prevent activation of promoters by inappropriate enhancers and/or block the spread of condensed chromatin (for a recent review see [1]). Insulators have been identified in various eukaryotic organisms, including vertebrates, Drosophila, sea urchin, and yeast [2-6]. Some insulators can function when transferred into phylogenetically distant organisms, like sea urchin, plants, and human [5, 7], and can interfere with heterologous enhancers [7, 8].
The activity of different insulators can depend [2, 9, 10] or not depend [11-13] on their orientation relative to the cognate promoters. If more than one insulator is located between enhancer and promoter, their combined enhancer-blocking activity can be much lower than that of a single insulator [14-16]. This neutralization effect can depend on the orientation of insulators relative to each other [17]. Consequently, insulators may not just subdivide a genome into domains but rather form, in cooperation with genes, promoters, enhancers, and other elements, a multilevel network regulating the transcriptional activity of the genome.
Functioning of many insulators depends on binding of transcription factor CTCF (reviewed in [18, 19]). However, it would be incorrect to consider all CTCF-binding sequences as insulators.
Quantitative characteristics of the insulator activity are not sufficiently studied. In most cases the activity of insulator is estimated by its ability to block enhancer–promoter interactions (enhancer-blocking insulators). There exist just a few general methods to analyze insulator activity and several methods appropriate only for specific organisms, like the assay developed by Kellum and Schedl [20] and other similar assays, which are applicable only for Drosophila [21-23].
Among general methods, the most frequently used are the following: 1) the approach of Chung et al. [3], which allows for the quantitation of the insulator activity. This approach is based on insulation of the NeoR gene by flanking sequences, which leads to death of cells whose genome contains constructs with insulators; 2) the negative–positive selection approach [24]. This approach, in contrast to the previous method, is based on survival of the insulator-containing cells, so their DNA can be isolated and analyzed. It also allows for the quantitation of the insulator activity [13]; 3) the transient transfection assay using constructs expressing the luciferase or GFP reporter genes and containing insulator between enhancer and promoter. Comparison of expression of the reporter gene by this construction and similar construction containing the same insulator positioned outside of the promoter–enhancer pair allows for estimation of the insulator’s enhancer-blocking activity. To avoid interaction of enhancer and promoter along the circular plasmid, these elements are either separated by two potential insulators [25] or the measurements are performed using linearized plasmid [26].
This last method is the simplest and fastest of all mentioned. However, it is also the most artificial and is not allowed for chromatin structure and genome neighborhood on the regulatory elements. Despite this, it is widely used for evaluation of the insulator activity (for recent examples see [27-29]). Here we used the linearized plasmid transfection approach to check for enhancer-blocking and other activities of several potential insulators identified by different methods.
MATERIALS AND METHODS
Basic protocols. Growth and transformation of E. coli cells, preparation of plasmid DNA, agarose gel electrophoresis, and other standard manipulations were performed as described [30].
Cell culture. HeLa (human epithelial cervical carcinoma), HepG2 (human hepatocellular carcinoma), and CHO (Chinese hamster ovary) cells were grown at 37°C and 5% CO2 in DMEM/F12 (1 : 1) medium containing 10% fetal calf serum.
Plasmid constructions. To construct the pGL4PV plasmid containing the luciferase gene under control of SV40 minimal promoter, the 215-bp promoter fragment was cut out from pGL3-Promoter Vector (Promega, USA) with XhoI and HindIII and inserted into pGL4.10 plasmid (Promega) cut with the same enzymes.
Plasmid pGL4EPV, containing SV40 minimal promoter and enhancer, was made from the pGL4PV plasmid by digestion of the latter with XbaI and BamHI and ligation with the enhancer fragment obtained from pGL3-Control Vector (Promega) by digestion with XbaI and BamHI. pGL4EPV was used for cloning of the potential insulators into the unique SalI site located outside of the promoter–enhancer pair.
To obtain the pGL4EPV2 plasmid, the pGL4PV plasmid was linearized by SalI digestion, and the resulting ends were filled-in with Klenow enzyme. Next, the 353-bp enhancer fragment was cut out from pGL3-Control Vector with HpaI and BamHI, blunt-ended with Klenow enzyme, purified by electrophoresis in 1.5% agarose gel, and ligated with the linearized pGL4PV. The pGL4EPV2 plasmid was used for cloning of the potential insulators into the unique BamHI site located between SV40 promoter and enhancer.
Constructions for enhancer activity assay. Six DNA fragments containing potential enhancers and five fragments containing potential insulators were selected from the human genome (see “Results and Discussion” for detail), PCR-amplified using human genome DNA as a template (structure of the primers are presented in Table 1), and cloned into pGEM-T plasmid (Promega). The fragments were next cut out from the plasmids with PstI and Cfr42I (isoschizomer of SacII) and, after filling-in of the sticky ends, ligated into pGL4PV cut with SalI and treated with Klenow enzyme.
Constructions for enhancer-blocking assay. As a standard enhancer-blocking element, we employed the well-characterized cHS4 insulator from chicken β-globin locus [31]. The pJC13-1 plasmid (kindly provided by Gaelle M. Lefevre and Gary Felsenfeld, NIDDK, National Institutes of Health, USA) was digested with KpnI, treated with Klenow enzyme to fill-in the ends, and 1.2-kb fragment was purified by electrophoresis in 1.5% agarose gel. The fragment was cloned in both orientations into pGL4EPV and pGL4EPV2 previously cut with correspondingly SalI and BamHI and treated with Klenow enzyme to fill-in the ends. Orientation of the insert relative to SV40 promoter was determined by PCR-amplification with the insert-specific primer and primer complementary to the plasmid sequence.
Phage λ fragment PCR-amplified using phage genome DNA template and primers TCCGTGAGGTGAATGTGGTG and TAGTCGGCTCAACGTGGGTT was used as a negative control. The fragment was first ligated into pGEM-T Easy vector (Promega), cut out with EcoRI, and made blunt-ended with Klenow enzyme. Next, the insert (~200 bp) was purified by electrophoresis in 1.5% agarose gel and ligated into pGL4EPV and pGL4EPV2 plasmids as described above for the cHS4 insulator.
The potential insulators, which we identified earlier and cloned in pGEM-T Easy vector [24, 32, 33], were cut out with SalI (its recognition site located in primer sequence flanking the insert [24]), isolated, and cloned into pGL4EPV and pGL4EPV2 plasmids in both orientations, as described above for the cHS4 insulator.
Before transfection, the plasmids were linearized with PstI and purified from the traces of non-digested DNA in agarose gel.
Transfection and luciferase activity measurement. Transfection was performed in 24-well plates using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s protocol. For transfection, 200 ng of linearized plasmid DNA and 75 ng of reference pRL-TK plasmid (Promega) were added to each well. After 48-h incubation, the cells were lysed in PLB buffer (Promega), and the activity of Photinus pyralis and Renilla reniformis luciferases was assessed using a GENios Pro luminometer (Tecan) and Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase activity was normalized to that of the Renilla luciferase. Three or four independent transfections with two parallel measurements of each luciferase activity were carried out, and the standard error of the mean was calculated.
RESULTS AND DISCUSSION
Selection of DNA fragments. Based on the ENCODE project ChIP-seq data as presented in the UCSC Human Genome Browser [34], we selected six characteristic DNA fragments with potential enhancer and five fragments with potential insulator properties. The selection was made based on the ability of the fragments to bind transcription factors p300 and CTCF, their chromatin DNase I hypersensitivity, and on the state of their chromatin structure determined by nine epigenetic markers including CTCF binding and different types of histone modification [35, 36]. The fragments defined in [35, 36] as “strong enhancers” and “insulators” were selected. Positions in the human genome and properties of the selected fragments are summarized in Table 1.
Table 1. Properties of genomic fragments
selected by their functional characteristics
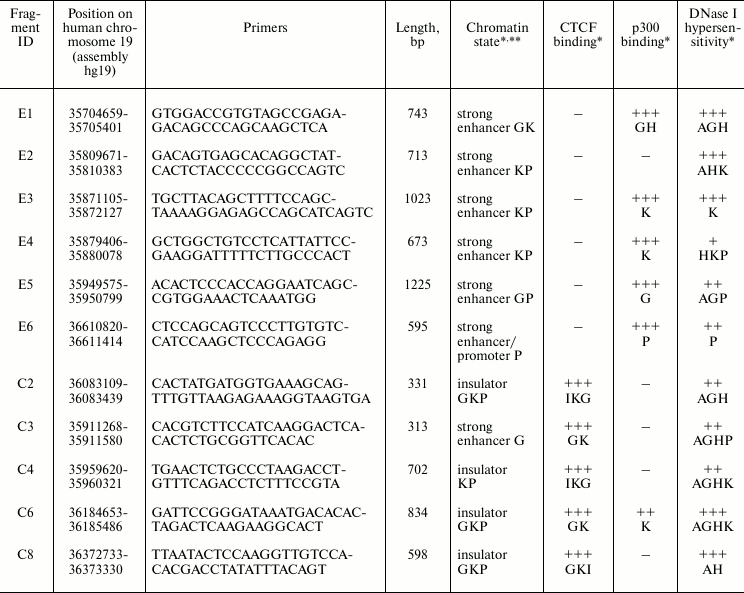
* Letters in the corresponding cells indicate the cell lines where this
activity was detected: H – HeLa, G – GM12878, P –
HepG2, K – K562, I – IMR90, A – A549. The binding
efficiency was estimated basing on the ENCODE ChIP-seq data [40]: from (–) – no binding to (+++)
– strong binding.
** Chromatin state was defined by nine epigenetic markers, including
binding of CTCF and different types of histone modifications. The
fragments referred by the authors of [35, 36] as “strong enhancer” or
“insulator” types were selected.
In addition, we selected four DNA fragments previously characterized experimentally as enhancer-blockers by means of positive–negative selection assay [24, 33]. The properties of these fragments are summarized in Table 2.
Table 2. Properties of potential insulators
identified by positive–negative selection
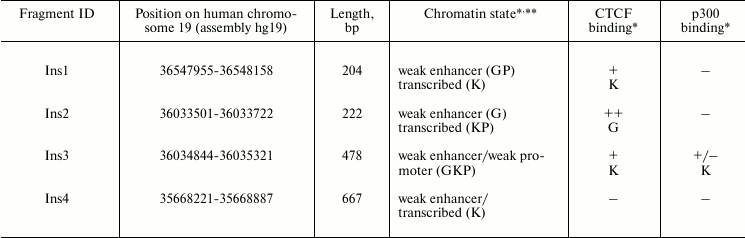
* Letters in the corresponding cells indicate the cell lines where this
activity was detected: H – HeLa, G – GM12878, P –
HepG2, K – K562, I – IMR90, A – A549. The binding
efficiency was estimated basing on the ENCODE ChIP-seq data [40]: from (–) – no binding to (+++)
– strong binding.
** Chromatin state was defined by nine epigenetic markers, including
binding of CTCF and different types of histone modifications [35, 36].
Enhancer activity of genome fragments. To test for their enhancer activity, the DNA fragments selected from the human genome were PCR-amplified on the genome DNA template (primer structures are presented in Table 1), cloned into pGL4PV plasmid downstream to the luciferase reporter gene (Fig. 1), and transfected into HeLa and HepG2 cells. This transfection was performed using circular plasmids, and all other transfections with linearized plasmids.
Fig. 1. Enhancer activity in HeLa and HepG2 cells of DNA fragments selected from the human genome. The fragments were selected as a potential enhancer (E1-E6) or potential insulators (C2-C8) (for their properties, see Table 1). The selection was made according to the criteria indicated in the “Results and Discussion” section. The DNA fragments were cloned into pGL4PV plasmid as shown in the figure (top). Circular plasmids were used for the luciferase activity assay. Control plasmids: PV – pGL4PV (contains SV40 promoter and no enhancer); EPV – pGL4EPV (contains SV40 promoter and enhancer). The activity was normalized to that of pGL4PV.
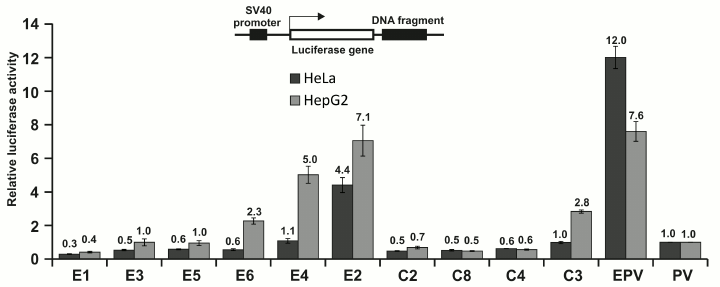
Of six fragments selected as potential enhancers (E1-E6, see Fig. 1 and Table 1), three were able to activate SV40 promoter under the conditions used in HepG2 cells, and only one (E2) was active also in HeLa cells. Of five fragments selected as potential insulators (CTCF binders), one (C3) demonstrated relatively low enhancer activity in HepG2 cells. Other fragments, as expected, were not active as enhancers. It should be noted that fragment C3, except for binding CTCF, had also properties of potential enhancer (see Table 1).
Therefore, four out of seven (60%) of the potential enhancers demonstrated the expected activity in the system employed in this study, indicating significant (but not complete) correlation between the properties used for their selection and their enhancer activity.
Enhancer-blocking activity of genome fragments. To check for the ability of the selected DNA fragments to block the promoter activation by nearby enhancer, we cloned these fragments between SV40 promoter and enhancer. Control constructions containing the same fragments downstream from the enhancer (outside of the promoter–enhancer pair, schematically presented in Fig. 2a) allowed assaying silencer activity of the fragments, which could interfere with the enhancer-blocking activity assay. The constructed plasmids were linearized, transfected into mammalian cells, and luciferase reporter gene activity was assessed using the Dual-Luciferase Reporter Assay System (Promega).
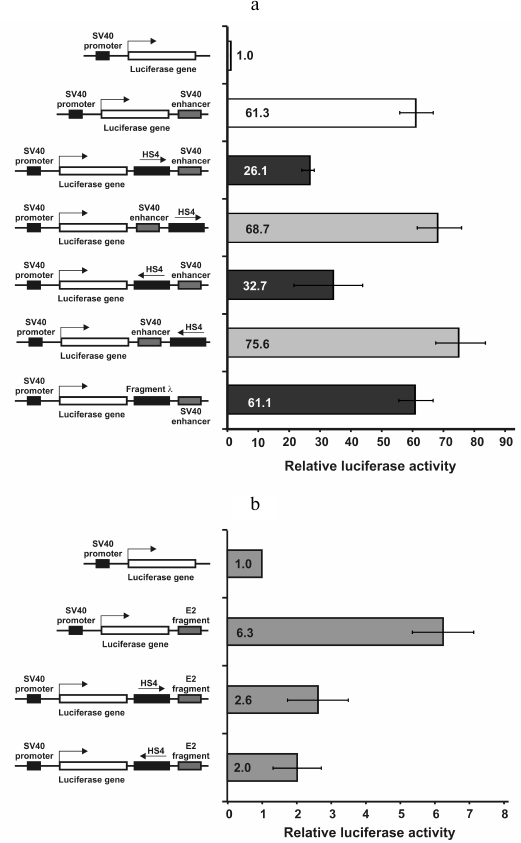
Fig. 2. Enhancer-blocking activity in HeLa cells of DNA fragments from the human genome. a) The DNA fragments were cloned either into the pGL4EPV2 plasmid between enhancer and promoter (dark columns) or into pGL4EPV plasmid outside of the promoter–enhancer pair (light columns) in both orientations. “+”, orientation of the fragment coincide with its orientation in the genome; “–”, orientation of the fragment is opposite to its orientation in the genome. Linearized plasmids PV and EPV were used for the transfection (see legend to Fig. 1). The luciferase activity was normalized to that of the pGL4EPV. b) Alignment of the CTCF binding sites found in the studied DNA fragments. In the right column, the extents of the enhancer blocking by the corresponding fragments in both orientations relative to promoter are shown. Nucleotides that coincide with the consensus sequence (shown as a Logo) are underlined.
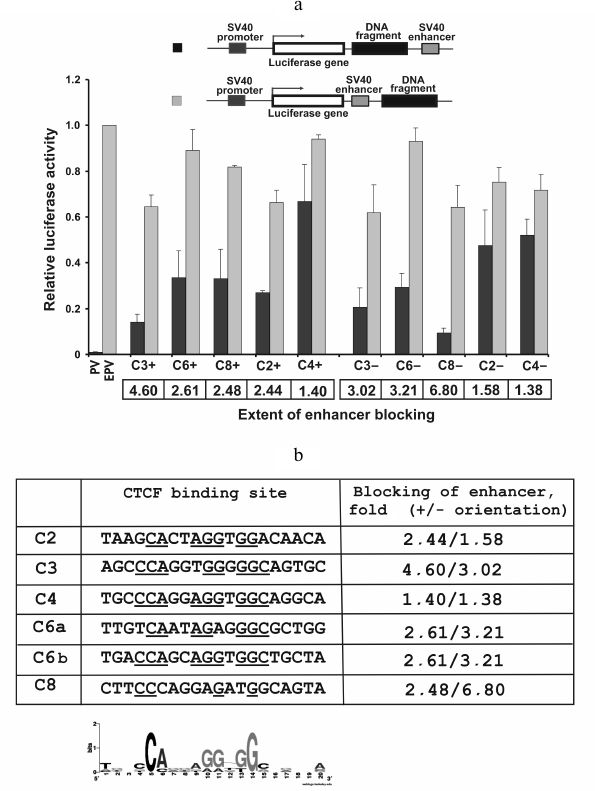
Four out of five (80%) genome fragments selected as potential insulators (see Table 1) showed evident ability to reduce the extent of activation of SV40 promoter by SV40 enhancer in HeLa cells only when placed between these elements (Fig. 2a). A possible exception is fragment C4, the enhancer-blocking activity of which was not detected despite strong binding of CTCF and chromatin state typical for insulators (Table 1). It was reported that CTCF binding is not necessary for a sequence to display enhancer-blocking activity [37, 38], whereas the reverse situation, when CTCF-binding sequence does not block promoter activation by enhancer, is not documented. However, the existence of this kind of sequences is not unlikely since CTCF protein has multiple functions not necessarily connected with blocking of enhancers (see [18] for review).
The extent of blocking of promoter activation by enhancer corrected for silencer activity of the fragment varies from 2.4-fold (fragment C2 in plus orientation) to almost 7-fold (fragment C8 in minus orientation). Besides, there are fragments whose enhancer-blocking activity depend on orientation relative to promoter (C8) and have practically no dependence on the orientation (C3, C6).
It is interesting that fragment C3, which was not identified as an insulator by its chromatin structure properties [35, 36], but able to strongly bind CTCF (Table 1), also displayed high enhancer-blocking activity.
Using the CTCF binding site database (http://insulatordb.uthsc.edu/storm.php), we identified within five selected DNA fragments the most probable 20 bp binding sites for CTCF. Fragment C6 contained two such sites (Fig. 2b). Using these sequences and the WebLogo server (http://weblogo.berkeley.edu/logo.cgi), we constructed the consensus sequence logo for CTCF binding qualitatively coinciding with a similar consensus constructed based on the whole genome data [39]. However, we found no correlation between the number of nucleotides that coincide with the consensus and enhancer-blocking activity of the DNA fragment. Thus, within sequence C8 with the highest enhancer-blocking efficiency, four nucleotides out of 20 was found to coincide with the consensus, whereas within sequence C4 with the lowest blocking efficiency there were nine such nucleotides. Most likely the enhancer-blocking efficiency is determined not only by CTCF binding, but also by other factors, e.g. by the ability to bind additional proteins.
Enhancer-blocking activity of cHS4 insulator from chicken β-globin locus. The full-length (1.2 kb) cHS4 chicken β-globin insulator [3, 31] is a typical CTCF-binding element that displays enhancer-blocking activity in different systems, including transient transfection assay with constructions containing SV40 promoter and enhancer [26].
We inserted the cHS4 insulator in both orientations both between SV40 enhancer and promoter and between promoter SV40 and enhancer E2 identified in this work (see above). In parallel, control constructs were prepared with the cHS4 insulator inserted outside of the promoter–enhancer pair and constructs not containing insulator. The plasmids were transfected into HeLa cells, and luciferase activity was measured (Fig. 3). As seen in Fig. 3a, the cHS4 insulator is able to reduce the activity of the SV40 promoter–enhancer pair 2-3-fold only when located between these elements. The enhancer-blocking effect was not observed when phage λ DNA fragment was cloned between promoter and enhancer. Similar results were obtained for the SV40 promoter–E2 enhancer pair. In both cases, the enhancer-blocking activity barely depended on the orientation of the insulator relative to the promoter (Fig. 3b).
Fig. 3. Enhancer-blocking activity of the cHS4 insulator from chicken β-globin locus with respect to SV40 promoter–SV40 enhancer (a) and SV40 promoter–E2 enhancer (b) combinations. The luciferase activity was normalized to that of pGL4PV.
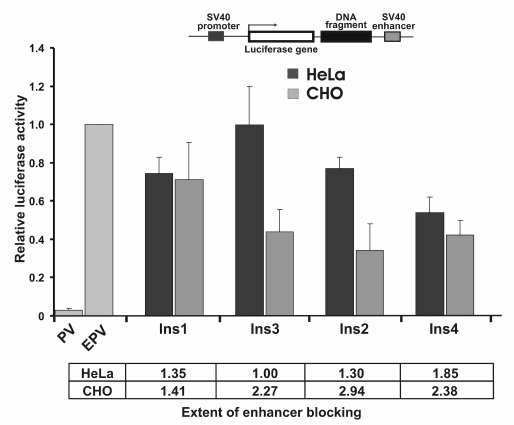
Enhancer-blocking activity of fragments identified by positive–negative selection. Four fragments identified earlier (see Table 2) were inserted into pGL4EPV2 plasmid between SV40 promoter and enhancer. Since the selection of the enhancer-blocking fragments was initially done in CHO cells, the constructions were linearized and transfected in CHO and, for comparison, into HeLa cells. The results are presented in Fig. 4. As seen, three out of four (75%) of the selected fragments significantly (2-3-fold) reduced the activity of the SV40 promoter–enhancer pair independent of their orientation relative to the promoter (not shown). Moreover, the enhancer-blocking activity of two elements (Ins2 and Ins3) was detected only in CHO cells and was practically absent in HeLa, while the activity of Ins4 was identical in these cell lines. Notably, the enhancer-blocking activity of the CTCF-binding elements Ins2 and Ins3 (Table 2) was dependent on cell type, whereas the activity of Ins4, which does not bind CTCF, was independent of cell type. However, more study is necessary to confirm this correlation.
Fig. 4. Enhancer-blocking activity of potential insulators identified in the human genome by positive–negative selection with respect to SV40 promoter–enhancer pair. Data for cell lines HeLa and CHO are shown. The luciferase activity was normalized to that of pGL4EPV.
The results obtained here show that the transient transfection of cultured cells with linearized plasmids allows revealing the enhancer-blocking activity of potential insulators including both the standard cHS4 chicken β-globin insulator and several DNA fragments selected from the human genome sequence. The activity of different sequences is characterized by certain tissue specificity and by dependence on orientation of the fragments relative to the promoter. Thus, the transfection model can be used for quantitative analysis of the enhancer-blocking activity of potential insulators. The enhancer-blocking activity of different sequences can be characterized by tissue-specificity and orientation-dependence relative to the promoter. We conclude that the system used is in many cases an adequate instrument for quantitative analysis of enhancer-blocking activity.
The authors are grateful to Gaelle M. Lefevre and Gary Felsenfeld (NIDDK, National Institutes of Health, USA) for gift of pJC13-1 plasmid containing cHS4 insulator.
This work was supported by the Scientific School program (project NSh 2395.2008.4), the Program of the Russian Academy of Sciences on Molecular and Cellular Biology, and by the Russian Foundation for Basic Research (project 10-04-01472).
REFERENCES
1.Yang, J., and Corces, V. G. (2011) Adv Cancer
Res., 110, 43-76.
2.Bi, X., and Broach, J. R. (1999) Genes Dev.,
13, 1089-1101.
3.Chung, J. H., Whiteley, M., and Felsenfeld, G.
(1993) Cell, 74, 505-514.
4.Melfi, R., Palla, F., Di Simone, P., Alessandro,
C., Cali, L., Anello, L., and Spinelli, G. (2000) J. Mol. Biol.,
304, 753-763.
5.Nagaya, S., Yoshida, K., Kato, K., Akasaka, K., and
Shinmyo, A. (2001) Mol. Genet. Genom., 265, 405-413.
6.Zhong, X. P., and Krangel, M. S. (1997) Proc.
Natl. Acad. Sci. USA, 94, 5219-5224.
7.Di Simone, P., Di Leonardo, A., Costanzo, G.,
Melfi, R., and Spinelli, G. (2001) Biochem. Biophys. Res.
Commun., 284, 987-992.
8.Verona, R. I., and Bartolomei, M. S. (2004)
Genomics, 84, 59-68.
9.Akasaka, K., Nishimura, A., Takata, K., Mitsunaga,
K., Mibuka, F., Ueda, H., Hirose, S., Tsutsui, K., and Shimada, H.
(1999) Cell Mol. Biol. (Noisy-le-grand), 45, 555-565.
10.Gruzdeva, N., Kyrchanova, O., Parshikov, A.,
Kullyev, A., and Georgiev, P. (2005) Mol. Cell Biol., 25,
3682-3689.
11.Belozerov, V. E., Majumder, P., Shen, P., and
Cai, H. N. (2003) EMBO J., 22, 3113-3121.
12.Yannaki, E., Tubb, J., Aker, M.,
Stamatoyannopoulos, G., and Emery, D. W. (2002) Mol. Ther.,
5, 589-598.
13.Didych, D. A., Kotova, E. S., Akopov, S. B.,
Nikolaev, L. G., and Sverdlov, E. D. (2012) BMC Res. Notes,
5, 178.
14.Kuhn, E. J., Viering, M. M., Rhodes, K. M., and
Geyer, P. K. (2003) EMBO J., 22, 2463-2471.
15.Maksimenko, O., Golovnin, A., and Georgiev, P.
(2008) Mol. Cell Biol., 28, 5469-5477.
16.Mongelard, F., and Corces, V. G. (2001) Nat.
Struct. Biol., 8, 192-194.
17.Kyrchanova, O., Toshchakov, S., Parshikov, A.,
and Georgiev, P. (2007) Mol. Cell Biol., 27,
3035-3043.
18.Nikolaev, L. G., Akopov, S. B., Didych, D. A.,
and Sverdlov, E. D. (2009) Curr. Genom., 10, 294-302.
19.Ohlsson, R., Bartkuhn, M., and Renkawitz, R.
(2010) Chromosoma, 119, 351-360.
20.Kellum, R., and Schedl, P. (1991) Cell,
64, 941-950.
21.Handoko, L., Xu, H., Li, G., Ngan, C. Y., Chew,
E., Schnapp, M., Lee, C. W., Ye, C., Ping, J. L., Mulawadi, F., et al.
(2011) Nat. Genet., 43, 630-638.
22.Raab, J. R., and Kamakaka, R. T. (2010) Nat.
Rev. Genet., 11, 439-446.
23.Sanyal, A., Lajoie, B. R., Jain, G., and Dekker,
J. (2012) Nature, 489, 109-113.
24.Akopov, S. B., Ruda, V. M., Batrak, V. V.,
Vetchinova, A. S., Chernov, I. P., Nikolaev, L. G., Bode, J., and
Sverdlov, E. D. (2006) Mamm. Genome, 17, 1042-1049.
25.Sass, A. V., Ruda, V. M., Akopov, S. B.,
Snezhkov, E. V., Nikolaev, L. G., and Sverdlov, E. D. (2005) Russ.
J. Bioorg. Chem., 31, 70-73.
26.Recillas-Targa, F., Bell, A. C., and Felsenfeld,
G. (1999) Proc. Natl. Acad. Sci. USA, 96,
14354-14359.
27.Takagi, H., Inai, Y., Watanabe, S., Tatemoto, S.,
Yajima, M., Akasaka, K., Yamamoto, T., and Sakamoto, N. (2011) J.
Biochem., 151, 75-87.
28.Jin, Y., Oomah, K., and Cattini, P. A. (2011)
DNA Cell Biol., 30, 995-1005.
29.Petrov, A., Allinne, J., Pirozhkova, I., Laoudj,
D., Lipinski, M., and Vassetzky, Y. S. (2008) Genome Res.,
18, 39-45.
30.Sambrook, J., and Russell, D. W. (eds.) (2001)
Molecular Cloning. A Laboratory Manual, 3rd Edn., CSHL Press,
Cold Spring Harbor.
31.Bell, A. C., West, A. G., and Felsenfeld, G.
(1999) Cell, 98, 387-396.
32.Vetchinova, A. S., Akopov, S. B., Chernov, I. P.,
Nikolaev, L. G., and Sverdlov, E. D. (2006) Anal. Biochem.,
354, 85-93.
33.Didych, D. A., Akopov, S. B., Snezhkov, E. V.,
Skaptsova, N. V., Nikolaev, L. G., and Sverdlov, E. D. (2009)
Biochemistry (Moscow), 74, 728-733.
34.Rosenbloom, K. R., Dreszer, T. R., Pheasant, M.,
Barber, G. P., Meyer, L. R., Pohl, A., Raney, B. J., Wang, T.,
Hinrichs, A. S., Zweig, A. S., et al. (2010) Nucleic Acids Res.,
38, D620-625.
35.Ernst, J., and Kellis, M. (2010) Nat.
Biotechnol., 28, 817-825.
36.Ernst, J., Kheradpour, P., Mikkelsen, T. S.,
Shoresh, N., Ward, L. D., Epstein, C. B., Zhang, X., Wang, L., Issner,
R., Coyne, M., et al. (2011) Nature, 473, 43-49.
37.Gomos-Klein, J., Harrow, F., Alarcon, J., and
Ortiz, B. D. (2007) J. Immunol., 179, 1088-1095.
38.Magdinier, F., Yusufzai, T. M., and Felsenfeld,
G. (2004) J. Biol. Chem., 279, 25381-25389.
39.Cuddapah, S., Jothi, R., Schones, D. E., Roh, T.
Y., Cui, K., and Zhao, K. (2009) Genome Res., 19,
24-32.
40.ENCODE Project consortium (2012) Nature,
489, 57-74.