REVIEW: Nano-Sized Manganese–Calcium Cluster in Photosystem II#
M. M. Najafpour1,2*, M. Z. Ghobadi1, B. Haghighi1,2, J. J. Eaton-Rye3, T. Tomo4,5, J.-R. Shen6, and S. I. Allakhverdiev7,8*
# This review is dedicated to the memory of Academician Alexander A. Krasnovsky, one of the great pioneering figures in the field of photosynthesis research who was the Ph.D. supervisor of one of the authors (SIA).1Department of Chemistry, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan, 45137-66731, Iran; fax: +98-241-415-3232; E-mail: mmnajafpour@iasbs.ac.ir
2Center of Climate Change and Global Warming, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan, 45137-66731, Iran
3Department of Biochemistry, University of Otago, P. O. Box 56, Dunedin 9054, New Zealand
4Department of Biology, Faculty of Science, Tokyo University of Science, Kagurazaka 1-3, Shinjuku-ku, Tokyo 162-8601, Japan
5PRESTO, Japan Science and Technology Agency (JST), 4-1-8 Honcho Kawaguchi, Saitama 332-0012, Japan
6Graduate School of Natural Science and Technology, Faculty of Science, Photosynthesis Research Center, Okayama University, Okayama, 700-8530, Japan
7Controlled Photobiosynthesis Laboratory, Institute of Plant Physiology, Russian Academy of Sciences, ul. Botanicheskaya 35, 127276 Moscow, Russia; fax: +7-496-7330-532; E-mail: suleyman.allakhverdiev@gmail.com
8Institute of Basic Biological Problems, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia
* To whom correspondence should be addressed.
Received December 18, 2013; Revision received January 15, 2014
Cyanobacteria, algae, and plants are the manufacturers that release O2 via water oxidation during photosynthesis. Since fossil resources are running out, researchers are now actively trying to use the natural catalytic center of water oxidation found in the photosystem II (PS II) reaction center of oxygenic photosynthetic organisms to synthesize a biomimetic supercatalyst for water oxidation. Success in this area of research will transcend the current bottleneck for the development of energy-conversion schemes based on sunlight. In this review, we go over the structure and function of the water-oxidizing complex (WOC) found in Nature by focusing on the recent advances made by the international research community dedicated to achieve the goal of artificial water splitting based on the WOC of PS II.
KEY WORDS: manganese, calcium, nano-sized manganese–calcium cluster, oxygen, photosynthesis, water oxidation, water-oxidizing complexDOI: 10.1134/S0006297914040026
Photosynthesis is one of the most important reactions that take place in Nature, and it is carried out by three major groups of organisms: green plants, algae, and some bacteria. There are five main bacterial photosynthetic organisms: cyanobacteria, purple bacteria, heliobacteria, green sulfur bacteria, and green non-sulfur bacteria. However, only cyanobacteria produce the byproduct of oxygen as a result of water splitting [1, 2]. The name of cyanobacteria is based on the bluish pigment phycocyanin, which is used to harvest light for photosynthesis in these cells. It is accepted that chloroplasts in plants and eukaryotic algae arose from the endosymbiotic uptake of an ancient cyanobacterial cell by an early non-photosynthetic eukaryote. The transition from anoxygenic to oxygenic photosynthesis in the eubacterial lineage was one of the most important innovations in evolution, and the vast majority of scientific communities are in agreement regarding the correlation between this event and the beginning of oxygen accumulation on Earth [3]. The evidence from chemical markers [4, 5], stromatolite fossils [6], and microfossils [7] attest that cyanobacteria arose before 2.5 billion years ago [6, 8].
As a result of the endosymbiotic uptake of cyanobacteria, algae and plants also use photosynthesis to obtain energy from sunlight to sustain life. Generally, photosynthetic organisms convert sunlight to chemical energy stored in the form of chemical bonds in carbohydrate [9]. To achieve the conversion of sunlight into useful chemical energy, two connected photosystems act in series in photosynthesis: photosystem I (PS I) and photosystem II (PS II). Photosystem II is responsible for producing the oxidizing power to split water at the start of the photosynthetic process, while PS I is a strong reductant responsible for the production of the intermediate energy rich coenzyme nicotinamide adenine dinucleotide phosphate (NADPH) that is required in the reactions that “fix” carbon dioxide into carbohydrate.
Photosystem II is a large homodimeric protein complex inserted in the thylakoid membrane of oxygenic photosynthetic organisms [10]. Each monomer is made up of more than 20 proteins and many cofactors including chlorophylls, carotenoids, xanthophylls, pheophytins, plastoquinones, a Mn cluster, chloride and Ca2+ ions, non-heme Fe2+, a bicarbonate ion, lipids, and heme groups [11].
Overall, these proteins and cofactors are divided into two categories in terms of performance. Most of the chlorophylls and carotenoids are involved in light energy transfer between so-called antenna pigments and the specialized pigments that comprise the reaction centers of the photosystems where the primary photochemistry takes place [11, 12]. Other cofactors contribute in the transfer of electrons. Likewise, the PS I reaction center contains protein subunits, chlorophylls, β-carotenoids, Fe4S4 clusters, and phylloquinones.
As shown in Fig. 1 [13], the photosynthetic process in PS II begins with light absorption by chlorophyll and other pigments, and as a consequence of the primary photochemistry the P680/Pheo– charge pair is created. It is important to note that Academician Alexander A. Krasnovsky, to whom this paper is dedicated, played an important role in the elucidation of the photochemical reactions of pigments and especially chlorophylls in photosynthetic organisms [14-18]. Here we review the water oxidation and detailed structure of the water-oxidizing complex (WOC) in natural photosynthesis.
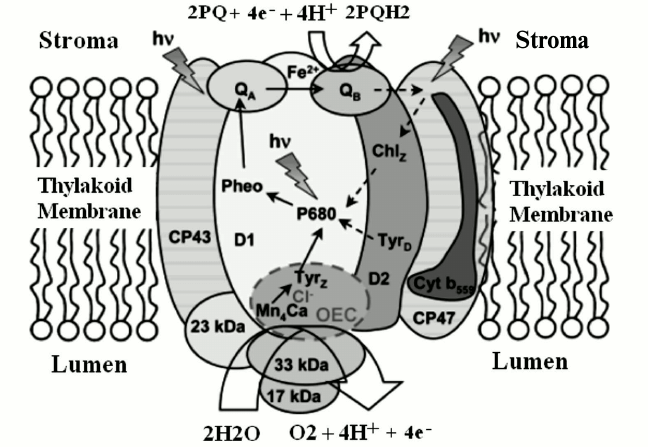
Fig. 1. PS II complex and its antenna system, consisting of more than 20 protein subunits, either embedded in the thylakoid membrane or associated with its lumenal surface. Light energy is trapped predominantly by the outer antenna and transferred to the photochemically active reaction centers, via chlorophyll-binding proteins CP47 and CP43, where it is used to drive the water-splitting reaction at the WOC. The electrons extracted from water are passed from the lumenally located Mn4CaO5 cluster to P680+ via D1-Y 161 (TyrZ), a process that is coupled to electron transfer (ET) from P680 to pheophytin (Pheo), QA, and QB, defining the ET pathway marked by the solid arrows. Broken arrows indicate secondary ET pathways that may play a photoprotective role. The protons and molecular oxygen produced during the water-splitting reaction are released into the lumen [13]. The image is reprinted with permission from [13] (Copyright (2008) by American Chemical Society).
CHLOROPHYLLS AND β-CAROTENES
Chlorophyll is a necessary biomolecule for light absorption and energy transfer in cyanobacteria and the chloroplast of plants and algae. The term “chlorophyll” was first proposed in 1817 by P. Pellitier and J. Caventou for the alcohol-extracted pigment from leaves, without knowing its composition and biological roles [19].
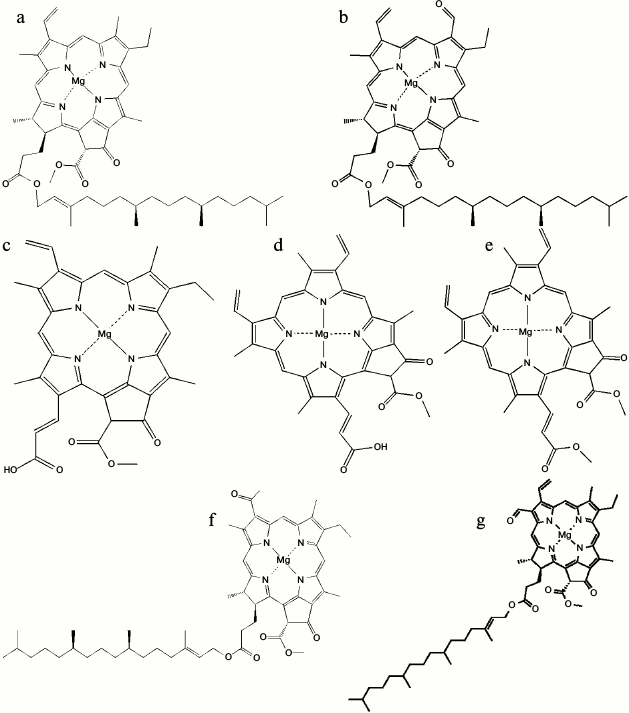
The main tasks of chlorophyll in photosynthesis are: optimal absorption of light; excitation energy transfer with high quantum efficiency to the reaction centers; accomplishing the primary charge separation along the photosynthetic membranes; and generating membrane potential [20]. So far, five different forms of chlorophylls (Chl) are recognized: Chl a, b, and c in the 19th century [21], Chl d in 1943 [22], and Chl f in 2010 [23] (Fig. 2). Chlorophyll a is the common pigment in all oxygenic photosynthetic organisms. It is composed of a central magnesium ion surrounded by a four nitrogen ion ring known as a chlorin ring. Also, a long tail is appended to the porphyrin ring of the chlorophyll. This hydrophobic tail helps the Chl a molecule when connecting to hydrophobic proteins in the thylakoid membrane.
Chlorophyll a can absorb light within the violet, blue, and red wavelengths. The presence of accessory pigments along with Chl a extends the absorbed light spectrum.
Fig. 2. Molecular structures of: a) Chl a; b) Chl b; c) Chl c1; d) Chl c2; e) Chl c3; f) Chl d; g) Chl f [34, 77].
In PS II, the core antenna proteins CP47 (~500 amino acids) and CP43 (470 amino acids) contain Chl a, which functions in both light harvesting and energy transfer to the reaction center. The ratio of these polypeptides per reaction center is 1 : 1 : 1. Also, they include 10-12 conserved histidine residues that connect to the chlorophyll molecules via ligation to the Mg atoms. Hydropathy-plot analysis of amino acid sequences reveals the presence of six transmembrane α-helices in both proteins and a large hydrophilic domain placed between the last two helices. These large loops are exposed to the inner thylakoid surface on the lumen side and may have a role in anchoring the three extrinsic proteins and the Mn cluster that are involved in oxygen evolution.
P680, which is known to be the PS II primary donor, consists of two Chl a molecules (PD1 or PD2) that absorb light at an optimal wavelength of 680 nm [24]. The 680 number is because of its absorption maximum in the red part of the visible spectrum. Energy transfer from the core antenna pigments of CP43 and CP47 leads to the excitation of P680, and this initiates photochemistry. The photooxidation of P680 occurs in a few picoseconds, and the removed electron is then captured by the primary electron acceptor, a pheophytin molecule located within PS II near P680. Eventually, the oxidized P680 (P680+) is reduced by an electron originating from water.
The reaction center of P700, the PS I primary donor, absorbs light at wavelength of 700 nm. Upon the absorption of a photon by P700, the electron from PS II is passed through a series of redox carries to ferredoxin (Fd).
The only structural difference between Chl b and Chl a is the replacement of the methyl group on ring B of the chlorin with a formyl group. The role of Chl b in photosynthesis is as a light energy absorbing pigment that transfers the absorbed energy to Chl a. It absorbs well at wavelengths of 450-500 and 600-650 nm. In addition to the CP43 and CP47, core antenna the photosystems of oxygenic photosynthetic organisms have peripheral antennas composed of a family of pigment-binding proteins that bind both Chl a and Chl b. The Chl a/b-binding proteins can be divided into two groups: the minor and major Chl a/b-binding complexes.
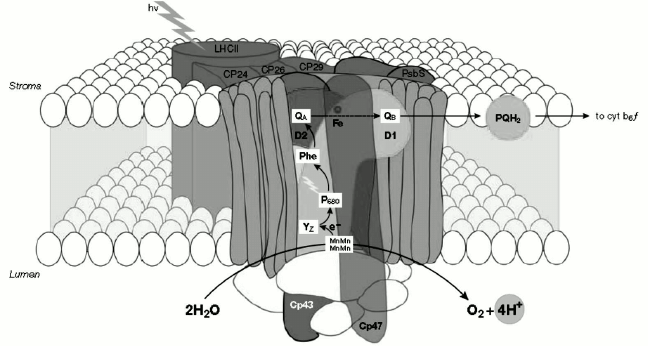
The minor Chl a/b-binding proteins located in the intermediate position of antennas include three CP29, CP26, and CP24 polypeptides, the first two being closer to the reaction center than CP24 (Fig. 3). Each of these complexes binds ~5% of the total PS II chlorophylls. They are also sometimes referred to as “accessory chlorophyll proteins” or “ACP”. The major Chl a/b-binding protein, named LHC II, is the main light-harvesting antenna of PS II and the major component of the thylakoid membrane placed in the farthest position from the reaction center (Fig. 3). Another known role for LHC II is membrane stacking, which leads to the regulation of the energy distribution between the two photosystems. The molecular mass of each monomeric subunit of LHC II is ~25 kDa, and it contains 15 Chl molecules (eight Chl a and seven Chl b) plus two xanthophyll molecules called luteins.
Fig. 3. Organization of PS II and light-harvesting complex II in the thylakoid membrane. CP43 and CP47, internal antenna chlorophyll–protein complexes; D1 and D2, main components of reaction centers (RCs) with binding sites for electron acceptor quinones (QB, QA); P680, chlorophyll special pair. Other cofactors associated with D1/D2: pheophytin (Phe), non-heme iron (Fe), Mn cluster. Accessory chlorophylls and β-carotene are not shown. Chl, chlorophyll; PQH2, plastoquinone pool; Cyt b6f, cytochrome b6f complex; YZ, D1-Tyr161 [78]. The image is reprinted with permission from [78] (Copyright (2005) by Wiley and Sons).
Chlorophyll c variants are additional pigments, which differ from Chl a and b in being Mg-phytoporphyrins rather than Mg-chlorins. In addition to Chls c1, c2, and c3 (Fig. 2, c-e), many new Chl c-like pigments have recently been isolated and characterized [25]. For about 60 years, it was thought that Chl d is also an accessory chlorophyll in some organisms until the discovery of Acaryochloris marina in 1996 [26]. Surprisingly, A. marina uses Chl d as the major photopigment. Also, Chl d can perform all functions of Chl a in A. marina, including light-harvesting complexes [27, 28] and the reaction center pigment [29-33]. The most recently discovered chlorophyll is Chl f, which can convert light of a much longer wavelength (720 nm) than any other known chlorophyll. Chlorophyll f, for example, allows the cyanobacteria that are found in stromatolites to perform photosynthesis using low-energy infrared light as the only light that can pierce these rock-like structures. The chemical structure of Chl f was found to be [2-formyl]-Chl a (C55H70O6N4Mg) [34].
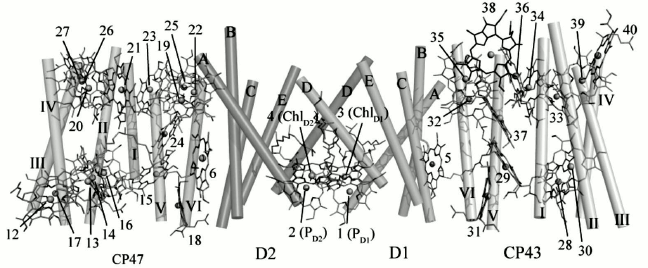
Recent high-resolution crystallography of PS II revealed 35 Chl a molecules in PS II. Interestingly, water ligands were found for a number of PS II chlorophylls. From the structural data, it can be seen that most of the vinyl groups of the CP43 and CP47 Chls are positioned in or near the same plane of the tetra pyrrole ring, which would be expected to facilitate energy transmission among adjoining chlorophylls (Fig. 4). The role of the Chls and β-carotenes (β-Car) bound to CP43 and CP47 is to capture photons and transfer excitation energy to the reaction center. Also, an additional function of β-Car is quenching of harmful Chl triplet states and any singlet oxygen that may be generated [35]. The only exceptions are the two β-Car molecules bound to the D1/D2 heterodimer, where the probable role is as electron transfer agents and possibly as singlet oxygen quenchers.
Fig. 4. Arrangement of chlorophylls in a PS II monomer viewed from a direction perpendicular to the membrane normal. Transmembrane helices of D1 and D2 are labeled A-E, and transmembrane helices of CP47 and CP43 are labeled I-VI [45]. The image is reprinted with permission from [45] (Copyright (2011) by Nature).
The electron required for reduction of P680+ is supplied by abstraction of one electron from the WOC (also known as the oxygen-evolving complex, often abbreviated as OEC) mediated by the redox-active tyrosine 161 residue of the D1 subunit. Finally, the extracted electrons are passed to the membrane redox mediator plastoquinone (PQ) located on the acceptor side of PS II after the primary pheophytin electron acceptor. These single electron steps are repeated four times until at the final stage in which molecular dioxygen is formed [11, 36]. The overall reaction equation is given below.
2 H2O + 2 PQ + 4 H+ → O2 + 4 H+ + 2 plastoquinol
The protons released from the oxidation of water contribute to a proton gradient, which is used by ATP synthase to generate ATP. Electrons are transferred from the reduced plastoquinone (plastoquinol) to PS I [37] via a cytochrome b6f complex [38] and next to a soluble electron carrier like plastocyanin or cytochrome c6, depending on the organism. These electrons are used for reduction of oxidized Chl a (P700). The formation of oxidized P700 due to light absorption and an electron transfer to Fd is ultimately used to reduce NADP+ to NADPH or in cyclic photophosphorylation around PS I [9, 39].
The structure and function of PS II and especially that of the WOC, as well as the pigments, are now intensively studied due to the developing world-wide energy crisis, since PS II may be a model for the synthesis of new compounds for the catalysis of water oxidation, which is a prerequisite for economic production of hydrogen [40].
WATER OXIDATION AND THE WATER-OXIDIZING COMPLEX IN NATURAL
PHOTOSYNTHESIS
Photosynthetic water oxidation is the main source of atmospheric oxygen and has been for about three billion years. This reaction is performed at a catalytic site (i.e. the WOC) that includes a Mn4O5Ca cluster surrounded by a protein matrix. Many researchers are actively seeking to establish the structural information and the mechanism of oxygen evolution by the WOC. In the next part of this brief overview, we go over the recent structural information of the WOC and the major suggested mechanism of water oxidation by the Mn4O5Ca cluster.
Structure of the Water-Oxidizing Complex
So far, various instruments and methods like extended X-ray absorption fine structure (EXAFS), X-ray diffraction (XRD), and X-ray crystallography have been used for determination of the constituent atoms and their arrangement in the cluster. In 2001, Witt, Saenger, and their colleagues presented the first three-dimensional structure of the WOC from a thermophilic cyanobacterium at 3.8 Å resolution [41]. In 2003, Nobuo Kamiya and Jian-Ren Shen isolated PS II from Thermosynechococcus vulcanus and gained a similar structural analysis result [42]. James Barber and So Iwata in 2004 proposed the first structure that included a Ca ion and proposed a cubane-like Mn3CaO4 cluster [43]. The most plausible model for the cluster is the “dangler model” in which three Mn ions are tightly coupled. In this model, the fourth Mn ion is connected to the trimer as a “dangler” [41, 42, 44-46]. Until 2011, however, the precise details of the Mn4Ca cluster structure were not known, and many questions remained concerning the location of the substrate water molecules and the accurate order of the amino acid side chains and cofactors of the WOC that were required to reach an accurate mechanism of water oxidation.In 2011, the research teams of Jian-Ren Shen and Nobuo Kamiya succeeded in obtaining the PS II crystal structure at 1.9 Å resolution, a major advance towards to the determination of the Mn4CaO5 cluster structure [45]. They resolved the electron densities and found that the WOC cluster is composed of four Mn atoms, one Ca atom, and five oxygen atoms (Fig. 5). They demonstrated that the four corners of the cluster are filled by three Mn atoms and one Ca atom. Four oxygen atoms occupy the other four corners of a cubane-like structure. The location of Mn(4) is outside of the cubane, and it is connected to two Mn atoms within the cubane by one oxygen (O5). Also, four water molecules are bound to the Mn4CaO5 cluster (Mn4O5Ca(H2O)4). Two of them coordinate to Ca, and the other two to Mn(4). Also, each Mn and Ca ion is surrounded by six and seven ligands, respectively. In the next section, we depict the detailed structure of the WOC determined at 1.9 Å resolution.
Fig. 5. The entire structure of the Mn4CaO5 cluster resembles a distorted chair, with an asymmetric cubane [45]. The image was reprinted with permission from [45] (Copyright (2011) by Nature).
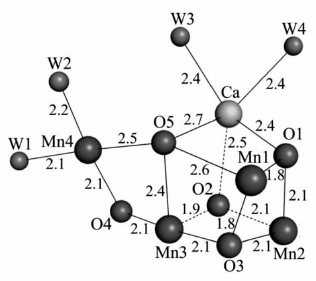
Mn ions. As noted above, the essential participation of Mn in photosynthesis was originally presented by Pirson in 1937 [47]. He showed that plants and algae cannot release O2 in the absence of Mn in their growth medium [47]. In 1977, J. Jaklevic and coworkers confirmed Pirson’s observation by recording the X-ray absorption spectrum of the Mn in a leaf [48]. Despite the widespread presence of Mn in Nature, it is found at a low level in human tissues. The available oxidation states of Mn in biological systems are II, III, and IV. Mn compounds with oxidation states V and VI are strong oxidizing agents that undergo disproportionation reactions.
There are several reasons why Nature selected Mn for catalyzing water splitting in photosynthetic organisms. Mn is the third most abundant transition element on Earth. It has a particularly rich redox chemistry compared with other d-block elements, with several available oxidizing states. The most stable-state Mn(II) can be absorbed by cells. Also, the oxidation of Mn(II) is facilitated by appropriate ligands, so it provides a simple construction unit for the WOC. The intermediate oxidation states Mn(III) and Mn(IV) can form a strong complex with O2– and constitute robust mixed-valence poly-oxo clusters [49]. Mn(III) and Mn(IV) are usually stabilized by many hard ligands in living organisms. Hard ligand refers to ones that are small, have high charge states and electronegativity, and are weakly polarizable [50].
In the following sections, the coordination chemistry of all components of the Mn4CaO5 cluster and some of its surrounding groups based on reference [45] are surveyed. It should be mentioned that charge on the Mn4CaO5(H2O)4 cluster is distributed, and charge on each ion is lower than suggested by its oxidation state. So, the whole system should be studied together.
Mn1. There are six ligands around the Mn1 atom that stabilize the oxidation state of III or IV for the Mn ion: three μ3-O (μn-O means an oxo bridge linking n atoms together), one monodentate carboxylate, one bridging carboxylate, and one imidazole ligand (Fig. 6a).
Fig. 6. Mn1 (a), Mn2 (b), Mn3 (c), Mn4 (d), and Ca (e) and their surrounding ligands [45].
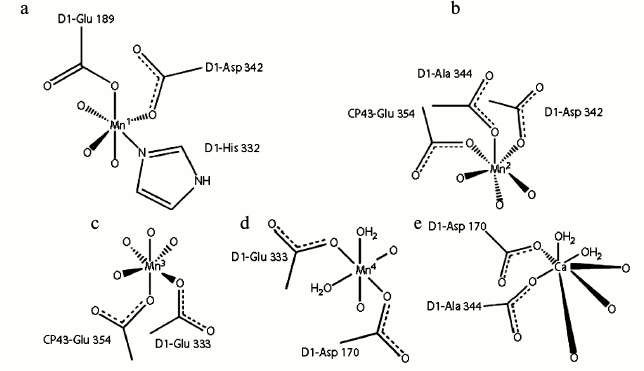
Mn2. As the coordination number of this ion is 6, it is surrounded by six ligands: three μ3-O and three bridging COO– (Fig. 6b). Similar to Mn1, the oxidation state of III or IV can be stabilized by this ion. The ion is linked to the Ca, Mn1, and Mn3 ions with a bridging carboxylate and three oxo groups.
Mn3. The six ligands around this ion are three μ3-O, one μ2-O, and two bridging COO− ions. Four hard μ-O would be able to better stabilize Mn(IV) than Mn(III) (Fig. 6c).
Mn4. Mn4 is encompassed by one μ4-O, one μ2-O, two bridging COO−, and two H2O (Fig. 6d). These ligands are able to stabilize the oxidation state of (III); however, deprotonation of water molecules could stabilize the oxidation state of Mn(IV) as well as higher oxidation states (e.g. Mn(V)).
Ca ion. The requirement of a Ca ion in the electron transfer of the WOC was discovered in 1984 [51]. Subsequent studies revealed the important role of this ion in oxygen evolution [45, 52, 53]. Crystallographic data on WOC at 1.9 Å clearly showed that the Ca ion has seven ligands, three μ3-O, two bridging COO−, and two H2O molecules (Fig. 6e). Here, analogously with water molecules coordinated to Mn4, one of the water molecules may serve as the substrate for water oxidation. Variable coordination numbers of Ca from 6 to 10 can lead to coordination or de-coordination of its surrounding ligands during water oxidation. Different studies have demonstrated that the only functional substitution for Ca is Sr, as Mn4OxSr can behave similarly to Mn4OxCa in water oxidation [51, 54]. However, the structural changes due to replacement of Ca by Sr were unclear until 2013, because the crystal structure containing Sr had only been recorded at low resolution. In 2013, Jian-Ren Shen and his colleague were able to crystallize and analyze the Mn4(Sr/Ca)O5 cluster of Thermosynechococcus vulcanus at 2.1 Å resolution [55]. They compared the structure of Sr-PS II and Mn4CaO5 and used the results for the investigation of conceivable mechanisms of photosynthetic water oxidation. It was found that the general shape of the Sr-PS II cluster as a distorted chair form is similar to the native Mn cluster. Although the position of four Mn ions were intact relative to the original PS II, the average Mn1–Mn4 distance of two monomers was somewhat (0.1 Å) longer, and the average Mn3–Mn4 distance was somewhat (0.1 Å) shorter due to a slight shift in the position of the dangler Mn4 atom in the Mn4SrO5 cluster (Fig. 7). The location of Sr was moved into a more strained position in the cubane unit than that found with Ca. This shift leads to lengthening of the Sr–Mn distances compared with the Ca–Mn distances because of larger ionic radius of Sr ion (1.12 Å) compared to Ca ion (0.99 Å). Thus, the average distances of two monomers between Sr and Mn2–Mn4 were slightly (0.2 Å) longer than the corresponding Ca and Mn distances, and the Mn1–Sr distance was slightly (0.1 Å) longer than the Mn1–Ca distance. As a result, the greater distortion of the Mn4SrO5 cluster structure causes its instability. This may be the reason of oxygen evolution decline due substitution of Ca by Sr.
Fig. 7. Average distances between metal and oxo-ligands of the Mn4SrO5 cluster [55]. The image is reprinted with permission from [55] (Copyright (2011) by Proc. Natl. Acad. Sci. USA).
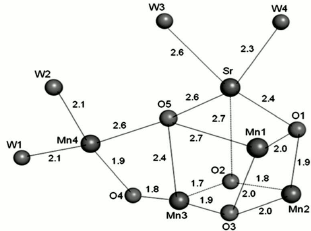
The detection of oxo-bridged oxygens in the Sr-PS II was difficult because the electron density of oxygen atoms was much weaker than those of Mn and Sr ions. So, the position of oxygens in the Mn4SrO5 cluster was specified based on their omit-map that was coincident with the oxygen atom location within the Mn4CaO5 cluster. The lower occupancy of Sr is probably a further reason for the observed differences, as there were five Mn–O bonds that became 0.2 Å shorter and one bond that became 0.3 Å shorter, whereas only two bonds became 0.2 Å longer upon Sr substitution. All O–Sr distances were the same as O–Ca distances except for O2–Sr, which was 0.2 Å longer, revealing the movement of Sr relative to Ca. The locations of three water molecules (W1, W2, W4) of four in the Sr-PS II cluster were similar to those in the Mn-Ca cluster. But the position of W3 was shifted by 0.4-0.6 Å relative to its position in Ca-PS II in the two monomers. Also, the average bond length of W3–Sr was 2.6 Å, offering a weaker binding of W3 than that of other water molecules in Sr-PS II. This substantial elongation in the W3-Sr bond length suggests that W3 may confer the “slowly exchanging” water with a fast exchange rate. So, the possibility of W3 motion toward the O5 at some steps of the S-state cycle was presented. This allows the formation of an O–O bond between W3 and O5 resembling the proposed mechanism of O–O bond formation between a Ca-bound water/hydroxo and a Mn-bridged oxygen in the WOC [56, 57].
The lengthening of the W3–Sr distance changes the W2–W3 distance from 3.3 to 3.5 Å. This leads to weak interaction between these two water molecules in Sr-PS II. The breakage of the hydrogen bond between W2 and W3 is likely to lead to a fast exchange rate of W2, which suggests that W2 may be involved in the O–O bond formation. In this case, the O–O bond formation may occur either between W2 and W3, or between W2 and O5.
These changes may be another reason for reduction in the rate of oxygen evolution. The surrounded ligands of the Mn4SrO5 cluster were the same as for the Mn4CaO5 cluster. There are a few differences regarding bond distances between ligands and the metal ions of the Sr-PS II cluster: (i) the side-chain of D1-Asp170 is shifted, so the average Sr–D1-Asp170 distance is 2.6 Å, which is 0.2 Å longer than the corresponding distance in the Mn-Ca cluster; (ii) the distance of Sr–Glu189-D1 is 0.2 Å shorter than that in the Mn4CaO5 cluster; (iii) as D1-Asp170 is a bidentate ligand to Ca and Mn4, the elongation of the Sr–Asp170 distance represents the larger ionic radius of the Sr ion compared with the Ca ion, and it may cause weaker binding of this residue to the Sr ion, and (iv) the distance between D1-Glu189 and Sr was shorter (3.1 Å) than that in the Mn4CaO5 cluster (3.3 Å). This outcome suggests that the carboxylate group of D1-Glu189 may have a stronger interaction with Sr relative to Ca.
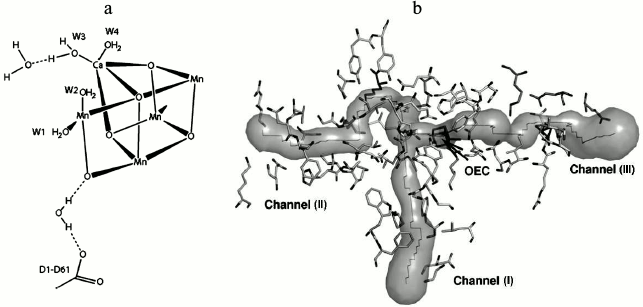
Water. The accurate study of the mechanism of water oxidation requires the position of the substrate water binding sites on Mn4CaO5 to be established. Based on the high-resolution crystallographic structure of PS II [45], it was found that 2795 water molecules are present mainly in two layers on the surfaces of the stromal and lumenal sides of the photosystem. The hydrogen-bonding formation by some of these molecules may serve as channels for protons, water, or oxygen molecules. Also, a small number of water molecules are located within the membrane region, which are attached as ligands to the chlorophylls. By focusing on the Mn cluster, four water molecules (W1 to W4) are detected in association with the Mn4CaO5 cluster, so that W1 and W2 are coordinated to Mn4 with distances of 2.1 and 2.2 Å, and W3 and W4 are coordinated to Ca with a similar distance of 2.4 Å. It was proposed that some of these water molecules may act as substrates for water oxidation (Fig. 8).
Fig. 8. a) Location of substrate water binding sites on the WOC [45]. b) The figure shows channels for hydrophobic oxygen (I), water (II), and protons (III), all leading to the catalytic Mn4CaO5 cluster of PS II. OEC stands for oxygen-evolving complex (i.e. the WOC) [79]. Image (b) is reprinted with permission from [79] (Copyright (2008) by American Chemical Society).
Amino acids. There are two amino acid side-chains that are bound to the WOC as either direct or indirect ligands. The direct ligands and their positions include: monodentate ligand of D1-Glu189 to Mn1, bidentate ligands of D1-Asp170 to Mn4 and Ca, D1-Glu333 to Mn3 and Mn4, D1-Asp342 to Mn1 and Mn2, D1-Ala344 to Mn2 and Ca, and CP43-Glu354 to Mn2 and Mn3. Furthermore, D1-His332 is coordinated to Mn1, whereas D1-His337 is not directly coordinated to the metal cluster. Also, D1-Asp61, D1-His337, and CP43-Arg357 as indirect ligands were found in the second coordination sphere. Regarding the location of D1-Asp61 in the entrance of a proposed proton exit channel, it was suggested that this residue may facilitate the proton exit from the Mn4CaO5 cluster.
The imidazole nitrogen of D1-His337 is hydrogen-bonded to one μ-O (O3). The role of this hydrogen bond may be as a stabilizer for the WOC. One of the guanidinium η-nitrogens of CP43-Arg357 is hydrogen-bonded to two of the μ-O in the Mn4CaO5 cluster (O2 and O4), while the other one is hydrogen bonded to the carboxylate oxygen of both D1-Asp170 and D1-Ala344. Overall, the proposed roles for these residues are in maintaining the structure of the metal cluster, stabilizing the cubane structure by providing partial positive charges to compensate the negative charges induced by the oxo bridges and carboxylate ligands of the WOC.
Chloride. In 1949, Arnon and Whatley demonstrated the requirement of isolated photosynthetic membranes for Cl– to support the production of O2 [58, 59]; subsequently, various studies have revealed that Cl– is involved in the electron donor reactions of the WOC [59-61].
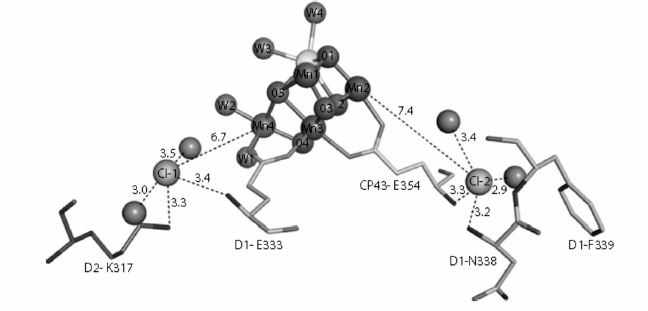
In 2011, Rivalta and coworkers found that removal of Cl– caused salt-bridge formation between D2-K317 and D1-D61, and this may result in the repression of proton transfer to the lumen [61]. Jian-Ren Shen’s group in 2011 demonstrated the presence of two chloride ions in the WOC structure. Both Cl– ions are surrounded by water molecules and amino acids as follows: the amino group of D2-Lys317 and the backbone nitrogen of D1-Glu333 around Cl–(1) and the backbone nitrogens of D1-Asn338 and CP43-Glu354 around Cl–(2). Also, both Cl– ions are surrounded by two water molecules (Fig. 9). According to the direct coordination of the side chain of D1-Glu333 and CP43-Glu354 to the Mn cluster, Jian-Ren Shen’s group proposed that the chloride ions are involved in the preservation of the coordination environment of the Mn4CaO5 cluster and help the progress of the oxygen-evolving reactions.
Fig. 9. Structure of the two Cl−-binding sites near the Mn4CaO5(H2O)4 cluster. Hydrogen bond distances are in Å [45]. The image is reprinted with permission from [45] (Copyright (2011) by Nature).
Tyrosine 161. The photosynthetic process in PS II is initiated by light absorption and oxidized Chl a (P680) formation. Then, the electron deficiency of P680+ is compensated by tyrosine 161 (Yz), and subsequently Yz+ is reduced by electrons resulting from the oxidation of water [62]. The last high-resolution crystallographic structure of PS II revealed that the position of YZ is between the Mn4CaO5 cluster and the PS II reaction center.
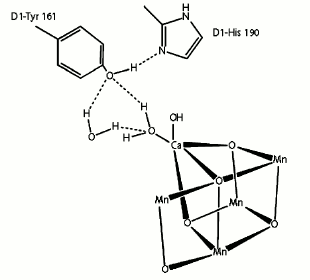
Tyrosine 161 forms a strong hydrogen bond to the two water molecules coordinated to Ca either directly (W4) or indirectly (W3) through another water molecule. Moreover, another strong hydrogen bond is found between D1-Tyr161 and the ε-nitrogen of D1-His190 on the opposite side of the Mn4CaO5 cluster. This histidine is further hydrogen bonded to other amino acids or water molecules to form a hydrogen bond network suggested as an exit channel for protons (Fig. 10).
Fig. 10. Location of tyrosine 161 [45].
Mechanism of Water Oxidation
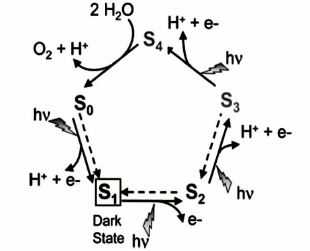
From the thermodynamic viewpoint, four-electron water oxidation is more facile than other possible reactions such as four sequential one-electron oxidation or two sequential two-electron oxidation reactions as their first steps are more endergonic. So, Nature preferred a four-electron oxidation mechanism for water oxidation with the lowest activation energy [63].P. Joliot in 1969 applied a superior method to activate a photosynthetic system, in which PS II was exposed to four-stage flash illumination and then the oxygen yield was measured on each flash. He showed that an oscillating pattern with a period of four persists such that the greatest yield occurs after the fourth flash [64]. The outcome is noteworthy because oxidation of two water molecules to produce one oxygen molecule requires the elimination of four electrons. According to this result, Kok and coworkers in 1970 suggested that in each stage of illumination, one electron is removed from the WOC [65]. When four oxidizing equivalents have been accumulated, an oxygen molecule is spontaneously evolved. Each increasing oxidation state was named an “S-state”, in which the degree of oxidation was progressively increased from S0 to S4 (Fig. 11). In the model the S1 state must be dark-stable, and oxygen evolution occurs in the light-independent transition of S4 to S0. All other S-state transitions are induced by the photochemical oxidation of P680. Based on the Kok cycle, numerous groups hypothesized that the proton transfer pattern from the WOC into the lumen is 1 : 0 : 1 : 2.
Fig. 11. Catalytic cycle of photosynthetic water oxidation proposed by Joliot and Kok. Dotted arrows describe reactions that relax the system back to the dark stable state S1 within minutes [13]. The image is reprinted with permission from [13] (Copyright (2008) by American Chemical Society).
Details of water oxidation mechanism in Nature. Perhaps the major issue discussed between researchers in the field of photosynthesis is determination of the water oxidation mechanism by the WOC of PS II. So, various proposals have been put forward. The most important suggested models are presented as follows.
1. Nucleophile–electrophile reaction: Pecoraro et al. in 1998 suggested the formation of a Mn-bound hydroperoxide due to the nucleophilic attack of a Ca-bound hydroxide ligand to a terminal Mn(V)=O [66]. Brudvig’s group proposed the nucleophilic attack of a Ca-bound water molecule to a Mn(V)=O species to form the O=O bond [36]. Umena et al. in 2011 suggested that the coordinated water molecule to the Ca near tyrosine 161 may serve as one of the substrate molecules, and the water molecule bound to Mn4 may serve as the second substrate in the O–O bond formation [45].
2. The interaction between an oxyl radical and Mn-binding µ-oxo-ligand: Siegbahn in 2012 proposed a proton transfer in the S3→S4 transition that precedes the oxidation step. Deprotonation is very important for the water oxidation and oxyl radical formation. After oxyl radical formation, another proton transfer before the S4 state causes O–O bond formation between an oxyl radical in the center of the cluster and a Mn-bridging μ-oxo ligand [67].
3. Reductive elimination of two bridging oxo-ligands: Ruttinger et al. suggested that a high-valency Mn-oxo-cluster collapses to form oxygen from two bridging oxo-ligands [68].
4. Radical coupling mechanism: This proposed mechanism includes radical coupling of two axial radicals (O•), which leads to production of the O–O bond [69]. Umena et al. suggested that bridging oxygen between Ca, Mn4, Mn3, and Mn1 may be present as a hydroxide ion in the S1 state and provide one of the substrates for dioxygen evolution [45]. Glatzel et al. in 2013 using resonant inelastic X-ray scattering spectroscopy showed delocalization of an electron in the Mn4CaO5 cluster and possible involvement of ligands in the redox chemistry. So, they suggested that the mechanisms including delocalization of charge on the ligands are possible [70].
Water oxidation in artificial photosynthesis. Nature is always inspiring humanity to achieve optimal conditions for life on Earth. Photosynthesis and specifically the Mn4CaO5 cluster in cyanobacteria, algae, and plants is a key model for researchers to synthesize new catalysts with water-splitting capability for production of hydrogen (Scheme) [71-74]. The recent high resolution crystallographic structure of the Mn4CaO5 cluster revealed in-depth structural information about the WOC structure [45] that can be used as a proper template for scientists. Based on this more detailed structural finding, it was revealed only 3-4 residues among the surrounding amino acids of the Mn4CaO5 cluster are directly coordinated to the WOC. The proposed roles of these residues are regulation of charges and stabilizing the cluster. On the other hand, the WOC activity is decreased in the absence of amino acids [75]. So, the presence of proper side chains is needed for facilitation of proton transfer, regulation of charges, and help in water molecule coordination at appropriate sites [76].
One of the most important goals of artificial photosynthesis is using
solar energy and water to obtain H2 as fuel
MMN and MZG are grateful to the Institute for Advanced Studies in Basic Sciences and the National Elite Foundation for financial support.
This work was supported by grants from the Russian Foundation for Basic Research (Nos. 13-04-91372, 14-04-01549, 14-04-92690) and by Molecular and Cell Biology Program of the Russian Academy of Sciences to SIA.
REFERENCES
1.Blankenship, R. E. (1992) Photosynth. Res.,
33, 91-111.
2.Knoll, A. H. (2003) Geobiology, 1,
3-14.
3.Canfield, D. E. (2005) Annu. Rev. Earth Planet.
Sci., 33, 1-36.
4.Kazmierczak, J., and Altermann, W. (2002)
Science, 298, 2351-2351.
5.Summons, R. E., Jahnke, L. L., Hope, J. M., and
Logan, G. A. (1999) Nature, 400, 554-557.
6.Olson, J. M., and Blankenship, R. E. (2004)
Photosynth. Res., 80, 373-386.
7.Schopf, J. W. (1975) Evolutionary Biology,
Springer, Dordrecht, pp. 1-43.
8.Konhauser, K. O., Lalonde, S. V., Planavsky, N. J.,
Pecoits, E., Lyons, T. W., Mojzsis, S. J., Rouxel, O. J., Barley, M.
E., Rosìere, C., and Fralick, P. W. (2011) Nature,
478, 369-373.
9.Najafpour, M. M., Nemati, A. M., Shen, J. R.,
and Govindjee (2013) in Stress Biology of Cyanobacteria
(Srivastava, A., Rai, A. N., and Neilan, B. A., eds.) CRC Press,
London, pp. 41-60.
10.Roose, J. L., Wegener, K. M., and Pakrasi, H. B.
(2007) Photosynth. Res., 92, 369-387.
11.McEvoy, J. P., and Brudvig, G. W. (2006) Chem.
Rev., 106, 4455-4483.
12.Amunts, A., Toporik, H., Borovikova, A., and
Nelson, N. (2010) J. Biol. Chem., 285, 3478-3486.
13.Sproviero, E. M., Gascon, J. A., McEvoy, J. P.,
Brudvig, G. W., and Batista, V. S. (2008) J. Am. Chem. Soc.,
130, 3428-3442.
14.Krasnovsky, A. A. (1960) Ann. Rev. Plant
Physiol., 11, 363-410.
15.Krasnovsky, A. A. (1965) Photochem.
Photobiol., 4, 641-655.
16.Krasnovsky, A. A. (1992) Photosynth. Res.,
34, 327-328.
17.Krasnovsky, A. A., Jr. (2007) Biochemistry
(Moscow), 72, 1065-1080.
18.Belyaeva, O. (2003) Photosynth. Res.,
76, 405-411.
19.Krasnovsky, A. A., Jr. (2003) Photosynth.
Res., 76, 389-403.
20.Krasnovsky, A. A., Jr. (2005) in Discoveries
in Photosynthesis, Springer, Dordrecht, pp. 1143-1157.
21.Krogmann, D. (2004) Photosynth. Res.,
80, 15-57.
22.Manning, W. M., and Strain, H. H. (1943) J.
Biol. Chem., 151, 1-19.
23.Chen, M., Schliep, M., Willows, R. D., Cai,
Z.-L., Neilan, B. A., and Scheer, H. (2010) Science, 329,
1318-1319.
24.Raszewski, G., Diner, B. A., Schlodder, E., and
Renger, T. (2008) Biophys. J., 95, 105-119.
25.Zapata, M., Garrido, J. L., and Jeffrey, S. W.
(2006) in Chlorophylls and Bacteriochlorophylls, Springer,
Dordrecht, pp. 39-53.
26.Miyashita, H., Ikemoto, H., Kurano, N., Adachi,
K., Chihara, M., and Miyachi, S. (1996) Nature, 383,
402-402.
27.Chen, M., Quinnell, R. G., and Larkum, A. W.
(2002) FEBS Lett., 514, 149-152.
28.Tomo, T., Allakhverdiev, S. I., and Mimuro, M.
(2011) J. Photochem. Photobiol. B, 104, 333-340.
29.Hu, Q., Miyashita, H., Iwasaki, I., Kurano, N.,
Miyachi, S., Iwaki, M., and Itoh, S. (1998) Proc. Natl. Acad. Sci.
USA, 95, 13319-13323.
30.Chen, M., Telfer, A., Lin, S., Pascal, A.,
Larkum, A. W., Barber, J., and Blankenship, R. E. (2005) Photochem.
Photobiol. Sci., 4, 1060-1064.
31.Tomo, T., Okubo, T., Akimoto, S., Yokono, M.,
Miyashita, H., Tsuchiya, T., Noguchi, T., and Mimuro, M. (2007)
Proc. Natl. Acad. Sci. USA, 104, 7283-7288.
32.Allakhverdiev, S. I., Tomo, T., Shimada, Y.,
Kindo, H., Nagao, R., Klimov, V. V., and Mimuro, M. (2010) Proc.
Natl. Acad. Sci. USA, 107, 3924-3929.
33.Allakhverdiev, S. I., Tsuchiya, T., Watabe, K.,
Kojima, A., Los, D. A., Tomo, T., Klimov, V. V., and Mimuro, M. (2011)
Proc. Natl. Acad. Sci. USA, 108, 8054-8058.
34.Willows, R. D., Li, Y., Scheer, H., and Chen, M.
(2013) Org. Lett., 15, 1588-1590.
35.Barber, J. (2012) Cold Spring Harbor Symp. on
Quantitative Biology.
36.Brudvig, G. (2008) Ser. B. Biol.
Sci., 363, 1211-1218.
37.Golbeck, J. H. (2006) Photosystem I: the
Light-Driven Plastocyanine: Ferredoxin Oxidoreductase,
Springer.
38.Kallas, T. (2012) in Photosynthesis
(Eaton-Rye, J. J., Tripathy, B. C., and Sharkey, T. D.,
eds.) Springer, Dordrecht, pp. 501-560.
39.Vermaas, W. F. (2001) Photosynthesis and
Respiration in Cyanobacteria, John Wiley and Sons, Singapore.
40.Jee, G., Kambara, T., and Coleman, W. (1985)
Photochem. Photobiol., 42, 187-210.
41.Zouni, A., Witt, H.-T., Kern, J., Fromme, P.,
Krauss, N., Saenger, W., and Orth, P. (2001) Nature, 409,
739-743.
42.Kamiya, N., and Shen, J.-R. (2003) Proc. Natl.
Acad. Sci. USA, 100, 98-103.
43.Ferreira, K. N., Iverson, T. M., Maghlaoui, K.,
Barber, J., and Iwata, S. (2004) Science, 303,
1831-1838.
44.Yano, J., Kern, J., Sauer, K., Latimer, M. J.,
Pushkar, Y., Biesiadka, J., Loll, B., Saenger, W., Messinger, J., and
Zouni, A. (2006) Science, 314, 821-825.
45.Umena, Y., Kawakami, K., Shen, J.-R., and Kamiya,
N. (2011) Nature, 473, 55-60.
46.Kawakami, K., Umena, Y., Kamiya, N., and Shen,
J.-R. (2011) J. Photochem. Photobiol. B, 104, 9-18.
47.Pirson, A. (1937) Z. Bot., 31,
193-267.
48.Jaklevic, J., Kirby, J., Klein, M., Robertson,
A., Brown, G., and Eisenberger, P. (1977) Solid State Commun.,
23, 679-682.
49.Armstrong, F. A. (2008) Biol. Sci.,
363, 1263-1270.
50.Pearson, R. G. (1963) J. Am. Chem. Soc.,
85, 3533-3539.
51.Ghanotakis, D. F., Babcock, G. T., and Yocum, C.
F. (1984) FEBS Lett., 167, 127-130.
52.Lee, C.-I., Lakshmi, K., and Brudvig, G. W.
(2007) Biochemistry, 46, 3211-3223.
53.Latimer, M. J., DeRose, V. J., Yachandra, V. K.,
Sauer, K., and Klein, M. P. (1998) J. Phys. Chem. B, 102,
8257-8265.
54.Boussac, A., and Rutherford, A. W. (1988)
Biochemistry, 27, 3476-3483.
55.Koua, F. H. M., Umena, Y., Kawakami, K., and
Shen, J.-R. (2013) Proc. Natl. Acad. Sci. USA,
110, 3889-3894.
56.Pushkar, Y., Yano, J., Sauer, K., Boussac, A.,
and Yachandra, V. K. (2008) Proc. Natl. Acad. Sci. USA,
105, 1879-1884.
57.Cox, N., Rapatskiy, L., Su, J.-H., Pantazis, D.
A., Sugiura, M., Kulik, L., Dorlet, P., Rutherford, A. W., Neese, F.,
and Boussac, A. (2011) J. Am. Chem. Soc., 133,
3635-3648.
58.Arnon, D. I., and Whatley, F. R. (1949)
Science, 110, 554-556.
59.Yocum, C. F. (2008) Coord. Chem. Rev.,
252, 296-305.
60.Izawa, S., Heath, R., and Hind, G. (1969)
Biochim. Biophys. Acta, 180, 388-398.
61.Rivalta, I., Amin, M., Luber, S., Vassiliev, S.,
Pokhrel, R., Umena, Y., Kawakami, K., Shen, J.-R., Kamiya, N., and
Bruce, D. (2011) Biochemistry, 50, 6312-6315.
62.Renger, G. (2008) Primary Processes of
Photosynthesis: Principles and Apparatus, Royal Society of
Chemistry, London.
63.Wiechen, M., Berends, H.-M., and Kurz, P. (2012)
Dalton Trans., 41, 21-31.
64.Joliot, P., Barbieri, G., and Chabaud, R. (1969)
Photochem. Photobiol., 10, 309-329.
65.Forbush, B., Kok, B., and McGloin, M. P. (1971)
Photochem. Photobiol., 14, 307-321.
66.Pecoraro, V. L., Baldwin, M. J., Caudle, M. T.,
Hsieh, W. Y., and Law, N. A. (1998) Pure Appl. Chem., 70,
925-930.
67.Siegbahn, P. E. M. (2013) Biochim. Biophys.
Acta, 1827, 1003-1019.
68.Ruettinger, W., Yagi, M., Wolf, K., Bernasek, S.,
and Dismukes, G. (2000) J. Am. Chem. Soc., 122,
10353-10357.
69.Yachandra, V. K., Sauer, K., and Klein, M. P.
(1996) Chem. Rev., 96, 2927-2950.
70.Glatzel, P., Schroeder, H., Pushkar, Y., Boron,
T., Mukherjee, S., Christou, G., Pecoraro, V. L., Messinger, J.,
Yachandra, V. K., Bergmann, U., and Yano, J. (2013) Inorg.
Chem., 52, 5642-5644.
71.Du, P., and Eisenberg, R. (2012) Energy
Environ. Sci., 5, 6012-6021.
72.Najafpour, M. M., and Allakhverdiev, S. I. (2012)
Int. J. Hydrogen Energ., 37, 8753-8764.
73.Najafpour, M. M., Abasi, M., and Allakhverdiev,
S. I. (2013) SOAJ NanoPhotoBioSciences, 1, 79-92.
74.Soriano-Lopez, J., Goberna-Ferron, S., Vigara,
L., Carbo, J. J., Poblet, J. M., and Galan-Mascaros, J. R. (2013)
Inorg. Chem., 52, 4753-4755.
75.Debus, R. J. (2008) Coord. Chem. Rev.,
252, 244-258.
76.Najafpour, M. M., Moghaddam, A. N., and
Allakhverdiev, S. I. (2012) Biochim. Biophys. Acta, 1817,
1110-1121.
77.Scheer, H. (ed.) (1991) Chlorophylls, CRC
Press, Boca Raton, pp. 3-30.
78.Szabo, I., Bergantino, E., and Giacometti, G. M.
(2005) EMBO Rep., 6, 629-634.
79.Barber, J. (2008) Inorg. Chem., 47,
1700-1710.