REVIEW: Modern Anti-Cytokine Therapy of Autoimmune Diseases
I. V. Astrakhantseva1*, G. A. Efimov2, M. S. Drutskaya2, A. A. Kruglov1,3,4, and S. A. Nedospasov1,2,3,4*
1Institute of Molecular Biology and Regional Ecology, Lobachevsky State University of Nizhni Novgorod, pr. Gagarina 23, 603950 Nizhni Novgorod, Russia; fax: +7 (831) 623-3085; E-mail: astrakhantsevairina@gmail.com2Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; fax: +7 (499) 135-1405; E-mail: isinfo@eimb.ru
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: +7 (495) 939-0338; E-mail: fxb@genebee.msu.su
4German Rheumatism Research Center, Leibnitz Institute, Chariteplatz 1, Berlin 10117, Germany; fax: +49-30-284-60-603; E-mail: info@drfz.de
* To whom correspondence should be addressed.
Received July 14, 2014
The emergence of genetically engineered biological agents opened new prospects in the treatment of autoimmune and inflammatory diseases. Cytokines responsible for regulation of a wide range of processes during development of the normal immune response are among the most successful therapeutic targets. Studies carried out in recent decades and accompanied by rapid development of biotechnology have promoted establishing in detail the role and place of cytokines in autoimmune and inflammatory pathologies. Nevertheless, mechanisms that underlie anti-cytokine therapy are still not fully understood. This review examines the role of such cytokines as TNF, IL-1, and IL-6 in the development of inflammatory processes and the action mechanisms of their inhibitors.
KEY WORDS: cytokines, TNF, IL-6, IL-1, autoimmunity, chronic inflammationDOI: 10.1134/S0006297914120049
Cytokines regulate a wide range of physiological processes during development of the normal immune response, and they are involved in the pathogenesis of autoimmune diseases. Disorders in pro- and anti-inflammatory cytokines’ balance and recognizing self tissues as foreign by the immune system cells can trigger the development of autoimmune pathologies. Anti-cytokine therapy alone or combined with different classes of immunosuppressive drugs has been shown to be highly effective. Some cytokines have been shown to be good therapeutic targets for treatment of autoimmune and inflammatory diseases. A great number of anti-cytokine drugs are already successfully used in clinical practice, and more biologics are under development. In the present review, we consider the biological effects of three main pro-inflammatory cytokines: TNF (tumor necrosis factor), IL-1 (interleukin 1), and IL-6 (interleukin 6), as well as the action mechanisms of their inhibitors.
TNF AND ITS BIOLOGICAL EFFECTS
Tumor necrosis factor (TNF-α) was discovered in 1975 in L. Old’s laboratory as a specific product of lymphocytes and macrophages that induces lysis of definite type cells including tumor cells [1]. TNF became the “founder” of a whole family of homologous cytokines: LTα, CD40L, CD27L, FASL, etc. These cytokines transmit a signal through TNF family receptors (TNFR), which include such molecules as the FAS receptor (CD95, Apo-1) and the co-stimulatory receptor CD40. TNF is able to bind with two receptors of this family: TNFR1 and TNFR2 (Fig. 1a). TNFR1 (p55) is present on the membrane of various cells, whereas TNFR2 (p75) is mainly expressed by immunocytes and endothelial cells [2].
Fig. 1. Mechanism of action of TNF inhibitors. a) Structure of the TNF–TNFR system. Two receptors TNFR1 and TNFR2 are shown, which bind both soluble and membrane-bound (not shown) TNF trimer. The binding of TNF by monoclonal antibodies or by a recombinant soluble receptor prevents the interaction with receptors. b) Schemes of existing TNF inhibitors.
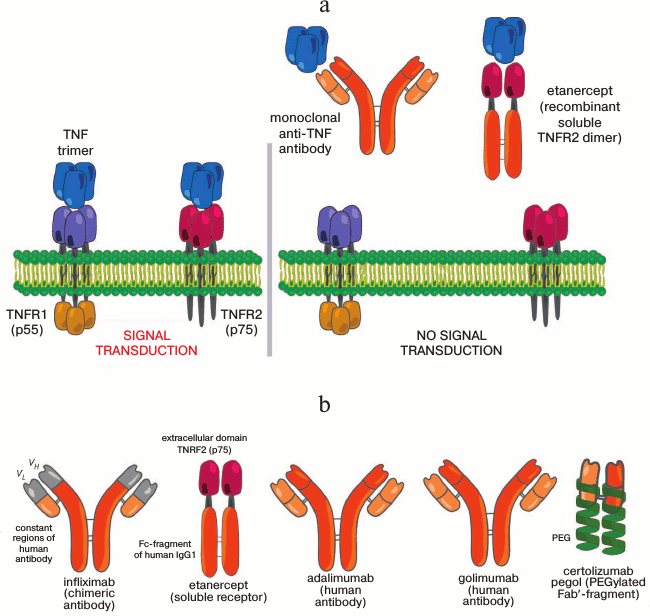
There are two forms of TNF and both are active as homotrimers: the membrane-bound one consisting of 26-kDa monomers and the soluble one consisting of 17-kDa monomers. Transmembrane TNF (mTNF) gives rise to the soluble form (sTNF) due to a specific cleavage of the polypeptide chain between amino acid residues 76 and 77 by the metalloproteinase ADAM-17 (TACE) [3]. The majority of proinflammatory functions of mTNF and sTNF are mediated through binding with TNFR1 and the downstream activation of transcription factors of the NF-κB and AP families, which, in turn, trigger the expression of the other proinflammatory cytokines including IL-1 and IL-6 [4]. The role of TNFR2 is not studied in detail, but the binding of mTNF to TNFR2 has been shown to contribute to the regulation of T-cell migration, proliferation, activation, antigen presentation, tissue repair, and angiogenesis [5-7]. During the development of inflammation, signal transduction through TNFR2 is necessary to stabilize the regulatory T-cell pool and prevent damage of self tissues [8].
The extracellular domains of TNFR1 and TNFR2 also can be shed under the influence of proteolytic enzymes and thus produce soluble forms of the receptors [9, 10]. For example, the soluble form of TNFR2 performs an immunoregulatory function and participates in pathogenesis of M. tuberculosis infection in mice [11].
The role of TNF in the development of inflammation is studied extensively. For instance, during pathogenesis of rheumatoid arthritis (RA), TNF stimulates endothelial cells to express integrins and adhesion molecules. TNF also induces the production and secretion of such chemokines as CXCL8, CCL2, and CCL5. The combination of these signals leads to extravasation of leukocytes and their subsequent accumulation in the site of inflammation. Moreover, TNF induces the differentiation of monocytes. The infiltrating, activated leucocytes in turn produce proinflammatory cytokines IL-1, IL-6, and TNF and thus provide a positive feedback regulation and promote inflammation. Furthermore, TNF activates synovial fibroblasts, which form an inflammatory pannus and destroy cartilage. Finally, TNF directly stimulates osteoclasts, which are responsible for resorption of bone tissue [12].
However, negative feedback mechanisms are also involved in these processes: signaling through TNFR2 suppresses osteoclastogenesis, and the transmembrane form of TNF acts as antagonist of osteoclast activation [13].
TNF INHIBITORS IN THERAPY OF RHEUMATOID ARTHRITIS AND OTHER
AUTOIMMUNE DISEASES
Clinical trials of TNF inhibitors have shown for the first time that inhibition of only one cytokine can break the vicious cycle of immune system activation, gain effective control of pathological inflammation symptoms, ameliorate deceleration of joint destruction, and in some cases can cause in patients a stable remission even after abolishing the anti-TNF therapy [14-17].
At present, TNF inhibitors are successfully used in the treatment of many autoimmune diseases. In the majority of developed countries, application of five different TNF inhibitors is approved (Fig. 1b and table).
Brief characteristics of modern anti-cytokine inhibitors
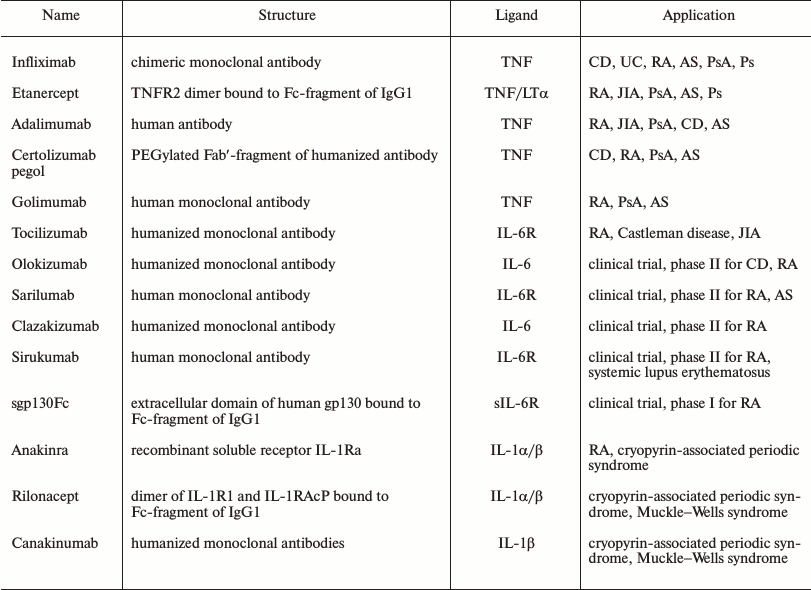
Notes: CD, Crohn’s disease; UC, ulcerative colitis; RA, rheumatoid
arthritis; AS, ankylosing spondyloarthritis; JIA, juvenile idiopathic
arthritis; PsA, psoriatic arthritis; Ps, psoriasis.
Infliximab was the first TNF inhibitor found useful for the treatment of patients with Crohn’s disease unresponsive to the conventional therapy [18, 19] and for the treatment of RA. Later, infliximab was shown to be effective also for the therapy of ankylosing spondyloarthritis [20, 21] and psoriasis [22].
Infliximab is a chimeric monoclonal antibody with constant regions of human IgG1 and variable domains of light and heavy chains of mouse origin. This antibody is highly specific and has a high affinity for both human sTNF (monomer and trimer) and human mTNF [23]. Its effects include suppression of cell migration, of infiltration of the synovial membrane, and of activation of leukocytes (including macrophages). Infliximab diminishes angiogenesis and osteoclastogenesis, which prevents damage to the bone tissue. Infliximab can induce lysis of cells carrying mTNF on their surface [24], i.e. participates in the antibody-dependent cellular and complement-mediated cytotoxicity.
Etanercept is another TNF inhibitor approved for the treatment of RA. Etanercept is a fusion protein consisting of the dimer of the extracellular TNF-binding domain of human TNFR2 bound with the Fc-fragment of human IgG1. Unlike the other inhibitors, etanercept inhibits lymphotoxin-α (LTα) as well as TNF. Physiological functions of this TNF-like cytokine are not yet studied in detail. However, LTα was recently shown to control IgA production in the intestine and influence the composition of intestinal microbiota independently of TNF and lymphotoxin-β [25]. Considering that etanercept, in contrast to infliximab, is inefficient in inflammatory intestinal diseases (including Crohn’s disease), this problem needs to be studied further. However, etanercept was shown to be effective in the treatment of ankylosing spondyloarthritis and psoriasis.
Later, adalimumab was also found useful for the treatment of patients with RA. Adalimumab is an antibody capable of binding and neutralizing human TNF (both the membrane-bound and soluble forms) that was prepared using a phage display method from the library of VL and VH fragments of human IgG1 [26, 27]. Adalimumab is now also used in the therapy of ankylosing spondyloarthritis, Crohn’s disease, and psoriasis.
In addition to the above-listed agents, two other TNF inhibitors are also approved: certolizumab pegol and golimumab [28]. Certolizumab pegol is a recombinant humanized Fab′-fragment specific to human TNF (hTNF) conjugated with a polyethylene glycol molecule of 40-kDa size [29] that ensures prolonged lifetime of the agents in the body. Because this inhibitor lacks the Fc-fragment, it cannot induce antibody-dependent cytotoxicity.
Golimumab is a monoclonal antibody against hTNF with a full human amino acid sequence that is already approved for the treatment of arthritis and ankylosing spondyloarthritis. This antibody was prepared using a unique technology in transgenic mice with the human immunoglobulin locus. Golimumab has the highest affinity for TNF among all inhibitors now used in clinical practice [30]. Due to absence of mouse antibody fragments, the immunogenicity of adalimumab and golimumab is lower than the immunogenicity of the chimeric antibody infliximab.
It should be noted that notwithstanding the about 20-year history of studies, the action mechanisms of anti-TNF preparations are still not well understood. Thus, it has been found only recently that inhibition of TNF mediated through non-immune mechanisms can lower pain in an inflamed joint, which is important in the treatment of arthritis [31]. In the collagen-induced model of arthritis in mice, it has been shown that inhibition of TNF leads, on one hand, to activation of Th1/Th17 cells, but, on the other hand, prevents their migration from lymph nodes into the inflammation locus [32]. Recent studies with RA patients also demonstrated that long-term anti-TNF therapy caused a decrease in the number of Th17 cells in joints [33]. Moreover, therapy with TNF inhibitors induced production of Th17 cells, which produced IL-10 [34], inhibited the development of germinal centers, decreased the number of memory B-cells [35], and restored the balance of regulatory T-cells [36, 37]. Notably, the list of potential complications associated with such therapy is increasing because TNF is involved in many physiological functions, and in some of them its role is unique.
IL-1 AND REGULATION OF ITS FUNCTIONS
Interleukin-1 (IL-1) was one of the first interleukins identified due to its biological activity [38]. Its molecular structure was determined in the middle 1980s, and thereafter some members of the IL-1 family were described and their important role in the development of the immune response was discovered.
IL-1 is represented by two proteins, IL-1α and IL-1β, which are encoded by two related genes, IL1A and IL1B. In addition to IL-1α and IL-1β, the family of IL-1 cytokines includes also the proinflammatory cytokine IL-18 and an antagonist of the IL-1 receptor (IL-1Ra) that has an anti-inflammatory action [39]. Recently, six new cytokines of this family have been described (from IL-1F5 to IL-1F10) [40-42]. Cytokines of the IL-1 family play an important role in immune response regulation and in the development of inflammation through the control of expression of many effector proteins, such as cytokines, chemokines, cytoplasmic metalloproteinases, etc. [43].
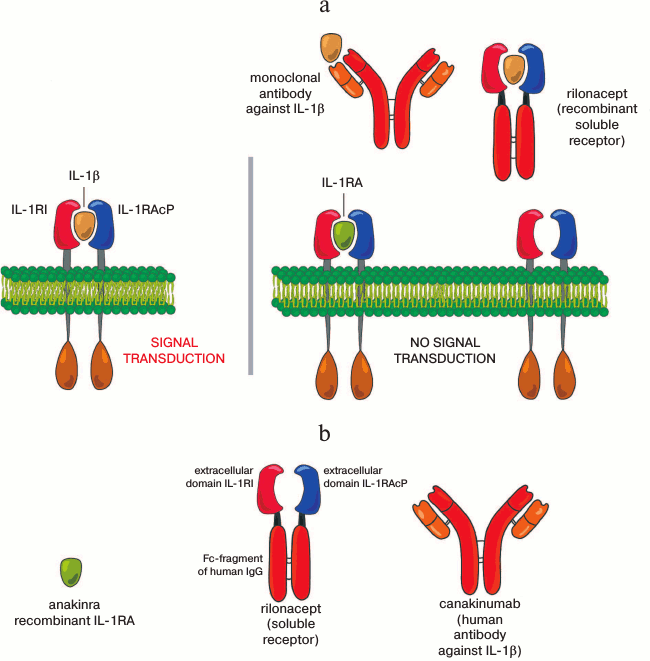
A signal from IL-1 cytokine family is transduced via a group of related receptors. These receptors contain in the cytoplasm an extracellular immunoglobulin domain and the Toll/IL-1 (TIR) receptor domain. In the case of IL-1α/β, the ligand initially binds to the primary subunit of the receptor (IL-1R1), and then the second subunit of the receptor (IL-1RAcP) binds to this complex (Fig. 2a). Formation of the heterodimer allows the TIR domain to bind the adaptor protein MyD88. The binding of MyD88 to the receptor heterodimer recruits signaling kinases IRAK1 and IRAK4. IRAK4 activates IRAK1, then the IRAK1–IRAK4 complex triggers a signaling cascade that results in the activation of the NF-κB and MAPK pathways [44, 45].
Although IL-1α and IL-1β use a common receptor for signaling, their effects are different. IL-1α is produced by the majority of cells of healthy humans. It is synthesized as a precursor associated with cytoskeleton elements (microtubules) mainly localized within the cell. Upon release from the cell, IL-1α acts locally. IL-1β cannot be detected in blood sera of healthy humans by standard tests. Components of the complement system, hypoxia, and blood coagulation can induce the transcription of IL-1β mRNA, but this is virtually not accompanied by translation of IL-1β because additional stimulation is necessary to initiate translation. Such stimulation can be carried out both by proteins of bacterial cells and by cytokines (TNF, IL-18, IL-1α) including IL-1β [43, 46]. In the presence of these factors, monocytes, macrophages, and dendritic cells produce IL-1β that circulates systemically.
The biological activity of IL-1 is strictly regulated at several levels. For activation of IL-1β, its precursor has to be processed by an intracellular proteinase – caspase 1. This proteinase, in turn, is activated on oligomerization of an intracellular complex of proteins that is called the “inflammasome” [47].
Earlier IL-1α was believed to be an endogenous signal of danger passively released during cell death. However, recently it was shown that the secretion of both IL-1α and IL-1β could be activated by the inflammasome both in vitro and in vivo [48].
When the active forms of these cytokines are released, their action can be downregulated through two pathways. First, IL-1RA can bind to the IL-1R1 receptor and thus prevent its binding with IL-1α and IL-1β. Moreover, the binding of IL-1RA with the receptor does not initiate signal transduction because no binding with IL-1RAcP occurs [39]. Second, the extracellular region of the second type IL-1 receptor (IL-1R2) is homologous to the IL-1R1 domain, but its short cytoplasmic domain is unable to transmit the intracellular signal. Thus, IL-1R2 acts as a decoy receptor, and it is supposed that its main role is to exclude excess of autocrine activation of the IL-1 signal [49, 50]. IL-1R2 can also be shed from the cell surface, and as a soluble protein it inhibits ligand binding with the receptor [51]. Finally, there is a soluble form IL-1RAcP that binds with soluble IL-1R2 and thus increasing its inhibitory activity [44, 52, 53].
ROLE OF IL-1 IN DEVELOPMENT OF CHRONIC INFLAMMATORY DISEASES AND
THERAPEUTIC INHIBITORS
Disorders in the regulation of synthesis and secretion of IL-1α/β can lead to serious pathologies. Many “classic” chronic inflammatory diseases are associated with increased level of IL-1α/β caused by constant activation of its synthesis and secretion due to activation of inflammasomes.
Note that cryopyrin-associated periodic syndrome is characterized by appearance of a mutation in a component of the inflammasome, cryopyrin, which stimulates the activity of caspase 1 and therefore leads to an increase in IL-1β secretion. Other chronic inflammatory diseases (gout, multiple sclerosis, hypertension) are characterized by hyperuricemia. Uric acid produces urate crystals that activate inflammasomes and also result in chronic inflammation mediated through IL-1. In type-2 diabetes, elevated glucose concentration increases the metabolic activity of Langerhans islets that leads to an increase in the concentration of reactive oxygen species and activation of the inflammasome. Moreover, the increase in glucose concentration stimulates the production of insulin, which induces stress of the endoplasmic reticulum that, in turn, activates the inflammasome. Altogether, this results in a closed cycle where the activation of the IL-1β pathway is stimulated by a positive feedback mechanism. Moreover, β-cells can produce an amyloid polypeptide that also activates synthesis of IL-1β through the inflammasome. Thus, the increased production of IL-1β can result in the loss of β-cells of the pancreas [54].
In the therapy of these chronic inflammatory diseases, the IL-1 inhibitors (anakinra, rylonacept, and canakinumab) are actively used [55-57]. These inhibitors block signal transduction through IL-1/IL-1R and thus interrupt the closed circle of inflammation.
Anakinra is a non-glycosylated recombinant soluble antagonist of the IL-1 receptor (IL-1RA) that differs from human IL-1RA by presence of an additional methionine. By binding to the IL-1 receptor, anakinra inhibits effects of both IL-1α and IL-1β. Rilonacept (IL-1 TRAP) is a recombinant dimeric chimeric protein consisting of extracellular domains of human proteins IL-1R1 and IL-1RAcP joined with the Fc-fragment of human IgG. This protein can bind with IL-1α/β and inhibit their functions. Canakinumab is a humanized monoclonal antibody against IL-1β (Fig. 2b and table).
Fig. 2. Mechanism of action of IL-1 inhibitors. a) Structure of the IL-1 receptor. The dimer IL-1RI and IL-1RAcP is represented in the complex with IL-1β. To the right: binding of the dimer IL-1RI and IL-1RAcP to the IL-1RA antagonist does not lead to signal transduction, whereas inhibition of IL-1β by monoclonal antibodies or by the recombinant soluble receptor prevents its binding with the receptor. b) Schemes of existing inhibitors of IL-1.
Similarly to TNF, IL-1 is a key mediator of pathological processes in tissues during RA [58]. IL-1β is secreted by macrophages, T-cells, fibroblasts, and chondrocytes of the synovial membrane, and its level is elevated in the synovial fluid of patients with RA [52, 59]. Along with TNF, IL-1 induces expression and production of some chemokines (CXCL12, MIP-1α, RANTES, and MCP-3) and adhesion molecules (VCAM-1, E-selectin, and ICAM-1) that bind to receptors on the surface of leukocytes and thus promote their penetration into the synovial membrane [60-62]. It seems that IL-1 can contribute to the development of RA through stimulating synovial fibroblasts to produce some cytokines, such as IL-15 and IL-18. In turn, IL-15 activates T-cells, macrophages (including TNF production), and fibroblasts of the synovial membrane. IL-18 promotes increase in IL-1 and TNF concentration by a positive feedback mechanism and also influences migration of T-cells and promotes angiogenesis [63]. Moreover, IL-1 lowers synthesis of intercellular matrix molecules, in particular, of proteoglycans in cartilaginous tissue [64], and introduction of the IL-1 receptor inhibits this effect [65]. Recently IL-1β was shown to induce proliferation of Th17 cells together with TGF-β, and on administration of anakinra the number of Th17 cells was significantly decreased, which correlated with a remission of patients with RA [66].
Clinical trial has shown that in RA anakinra abolishes the inflammation and reduces bone destruction, but these effects are weaker than effects of TNF inhibitors [67]. Neither TNF inhibitors nor anakinra are effective in sepsis [68, 69]. This might be because pathogenesis of RA and sepsis is associated not only with deregulation of the classical pathway of inflammation, but also with dysfunction of T-lymphocytes and humoral immune response [55]. However, in such diseases as gout and type-2 diabetes, therapy with IL-1 inhibitors is extremely efficient, whereas treatment with TNF antagonists is not only ineffective, but it can even aggravate the symptoms.
MECHANISMS OF SIGNALING AND PHYSIOLOGICAL FUNCTIONS OF
IL-6
IL-6 was discovered simultaneously in several laboratories and was initially known under different names, including interferon-β2. The cDNA of IL-6 was first cloned and expressed in 1986 [70].
IL-6 is a protein with length of 184 amino acids that forms a bundle of four antiparallel helices. It is a member of the cytokine family with similar three-dimensional structure [71]. The family includes IL-11, IL-27, IL-31, ciliary neurotrophic factor (CNTF), cardiotrophin-like cytokine (NNT-1), cardiotropin-1 (CT-1), neuropoietin, and oncostatin M [72] that contribute to the regulation of inflammation via the control of differentiation, proliferation, migration, and apoptosis of target cells. This cytokine family is characterized by signal transduction through the transmembrane protein gp130 (CD130), which is a common subunit of receptors of these cytokines.
The binding of IL-6 to the receptor causes dimerization of gp130, which leads to activation of tyrosine kinases of the JAK family. Activated JAKs, in turn, phosphorylate and activate transcription factors of the STAT family, e.g. STAT3 in the case of the IL-6 receptor [73, 74].
Note that the signal from IL-6 can be transmitted through both the “classical” mechanism and an alternative mechanism, so-called “trans-signaling”. In the classical pathway of signal transduction, IL-6 binds to the membrane-bound receptor of IL-6 (IL-6R), which induces association of this complex with the gp130 homodimer that then transmits the signal into the cell (Fig. 3a). Because IL-6R is expressed by limited cell types (hepatocytes, neutrophils, monocytes, macrophages, glia cells, neurons, and some lymphocytes), the influence of IL-6 is expected to be limited. On the other hand, it was known that IL-6 acts also on other cells. It occurred that during the proteolytic processing (shedding) of membrane-bound IL-6R by metalloproteinases ADAM10 and ADAM17 [71, 75], a soluble form of the IL-6 receptor (sIL-6R) can be produced. It seems that neutrophils [76], macrophages [77], and CD4+ lymphocytes [78] infiltrating inflamed tissue can be a source of sIL-6R. Moreover, sIL-6R can be produced also during alternative splicing of IL-6R mRNA. It is important that, unlike other soluble receptors (e.g. sTNFR), sIL-6R has features of an agonist and in complex with IL-6 can activate target cells if a sufficient number of protein gp130 molecules is present on their surface. Thus, even cells not expressing IL-6R on their surface can receive via sIL-R6 a signal from IL-6 due to “trans-signaling”, which increases the number of potential target cells for IL-6. Trans-signaling, in turn, can be inhibited by a soluble form of the gp130 protein (sgp130) that is produced by an alternative splicing of gp130 mRNA.
It was already noted that IL-6 was initially described as a factor of B-cell differentiation inducing their maturation to plasma cells [70]. At present, IL-6 is discovered to be a regulator of many other functions of both immune and non-immune cells and to be a connecting link between the immune, nervous, and endocrine systems. This cytokine influences the differentiation and growth of T-cells, activation of NK cells, maturation of megakaryocytes, development of osteoblasts, and synthesis of acute phase proteins in hepatocytes [79, 80]. IL-6 acts as a growth factor for myelomas, keratinocytes, mesaglia cells, renal carcinoma, and Kaposi sarcoma cells and stimulates the growth of hemopoietic stem cells [81, 82]. Moreover, IL-6 is essential for normal functioning of the brain; it is involved in the control of energy consumption and of pituitary–adrenal axis stimulation [83-85]. The role of IL-6 in blocking pain symptoms, regulation of sleep–wake cycles, emotional reactivity, and also in behavioral effects including learning and memory was described in IL-6-deficient mice (see review [86]).
Dysfunction of the regulatory network connected with this cytokine can lead to chronic and acute inflammations, autoimmune diseases, and neoplastic disorders. IL-6-deficient mice are resistant to development of collagen- and antigen-induced arthritis, indicating that IL-6 plays a key role in the development of chronic inflammation and autoimmune diseases [87].
THERAPEUTIC INHIBITION OF IL-6
For the defense of the body against such stresses as infections and traumas, IL-6 triggers a wide range of signaling events. When the “danger” disappears, the activation regulated by IL-6 is suppressed through a negative feedback system that normalizes the IL-6 level. However, the uncontrolled production of IL-6 can shift the balance to the Th17/Th1 side with reduction of regulatory T-cells (Treg) and can cause various autoimmune and chronic inflammatory diseases. It was shown in mouse models that IL-6 inhibition suppressed the differentiation of Th17/Th1 cells simultaneously with activation of Treg [88, 89]. Causes of IL-6 deregulation in vivo are not fully understood and are still being intensively studied.
It has been said already that IL-6 plays a key role in the development of collagen- and antigen-induced arthritis; moreover, overproduction of IL-6 in transgenic mice is associated with the development in them of a syndrome similar to Castleman disease, and the symptoms of inflammation decrease upon IL-6 blockade. These data and others have become a reasonable basis for anti-IL-6 therapy in chronic systemic inflammatory diseases.
Fig. 3. Action mechanism of IL-6 inhibitors. a) Structure of the ligand—receptor system for IL-6. The transmembrane form of IL-6R that binds IL-6 is represented in complex with two molecules of gp130. To the right: inhibition of IL-6 or IL-6R by monoclonal antibodies, as well as the capture of IL-6 by the soluble form of IL-6R in complex with recombinant soluble gp130-Fc prevents the activation of gp130 and subsequent transduction of the signal. b) Schematic figures of IL-6 inhibitors.
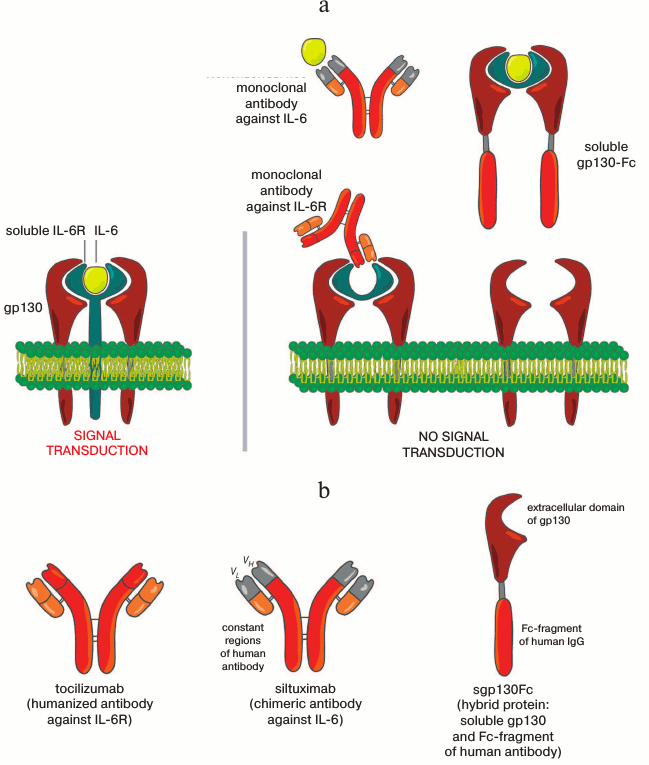
Tocilizumab was the first inhibitor of IL-6 approved for clinical application. It is a humanized monoclonal antibody to the IL-6 receptor (Fig. 3b and table). This antibody inhibits IL-6 due to binding with both IL-6R and sIL-6R. Tocilizumab was shown to improve the Th17/Treg balance in rheumatoid arthritis [90] and to normalize the blood serum level of amyloid A [91]. Roll et al. [92] found that tocilizumab restores the numbers of peripheral memory B-cells in patients with RA. Moreover, anti-IL-6 therapy was shown to induce the production of CD39+ Treg in a collagen-induced mouse model of RA [93]. Finally, therapy with tocilizumab decreased the number of pathologic CD38highCD19lowIgDneg plasma cells in patients with systemic lupus erythematosus and decreased the survival of plasmoblasts. Together, these data suggest that the positive clinical effect of tocilizumab may be partially due to inhibition of production of pathologic autoantibodies.
Tanaka et al. [94] compared the efficiency of tocilizumab with that of TNF inhibitors and concluded that their efficiencies were comparable if tocilizumab was used in combination with methotrexate. However, during monotherapy it is preferable to use tocilizumab. Moreover, in some cases patients who did not respond to therapy with TNF inhibitors reacted positively to treatment with tocilizumab. On the other hand, TNF inhibitors seem to be more effective in the treatment of ankylosing spondyloarthritis and inflammatory diseases of the intestine [88].
The successful clinical application of tocilizumab stimulated the development of other inhibitors of IL-6 (table). Data are now published on the second phase of clinical trial of such therapeutic antibodies as olokizumab, sarilumab, clazakizumab, and sirukumab [95].
Olokizumab and clazakizumab are humanized monoclonal antibodies capable of binding and neutralizing IL-6, whereas sarilumab and sirukumab are monoclonal antibodies against IL-6R. Sarilumab and sirukumab interact with both the membrane-bound and soluble IL-6 receptor, whereas anti-IL-6 antibodies bind with IL-6 and thus inhibit its binding with the receptors. All these therapeutic agents display similar clinical effects, and the efficiency and safety of these four new inhibitors of IL-6 are similar to those parameters of tocilizumab.
Still more data have been published recently showing that the IL-6 “trans-signaling” transmits signals about local and transient damage in the body but is poorly acting at the absence of stress conditions, whereas the classical pathway of signal transduction is responsible for maintaining homeostasis. Under pathological conditions, the levels of IL-6 and sIL-6R in the body are noticeably increased [96]. An important function of the IL-6 trans-signaling is to control the adaptive immunity including the differentiation of Th17 cells that are involved in the development of chronic inflammatory diseases, suppression of Treg proliferation, and the attraction of lymphocytes and neutrophils into an inflamed joint in arthritis. After extensive studies on the role and functions of the IL-6 trans-signaling, a chimeric protein sgp130Fc was constructed [87] consisting of an extracellular (soluble) domain of human gp130 protein and the Fc-fragment of human IgG1 (Fig. 3b). This protein can bind with the IL-6—sIL-6R complex but does not bind with sIL-6R. Thus, the protein sgp130Fc selectively inhibits the transduction of the IL-6 signal by “trans-signaling” without a noticeable effect on the classical pathway [97]. Injection of sgp130Fc into mice increased survival in the model of polymicrobial sepsis [98], reduced symptoms of collagen-induced arthritis [99], and lowered inflammation in Crohn’s disease, acute colitis, and systemic intestinal diseases [97, 100]. Now the IL-6 inhibitor based on the sgp130Fc protein is under clinical trial [87].
CONCLUSION
The introduction of anti-cytokine therapy in clinical practice for chronic inflammatory diseases has made a real revolution in medicine. However, such therapy affecting fundamental protective functions of the body mediated through particular cytokines has some limitations.
For instance, TNF is necessary for maintaining the integrity of granulomas on infection with intracellular pathogens such as Mycobacterium and Listeria; therefore, using TNF inhibitors is associated with an increased risk of development of latent opportunistic microbial infections [101]. There is a risk of activation of latent tuberculous infection on treatment with tocilizumab; however, up to now no case of reactivation of tuberculosis has been described during treatment with anakinra [102].
Cases of demyelinization were described during long-term therapy with TNF inhibitors, although it was believed that a systemically injected inhibitor could not penetrate across the blood–brain barrier. Studies on mice have shown that chronic inhibition of TNF action increases antigen-specific T-cellular response that can lead to survival of peripheral autoreactive myelin-specific T-cells. Moreover, suppression of TNF function through feedback mechanisms results in decrease in IL-10 secretion and consequently in increase in IL-12 and INFγ secretion that is specific for multiple sclerosis and leads to demyelinization [103].
The use of TNF inhibitors can lead to development of secondary autoimmune manifestations. Thus, binding of TNF inhibitors with the transmembrane TNF can result in the cell apoptosis, release of nuclear antigens, and production of autoantibodies. In some cases inhibition of TNF can lead even to appearance of symptoms of lupus erythematosus. Moreover, inhibition of TNF is associated with an abundant secretion of interferon α that, in turn, can result in thromboembolism. Concurrent infection can trigger these processes, as a result of inhibition of TNF [104].
New therapeutic blockers of all three main proinflammatory cytokines that would minimize adverse effects while maintaining the efficiency of current drugs are currently developing. In particular, studies on mouse models have shown that TNF secreted by different types of cells can execute both pathologic and protective functions [105]. We suppose that selective inhibition of TNF originating from particular cell sources would display better therapeutic effect if it did not affect the protective functions of TNF. Similarly to TNF, IL-6 also has a dual nature, but this is partially associated with different pathways of signal transduction. An inhibitor of IL-6 capable of interrupting the signal transduction only through trans-signaling [87] that is responsible for pathological effects of IL-6 is now already under the first phase of clinical trial. On the application of the first inhibitors of IL-1, the main side effect was inflammation at the injection site, partially caused by the need for frequent injections of the first inhibitor, anakinra, which had a short half-life (4-6 h) [55]. Inhibitors of the new generation (e.g. canakinumab) have a significantly more prolonged half-life.
The anti-cytokine therapy already actively used in various countries is based on comprehension of molecular mechanisms of pathological processes associated with expression of cytokines and undoubtedly will remain an innovative trend in clinical medicine for many years.
This work was supported by grants of the Russian Federation Government and Ministry of Education and Science of RF (contract No. 14.Z50.31.0008 of 19.03.2014), of the Russian Foundation for Basic Research (project No. 14-04-01656), and of the Russian Scientific Foundation (project No. 14-25-001600).
REFERENCES
1.Carswell, E. A., Old, L. J., Kassel, R. L., Green,
S., Fiore, N., and Williamson, B. (1975) An endotoxin serum factor that
causes necrosis of tumor, Proc. Natl. Acad. Sci. USA, 72,
3666-3670.
2.Tracey, D., Klareskog, L., Sasso, E. H., Salfeld,
J. G., and Tak, P. P. (2008) Tumor necrosis factor antagonist
mechanisms of action: a comprehensive review, Pharmacol.
Therap., 117, 244-279.
3.Black, R. A., Rauch, C. R., Kozlovsky, C. J.,
Peschon, J. J., Slack, J. L., Wolfson, M. F., Castner, B. J., Stocking,
K. L., Reddy, P., Srinivasan, S., Nelson, N., Boiani, N., Schooley, K.
A., Gerhart, M., Davis, R., Fitzner, J. N., Johnson, R. S., Paxton, R.
J., March, C. J., and Cerretti, D. P. (1997) A metalloproteinase
disintegrin that releases tumor-necrosis factor-alpha from cells,
Nature, 385, 729-733.
4.Simmonds, R. E., and Foxwell, B. M. (2008)
NF-κB and its relevance to arthritis and inflammation,
Rheumatology, 47, 584-590.
5.Grell, M., Douni, E., Wajant, H., Lohden, M.,
Clauss, M., Maxeiner, B., Georgopoulus, S., Lesslauer, W., Kollias, G.,
Pfizenmaier, K., and Scheurich, P. (1995) The transmembrane form of
tumor necrosis factor is the prime activating ligand of the 80 kDa
tumor necrosis factor receptor, Cell, 83, 793-802.
6.Grell, M., Becke, F. M., Wajant, H., Mannel, D. N.,
and Scheurich, P. (1998) TNF receptor type 2 mediates thymocyte
proliferation independently of TNF receptor type 1, Eur. J.
Immunol., 28, 257-263.
7.Carpentier, I., Coornaert, B., and Beyaert, R.
(2004) Function and regulation of tumor necrosis factor receptor type
2, Curr. Med. Chem., 11, 2205-2212.
8.Chen, X., Wu, X., Zhou, Q., Howard, O. M. Z.,
Netea, M. G., and Oppenheim, J. J. (2013) TNFR2 is critical for
stabilization of the CD4+FoxP3+ regulatory T cell phenotype in the
inflammatory environment, J. Immunol., 190,
1076-1084.
9.Redl, H., Schlag, G., Adolf, G. R., Natmessing, B.,
and Davies, J. (1995) Tumor necrosis factor (TNF)-dependent shedding of
the p55 TNF receptor in a baboon model of bacteremia, Infect.
Immun., 63, 297-300.
10.Aderka, D., Engelmann, H., Maor, Y., Brakebusch,
C., and Wallach, D. (1992) Stabilization of the bioactivity of tumor
necrosis factor by its soluble receptors, J. Exp. Med.,
175, 323-329.
11.Keeton, R., Allie, N., Dambuza, I., Abel, B.,
Hsu, N.-J., Sebesho, B., Randall, P., Burger, P., Fick, E., Quesniaux,
V. F. J., Ryffel, B., and Jacobs, M. (2014) Soluble TNFRp75 regulates
host protective immunity against Mycobacterium tuberculosis,
J. Clin. Invest., 124, 1537-1551.
12.Schett, G. (2009) Osteoimmunology in rheumatic
diseases, Arthr. Res. Therap., 11, 210-216.
13.Abu-Amer, Y., Erdmann, J., Kollias, G.,
Alexopoulou, L., Ross, P., and Teitelbaum, S. L. (2000) Tumor necrosis
factor receptors types 1 and 2 differentially regulate
osteoclastogenesis, J. Biol. Chem., 275, 27307-27310.
14.Requeiro, M., Kip, K. E., Baidoo, L., Swoqer, J.
M., and Schraut, W. (2014) Postoperative therapy with infliximab
prevents long-term Crohn’s disease recurrence, Clin.
Gastroenterol. Hepatol., 12, 1494-1502.
15.Verazza, S., Negro, G., Marafon, D., Consolaro,
A., Martini, A., and Ravelli, A. (2013) Possible discontinuation of
therapies after clinical remission in juvenile idiopathic arthritis,
Clin. Exp. Rheumatol., 31, S98-101.
16.Huang, Z., Yang, B., Shi, Y., Cai, B., Li, Y.,
Feng, W., Fu, Y., Luo, L., and Wang, L. (2012) Anti-TNF-α therapy
improves Treg and suppresses Teff in patients with rheumatoid
arthritis, Cell. Immunol., 279, 25-29.
17.Tanaka, Y., Hirata, S., Kubo, S., Fukuyo, S.,
Hanami, K., Sawamukai, N., Nakano, K., Nakayamada, S., Yamaoka, K.,
Sawamura, F., and Saito, K. (2013) Discontinuation of adalimumab after
achieving remission in patients with established rheumatoid arthritis:
1 year outcome of the HONOR study, Ann. Rheum. Dis.,
2013, Nov 28, doi: 10.1136/annrheumdis-2013-204016.
18.Targan, S. R., Hanauer, S. B., Van Deventer, S.
J., Mayer, L., Present, D. H., Braakman, T., DeWoody, K. L., Schaible,
T. F., and Rutgeerts, P. J. (1997) A short-term study of chimeric
monoclonal antibody cA2 to tumor necrosis factor alpha for
Crohn’s disease. Crohn’s disease cA2 study group, N.
Engl. J. Med., 337, 1029-1035.
19.Van Dullemen, H. M., Van Deventer, S. J., Hommes,
D. W., Bijl, H. A., Jansen, J., Tytgat, G. N., and Woody, J. (1995)
Treatment of Crohn’s disease with anti-tumor necrosis factor
chimeric monoclonal antibody (cA2), Gastroenterology,
109, 129-135.
20.Braun, J., Brandt, J., Listing, J., Zink, A.,
Alten, R., Golder, W., Gromnica-Ihle, E., Kellner, H., Krause, H.,
Schneider, M., Sorensen, H., Zeidler, H., Thriene, W., and Sieper, J.
(2002) Treatment of active ankylosing spondylitis with infliximab: a
randomized controlled multicentre trial, Lancet, 359,
1187-1193.
21.Kruithof, E., Van den Bosch, F., Baeten, D.,
Herssens, A., De Keyser, F., Mielants, H., and Veys, E. M. (2002)
Repeated infusion of infliximab, a chimeric anti-TNF-alpha monoclonal
antibody, in patients with active spondyloarthropathy: one years follow
up, Ann. Rheum. Dis., 61, 207-212.
22.Chaudhari, U., Romano, P., Mulcahy, L. D.,
Dooley, L. T., Baker, D. G., and Gottlieb, A. B. (2001) Efficacy and
safety of infliximab monotherapy for plaque-type psoriasis: randomized
trial, Lancet, 357, 1842-1847.
23.Knight, D. M., Trinh, H., Le, J., Siegel, S.,
Shealy, D., McDonough, M., Scallon, B., Moore, M. A., Vilcek, J., and
Daddona, P. (1993) Construction and initial characterization of a
mouse-human chimeric anti-TNF antibody, Mol. Immunol.,
30, 1143-1453.
24.Scallon, B. J., Moore, M. A., Trinh, H., Knight,
D. M., and Ghrayeb, J. (1995) Chimeric anti-TNF-alpha monoclonal
antibody cA2 binds recombinant transmembrane TNF-alpha and activates
immune effector functions, Cytokine, 7, 251-259.
25.Kruglov, A. A., Grivennikov, S. I., Kuprash, D.
V., Winsauer, C., Prepens, S., Seleznik, G. M., Ebert, G., Littman, D.
R., Heikenwalder, M., Tumanov, A. V., and Nedospasov, S. A. (2013)
Nonredundant function of soluble LTa3 produced by innate lymphoid cells
in intestinal homeostasis, Science, 342, 1243-1246.
26.Lukina, G. V., and Sigidin, Ya. A. (2008) Safety
of therapy with adalimumab, Nauch. Prakt. Revmatol., 2,
60-63.
27.Sigidin, Ya. A., and Lukina, G. V. (2008)
Adalimumab in therapy of early rheumatoid arthritis, Nauch. Prakt.
Revmatol., 2, 56-59.
28.Kay, J., and Rahman, U. (2009) Golimumab: a novel
human anti-TNF-α monoclonal antibody for the treatment of
rheumatoid arthritis, ankylosing spondylitis and psoriatic arthritis,
Core Evidence, 4, 159-170.
29.Lukina, G. V., and Sigidin, Ya. A. (2012)
Certolizumab in therapy of rheumatoid arthritis, Sovrem.
Revmatol., 2, 44-49.
30.Shealy, D., Cai, A., Staquet, K., Baker, A.,
Lacy, E. R., Johns, L., Vafa, O., Gunn III, G., Tam, S., Sague, S.,
Wang, D., Brigham-Burke, M., Dalmonte, P., Emmell, E., Pikounis, B.,
Bugelski, P. J., Zhou, H., Scallon, B., and Giles-Komar, J. (2010)
Characterization of golimumab, a human monoclonal antibody specific for
human tumor necrosis factor α, MAbs, 2,
428-439.
31.Schaible, H.-G., Von Anchet, G. S., Boettger, M.
K., Brauer, R., Gajda, M., Richter, F., Hensellek, S., Brenn, D., and
Natura, G. (2010) The role of proinflammatory cytokines in the
generation and maintenance of joint pain, Ann. NY Acad. Sci.,
1193, 60-69.
32.Notley, C. A., Inglis, J. J., Alzabin, S.,
McCann, F. E., McNamee, K. E., and Williams, R. O. (2008) Blockade of
tumor necrosis factor in collagen-induced arthritis reveals a novel
immunoregulatory pathway for Th1 and Th17 cells, J. Exp. Med.,
205, 2491-2497.
33.Szalay, B., Vasarhelyi, B., Cseh, A., Tulassay,
T., Deak, M., Kovacs, L., and Balog, A. (2013) The impact of
conventional DMARD and biological therapies on CD4+ cell subsets in
rheumatoid arthritis: a follow up study, Clin. Rheumatol.,
33, 175-185.
34.Evans, H. G., Roostalu, U., Walter, G. J.,
Gullick, N. J., Frederiksen, K. S., Roberts, C. A., Sumner, J., Baeten,
D. L., Gerwien, J. G., Cope, A. P., Geissmann, F., Kirkham, B. W., and
Taams, L. S. (2014) TNF-α blockade induces IL-10 expression in
human CD4+ T cells, Nat. Commun., 5, 3199-3211.
35.Anolik, J. H., Ravikumar, R., Barnard, J., Owen,
T., Almudevar, A., Milner, E. C., Miller, C. H., Dutcher, P. O.,
Hadley, J. A., and Sanz, I. (2008) Cutting edge: anti-tumor necrosis
factor therapy in rheumatoid arthritis inhibits memory B lymphocytes
via effects on lymphoid germinal centers and follicular dendritic cell
networks, J. Immunol., 180, 688-692.
36.Valencia, X., Stephens, G., Goldbach-Mansky, R.,
Wilson, M., Shevach, E. M., and Lipsky, P. E. (2006) TNF down-modulates
the function of human CD4+ CD25hi T-regulatory cells, Blood,
108, 253-261.
37.Nie, H., Zheng, Y., Li, R., Cuo, T. B., He, D.,
Fang, L., Liu, X., Xiao, L., Chen, X., Wan, B., Chin, Y. E., and Zhang,
J. Z. (2013) Phosphorylation of FOXP3 controls regulatory T cell
function and is inhibited by TNF-α in rheumatoid arthritis,
Nat. Med., 19, 322-328.
38.Dinarello, C. A. (1994) The interleukin-1 family:
10 years of discovery, FASEB J., 8, 1314-1325.
39.Arend, W. P., Malyak, M., Guthridge, C. J., and
Gabay, C. (1998) Interleukin-1 receptor antagonist: role in biology,
Ann. Rev. Immunol., 16, 27-55.
40.Magne, D., Palmer, G., Barton, J. L., Mezin, F.,
Talabot-Ayer, D., Bas, S., Duffy, T., Noger, M., Guerne, P.-A.,
Nicklin, M. J. H., and Gabay, C. (2006) The new IL-1 family member
IL-1F8 stimulates production of inflammatory mediators by synovial
fibroblast and articular, Arthr. Res. Ther., 8, R80.
41.Kumar, S., McDonnell, P. C., Lehr, R., Tierney,
L., Tzimas, M. N., Griswold, D. E., Capper, E. A., Tal-Singer, R.,
Wells, G. I., Doyle, M. L., and Young, P. R. (2000) Identification and
initial characterization of four novel members of the interleukin-1
family, J. Biol. Chem., 275, 10308-10314.
42.Smith, D. E., Renshaw, B. R., Ketchem, R. R.,
Kubin, M., Garka, K. E., and Sims, J. E. (2001) Four new members expand
the interleukin-1 family, J. Biol. Chem., 275,
1169-1175.
43.Dinarello, C. A. (1996) Biologic basis for
interleukin-1 in disease, Blood, 87, 2095-2147.
44.Sims, J. E., and Smith, D. E. (2010) The IL-1
family: regulators of immunity, Nat. Rev. Immunol., 10,
89-102.
45.O’Neill, L. A. J. (2008) The interleukin-1
receptor/Toll-like receptor superfamily: 10 years of progress,
Immunol. Rev., 226, 10-18.
46.Garlanda, C., Dinarello, C. A., and Mantovani, A.
(2013) The interleukin-1 family: back to the future, Immunity,
39, 1003-1018.
47.Martinon, F., Mayor, A., and Tschopp, J. (2009)
The inflammasomes: guardians of the body, Ann. Rev. Immunol.,
27, 229-269.
48.Gross, O., Yazdi, A. S., Thomas, C. J., Masin,
M., Heinz, L. X., Guarda, G., Quadroni, M., Drexler, S. K., and
Tschopp, J. (2012) Inflammasome activators induce interleukin-1α
secretion via distinct pathways with differential requirement for the
protease function of caspase-1, Immunity, 36,
388-400.
49.Colotta, F., Re, F., Muzio, M., Bertini, R.,
Polentarutti, N., Sironi, M., Giri, J. G., Dower, S. K., Sims, J. E.,
and Mantovani, A. (1993) Interleukin-1 type II receptor: a decoy target
for IL-1 that is regulated by IL-4, Science, 261,
472-475.
50.Colotta, F., Dower, S. K., Sims, J. E., and
Mantovani, A. (1994) The type II “decoy” receptor: a novel
regulatory pathway for interleukin 1, Immunol. Today, 15,
562-528.
51.Penton-Rol, G., Orlando, S., Polentarytti, N.,
Bernasconi, S., Muzio, M., Introna, M., and Mantovani, A. (1999)
Bacterial lipopolysaccharide causes rapid shedding, followed by
inhibition of mRNA expression, of the IL-1 type II receptor, with
concomitant up-regulation of the type I receptor and induction of
incompletely spliced transcript, J. Immunol., 162,
2931-2938.
52.Barksby, H. E., Lea, S. R., and Preshaw, P. M.
(2007) The expanding family of interleukin-1 cytokines and their role
in destructive inflammatory disorders, Clin. Exp. Immunol.,
149, 217-225.
53.Smith, D. E., Hanna, R., Friend, D., Moore, H.,
Chen, H., Farese, A. M., MacVittie, T. J., Virca, G. D., and Sims, J.
E. (2003) The soluble form of IL-1 receptor accessory protein enhances
the ability of soluble type II IL-1 receptor to inhibit IL-1 action,
Immunity, 18, 87-96.
54.Donath, M. Y. (2014) Targeting inflammation in
the treatment of type 2 diabetes: time to start, Nature Rev. Drug
Discov., 13, 465-476.
55.Dinarello, C. A., and van der Meer, J. W. M.
(2013) Treating inflammation by blocking interleukin-1 in humans,
Semin. Immunol., 25, 469-484.
56.Khanna, P., Gladue, H. S., Singh, M. K.,
FitzGerald, D., Bae, S., Prakash, S., Kaldas, M., Gogia, M., Berrocal,
V., Townsend, W., Terkeltaub, R., and Khanna, D. (2014) Treatment of
acute gout: a systematic review, Sem. Arthritis Rheum.,
44, 31-38.
57.Sterba, G., and Sterba, Y. (2013) Controlling
inflammation. Contemporary treatment for autoinflammatory diseases and
syndromes, Dermatol. Clin., 31, 507-511.
58.Gabay, C., and Arend, W. P. (1998) Treatment of
rheumatoid arthritis with IL-1 inhibitors, Springer Semin.
Immunopathol., 20, 229-246.
59.Bunning, R. A., Richardson, H. J., Crawford, A.,
Skiodt, H., Hughes, D., Evans, D. B., Gowen, M., Dobson, P. R., Brown,
B. L., and Russell, R. (1986) The effect of interleukin-1 on connective
tissue metabolism and its relevance to arthritis, Agents Actions
Suppl., 18, 131-152.
60.Volin, M. V., Shah, M. R., Tokuhira, M., Haines,
G. K., Woods, J. M., and Koch, A. E. (1998) RANTES expression and
contribution to monocyte chemotaxis in arthritis, Clin. Immunol.
Immunopathol., 89, 44-53.
61.Nakatsuka, K., Tanaka, Y., Hubscher, S., Abe, M.,
Wake, A., Saito, K., Morimoto, I., and Eto, S. (1997) Rheumatoid
synovial fibroblasts are stimulated by the cellular adhesion to T cells
through lymphocyte function associated antigen-1/intercellular adhesion
molecule-1, J. Rheumatol., 24, 458-464.
62.Garcia-Hernandez, M. H., Gonzalez-Amaro, R., and
Portales-Perez, D. P. (2014) Specific therapy to regulate inflammation
in rheumatoid arthritis: molecular aspects, Immunotherapy,
6, 623-636.
63.Arend, W. P., and Gabay, C. (2004) Cytokines in
the rheumatic diseases, Rheum. Dis. Clin. N. Am., 30,
41-67.
64.Chandrasekhar, S., and Phadke, K. (1988)
Interleukin-1-induced alterations in proteoglycan metabolism and matrix
assembly, Arch. Biochem. Biophys., 265, 294-301.
65.Murata, M., Bonassar, L. J., Wright, M., Mankin,
H. J., and Towle, C. A. (2003) A role for the interleukin-1 receptor in
the pathway linking static mechanical compression to decreased
proteoglycan synthesis in surface articular cartilage, Arch.
Biochem. Biophys., 413, 229-235.
66.Ikeda, S., Saijo, S., Murayama, M. A., Shimizu,
K., Akitsu, A., and Iwakura, Y. (2014) Excess IL-1 signaling enhances
the development of Th17 cells by down-regulating TGF-β-induced
Foxp3 expression, J. Immunol., 192, 1449-1458.
67.Brennan, F. M., and McInnes, I. B. (2008)
Evidence that cytokines play a role in rheumatoid arthritis, J.
Clin. Invest., 118, 3537-3545.
68.Opal, S. M., Fisher, C. J., Dhainaut, J. F.,
Vincent, J. L., Brase, R., Lowry, S. F., Sadoff, J. C., Slotman, G. J.,
Levy, H., Balk, R. A., Shelly, M. P., Pribble, J. P., LaBrecque, J. F.,
Lookabaugh, J., Donovan, H., Dubin, H., Baughman, R., Noeman, J.,
DeMaria, E., Matzek, K., Abraham, E., and Seneff, M. (1997)
Confirmatory interleukin-1 receptor antagonist trial in severe sepsis:
a phase III, randomized, double-blind, placebo-controlled, multicenter
trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group,
Crit. Care Med., 25, 1115-1124.
69.Fisher, C. J., Dhainaut, J. F., Opal, S. M.,
Pribble, J. P., Balk, R. A., Slotman, G. J., Iberti, T. J., Rackow, E.
C., Shapiro, M., and Greenman, R. L. (1994) Recombinant human
interleukin 1 receptor antagonist in the treatment of patients with
sepsis syndrome. Results from a randomized, double-blind,
placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study
Group, JAMA, 271, 1836-1843.
70.Hirano, T., Yasukawa, K., Harada, H., Taga, T.,
Watanabe, Y., Matsuda, T., Kashiwamura, S., Nakajima, K., Koyama, K.,
Iwamatsu, A., Tsunasawa, S., Sakiyama, F., Matsu, H., Takahara, Y.,
Taniguchi, T., and Kishimoto, T. (1986) Complementary DNA for a novel
human interleukin (BSF-2) that induces B lymphocytes to produce
immunoglobulin, Nature, 324, 73-76.
71.Rose-John, S. (2012) IL-6 trans-signaling
via the soluble IL-6 receptor: importance for the pro-inflammatory
activities of IL-6, Int. J. Biol. Sci., 8, 1237-1247.
72.White, U. A., and Stephens, J. M. (2011) The
gp130 receptor cytokine family: regulators of adipocyte development and
function, Curr. Pharm. Des., 17, 340-346.
73.Kishimoto, J., Akira, S., and Taga, T. (1992)
IL-6 receptor mechanism of signal transduction, Int. J.
Immunopharmacol., 14, 431-438.
74.Heinrich, P. C., Behrmann, I., Muller-Newen, G.,
Schaper, F., and Graeve, L. (1998) Interleukin-6-type cytokine
signaling through the gp130/JAK/STAT pathway, Biochem. J.,
334, 297-314.
75.Mullberg, J., Schooltink, H., Stoyan, T.,
Gunther, M., Graeve, L., Buse, G., Mackiewicz, A., Heinrich, P. C., and
Rose-John, S. (1993) The soluble interleukin-6 receptor is generated by
shedding, Eur. J. Immunol., 23, 473-480.
76.Hurst, S. M., Wilkinson, T. S., McLoughlin, R.
M., Jones, S., Horiuchi, S., Yamamoto, N., Rose-John, S., Fuller, G.
M., Topley, N., and Jones, S. A. (2001) IL-6 and its soluble receptor
orchestrate a temporal switch in the pattern of leukocyte recruitment
seen during acute inflammation, Immunity, 14,
706-714.
77.Horiuchi, S., Koyanagi, Y., Miyamoto, H., Tanaka,
Y., and Waki, M. (1994) Soluble interleukin-6 receptors released from T
cell or granulocyte/macrophage cell lines and human peripheral blood
mononuclear cells are generated through an alternative splicing
mechanism, Eur. J. Immunol., 24, 1945-1948.
78.Briso, E. M., Dienz, O., and Rincon, M. (2008)
Cutting edge: soluble IL-6R is produced by IL-6R ectodomain shedding
activates CD4 T cell, J. Immunol., 180, 7102-7106.
79.Lotz, M., Jirik, F., Kabouridis, P., Tsoukas, C.,
Hirano, T., Kishimoto, T., and Carson, D. A. (1988) B cell stimulating
factor 2/interleukin 6 is a costimulant for human thymocytes and T
lymphocytes, J. Exp. Med., 167, 1253-1258.
80.Sehgal, P. B. (1990) Interleukin-6: molecular
pathophysiology, J. Invest. Dermatol., 94, 2S-6S.
81.Hirano, T. (1998) Interleukin 6 and its receptor:
ten years later, Int. Rev. Immunol., 16, 249-284.
82.Striz, I., Brabcova, E., Kolesar, L., and
Sekerkova, A. (2014) Cytokine networking of innate immunity cells: a
potential target of therapy, Clin. Sci., 126,
593-612.
83.Marz, P., Cheng, J.-G., Gadient, R. A.,
Patterson, P. H., Stoyan, T., Otten, U., and Rose-John, S. (1998)
Sympathetic neurons can produce and respond to interleukin 6, Proc.
Natl. Acad. Sci. USA, 95, 3251-3256.
84.Streit, W. J., Hurley, S. D., McGraw, T. S., and
Semple-Rowland, S. L. (2000) Comparative evaluation of cytokine
profiles and reactive gliosis supports a critical role from
interleukin-6 in neuroglia signaling during regeneration, J.
Neurosci. Res., 61, 10-20.
85.Wallenius, V., Wallenius, K., Ahren, B., Rudling,
M., Carlsten, H., Dickson, S. L., Ohlsson, C., and Jansson, J. O.
(2002) Interleukin-6-deficient mice develop mature-onset obesity,
Nature Med., 8, 75-79.
86.Erta, M., Quintana, A., and Hidalgo, J. (2012)
Interleukin-6, major cytokine in the central nervous system, Int. J.
Biol. Sci., 8, 1254-1266.
87.Scheller, J., Garbers, C., and Rose-John, S.
(2014) Interleukin-6: from basic to selective blockade of
pro-inflammatory activities, Semin. Immunol., 26,
2-12.
88.Tanaka, T., and Kishimoto, T. (2012) Targeting
interleukin-6: all the way to treat autoimmune and inflammatory
disease, Int. J. Biol. Sci., 8, 1227-1236.
89.Kimura, A., and Kishimoto, T. (2010) IL-6:
regulator of Treg/Th17 balance, Eur. J. Immunol., 40,
1830-1835.
90.Samson, M., Audia, S., Janikashvili, N., Ciudad,
M., Trad, M., Fraszczak, J., Ornetti, P., Maillefert, J. F., Miossec,
P., and Bonnotte, B. (2012) Brief report: inhibition of interleukin-6
function corrects TH17/Treg cell imbalance in patients with rheumatoid
arthritis, Arthritis Rheum., 64, 2499-24503.
91.Nishida, S., Haqihara, K., Shima, Y., Kawai, M.,
Kuwahara, Y., Arimitsu, J., Hirano, T., Narazaki, M., Ogata, A.,
Yoshizaki, K., Kawase, I., Kishimoto, T., and Tanaka, T. (2009) Rapid
improvement of AA amyloidosis with humanized anti-interleukin 6
receptor antibody treatment, Ann. Rheum. Dis., 68,
1235-1236.
92.Roll, P., Muhammad, K., Schumann, M., Kleinert,
S., Einsele, H., Dorner, T., and Tony, H. P. (2011) In vivo
effect of the anti-interleukin-6 receptor tocilizumab on the B cell
compartment, Arthritis Rheum., 63, 1255-1264.
93.Thiolat, A., Semerano, L., Pers, Y. M., Biton,
J., Lemeiter, D., Portales, P., Quentin, J., Jorgensen, C., Decker, P.,
Boissier, M. C., Louis-Plence, P., and Bessis, N. (2014) Interleukin-6
receptor blockade enhances CD39+ regulatory T cell development in
rheumatoid arthritis and in experimental arthritis, Arthritis
Rheum., 66, 273-283.
94.Tanaka, T., Hishitani, Y., and Ogata, A. (2014)
Monoclonal antibodies in rheumatoid arthritis: comparative
effectiveness of tocilizumab with tumor necrosis factor inhibitors,
Biologics: Targets Therapy, 8, 141-153.
95.Tanaka, Y., and Mola, E. M. (2014) IL-6 targeting
compared to TNF targeting in rheumatoid arthritis: studies of
olokizumab, sarilumab and sirukumab, Ann. Rheum. Dis.,
73, 1595-1597.
96.Kallen, K.-J. (2002) The role of
trans-signaling via the agonistic soluble IL-6 receptor in human
disease, Biochim. Biophys. Acta, 1592, 323-343.
97.Chalaris, A., Schmidt-Arras, D., Yamamoto, K.,
and Rose-John, S. (2012) Interleukin-6 trans-signaling and
colonic cancer associated with inflammatory bowel disease, Digest.
Dis., 30, 492-499.
98.Barkhausen, T., Tschernig, T., Rosenstiel, T.,
van Griensven, M., Vonberg, R.-P., Dorsch, M., Mueller-Heine, A.,
Chalaris, A., Scheller, J., Rose-John, S., Seegert, D., Krettek, C.,
and Waetzig, G. (2011) Selective blockade of interleukin-6
trans-signaling improves survival in a murine polymicrobial
sepsis model, Crit. Care Med., 39, 1407-1413.
99.Nowell, M. A., Williams, A. S., Carty, S. A.,
Scheller, J., Hayes, A. J., Jones, G. W., Richards, P. J., Slinn, S.,
Ernst, M., Jemkins, B. J., Topley, N., Rose-John, S., and Jones, S. A.
(2009) Therapeutic targeting of IL-6 trans-signaling counteracts
STAT3 control of experimental inflammatory arthritis, J.
Immunol., 182, 613-622.
100.Atreys, R., Mudter, J., Finotto, S., Mullberg,
J., Jostock, T., Wirtz, S., Schutz, M., Bartsch, B., Holtmann, M.,
Becker, C., Strand, D., Czaja, J., Schlaak, J. F., Lehr, Y. A.,
Autschbach, F., Schurmann, G., Nishimoto, N., Yoshizaki, K., Ito, H.,
Kishimoto, T., Galle, P. R., Rose-John, S., and Neurath, M. F. (2000)
Blockade of interleukin 6 trans-signaling suppresses T-cell
resistance against apoptosis in chronic intestinal inflammation:
evidence in Crohn’s disease and experimental colitis in
vivo, Nature Med., 6, 583-588.
101.Ramiro, S., Gaujoux-Viala, C., Nam, J. L.,
Smolen, J. S., Buch, M., Gossec, L., Van Der Heijde, D., Winthrop, K.,
and Landewe, R. (2014) Safety of synthetic and biological DMARDs: a
systematic literature review informing the 2013 update of the EULAR
recommendations for management of rheumatoid arthritis, Ann. Rheum.
Dis., 73, 529-535.
102.Rubbert-Roth, A. (2012) Assessing the safety of
biologic agents in patients with rheumatoid arthritis,
Rheumatology, 51, 38-47.
103.Kaltsonoudis, E., Voulgari, P. V., Konitsiois,
S., and Drosos, A. A. (2014) Demyelination and other neurological
adverse events after anti-TNF therapy, Autoimmun. Rev.,
13, 54-58.
104.Prinz, J. C. (2011) Autoimmune-like syndromes
during TNF blockade: does infection have a role? Nat. Rev.
Rheumatol., 7, 429-434.
105.Winsauer, C., Kruglov, A. A., Chashchina, A.
A., Drutskaya, M. S., and Nedospasov, S. A. (2014) Cellular sources of
pathogenic and protective TNF and experimental strategies on
utilization of TNF humanized mice, Cytokine Growth Factor Rev.,
25, 115-123.