Epitope Mapping of Lymphocyte Phosphatase-Associated Phosphoprotein
A. V. Filatov1*, T. D. Meshkova2, and D. V. Mazurov1
1Institute of Immunology, Federal Medical-Biological Agency, Kashirskoe Shosse 24/2, 115478 Moscow, Russia; fax: (499) 617-7765; E-mail: avfilat@rambler.ru2Faculty of Biology, Lomonosov Moscow State University, 119234 Moscow, Russia; E-mail: meshkova.tatiana@yandex.ru
* To whom correspondence should be addressed.
Received July 12, 2014
Lymphocyte phosphatase-associated phosphoprotein (LPAP) is a transmembrane protein with unknown function. The available data on its close association with phosphatase CD45 and its phosphorylation depending on cell activation suggest that LPAP can play a significant role in the antigenic stimulation of lymphocytes. We have localized three antigenic epitopes of the LPAP molecule that can be detected using monoclonal antibodies prepared earlier. Experiments on reactions of antibodies with point mutants and shortened forms of the LPAP protein revealed regions of the amino acid sequence that correspond to the epitopes recognized by the antibodies.
KEY WORDS: LPAP, monoclonal antibodies, site directed mutagenesis, human lymphocytesDOI: 10.1134/S0006297914120153
Abbreviations: BSA, bovine serum albumin; CD45-AP, CD45-associated protein; FITC, fluorescein isothiocyanate; KLH, keyhole limpet hemocyanin; LPAP, lymphocyte phosphatase-associated phosphoprotein; PBS, phosphate buffered saline; PMSF, phenylmethylsulfonyl fluoride; R6G-SE, rhodamine 6G succinimidyl ester; SDS, sodium dodecyl sulfate.
In the 1990s, lymphocyte phosphatase-associated phosphoprotein (LPAP)
was described [1]. The mouse homolog of LPAP is
more frequently called CD45-associated protein (CD45-AP) [2]. LPAP was discovered due to its close association
with phosphatase CD45, which is reflected by its name. About 75% of the
total amount of CD45 and LPAP exist as an intermolecular complex [3]. Phosphatase CD45 is known to be a key regulator
that controls the efficiency of signal transduction from the T cell
receptor upon its binding with an antigen [4].
Because LPAP is in a complex with phosphatase CD45, it was supposed
that protein LPAP could control the activity of phosphatase CD45 and
thus have very important regulatory functions [5].
These hypotheses might be confirmed by experiments on knockout mice;
however, the data obtained on LPAP-deficient mice are contradictory.
Two groups of researchers found that mice with LPAP gene knockout
demonstrated normal development of the thymus and normal proliferative
response to mitogens [6, 7]. On
the contrary, in independent work [8] the
interaction between CD45 and Lck was shown to be decreased in
LPAP-deficient mice. The contradiction in these results was thought to
be due to the more accurate regulation of CD45 activity by LPAP than
was believed earlier [9]. Notwithstanding the
studies performed, the functions of LPAP are still unclear [10].
The functional potential of the LPAP may be more or less evaluated basing on its structural features. In LPAP, there are several functionally important domains. First, a transmembrane domain of LPAP is responsible for its association with phosphatase CD45. This was shown in experiments with chimeric CD45 and deletion-carrying mutants of LPAP [11-13]. The interaction with CD45 is also contributed to by an intracellular region of LPAP that is next to the transmembrane domain and consists of 15 amino acid residues (a.a.) [13]. In the region of a.a. 108-168, there is an “acidic domain” enriched with residues of glutamic and aspartic acids that is involved with the interaction of LPAP with kinase Lck [14]. The cytoplasmic part of LPAP contains a sequence that is reminiscent of the WW-domain [12]. Such a domain is involved in interactions of some proteins. However, the structure of LPAP is still poorly known. Partially, this is due to the absence of available monoclonal antibodies reacting with different regions of the LPAP protein. In the literature, only polyclonal antibodies against LPAP are described, and these were prepared by immunization with individual peptides [1] or with the entire cytoplasmic domain of LPAP [15]. But polyclonal antibodies do not allow detailed study of the epitope and domain organization of LPAP.
Earlier, we prepared a series of monoclonal antibodies against the LPAP protein [16]. In the present work, these antibodies have been used to study in more detail the antigenic topology of LPAP protein.
MATERIALS AND METHODS
Cell cultures, antibodies, and flow cytometry. Human cell cultures CCRF-CEM (T-lymphoblastoid line) and HEK293T (embryonal kidney cells) were cultured, respectively, in RPMI-1640 and DMEM media supplemented with 10% fetal calf serum, L-glutamine (4 mM), and gentamycin (80 mg/liter) in a humidified atmosphere with 5% CO2 at 37°C. Antibodies CL3, CL4, and CL7 against LPAP were prepared by us earlier [16]. The antibody OKT4 (IgG1, anti-CD4) was kindly presented by M. B. Zaitseva (Center for Biologics Evaluation and Research, USA). The M2 antibody (Sigma, USA) was used against the FLAG epitope.
Antibody binding was assessed using an immunofluorescence test. For intercellular staining, the cells were fixed with 1% solution of paraformaldehyde in PBS and permeabilized with 0.1% saponin solution supplemented with 5% defatted dry milk. All subsequent incubations and washings were performed in the permeabilizing solution. The cells (5·105) in volume 50 µl were treated with monoclonal antibodies (10 µg/ml) at 4°C for 30 min and then washed twice by centrifugation at 500g for 5 min. Then the cells in 50-µl volume were treated with goat antibodies against mouse Ig labeled with phycoerythrin (1 µg/ml) (Santa Cruz, USA) at 4°C for 30 min. The stained cells were analyzed with a FACScan flow cytometer (Becton Dickinson, USA) using the Cell Quest program software. The fluorescence intensity was measured in relative units by evaluating the mean fluorescence intensity. The cells treated only with goat antibodies were used as a negative control. To prepare antibodies directly labeled with FITC, the isolated antibodies at the concentration of 4 mg/ml in 500 mM bicarbonate buffer (pH 9.5) were supplemented with FITC (Invitrogen, USA) at ratio 100 µg dye per mg protein. The reaction was performed during 1 h at room temperature, and then the labeled antibodies were separated from the free dye by gel filtration on a PD-10 column (GE Healthcare, USA).
Fluorescent immunoprecipitation assay. The CCRF-CEM cells were washed in PBS and labeled with a fluorescent dye – rhodamine 6G succinimidyl ester (R6G-SE) (Syntol, Russia). To do this, the cells (4·106) were suspended in 1 ml of PBS and supplemented with 15 µl of R6G-SE solution in dimethylsulfoxide (10 mg/ml). After 10 min, 15 µl of R6G-SE solution was added. Ten minutes after the second addition of the fluorochrome, the cells were washed thrice in PBS. Cell lysate was prepared by suspending the cell precipitate in 1 ml of cold lysing buffer (20 mM Tris-HCl, pH 8.0, containing 1% detergent Triton X-100, 150 mM NaCl, 5 mM EDTA, 1 mM PMSF, 2 mM NaF, 0.5 mM Na2VO4) (Fluka, Switzerland). After 30 min of incubation on ice, the nuclei and cell debris were removed by centrifugation at 15,000g for 15 min at 4°C. The resulting supernatant was clarified twice by incubation during 4 h with 100 µl of BrCN-Sepharose (GE Healthcare) with covalently immobilized normal mouse IgG. The sample was immunoprecipitated with 40 µl of agarose AffiGel Hz (Bio-Rad, USA) containing covalently immobilized antibody CL7 during 4 h or overnight at 4°C. The immunoprecipitates were washed thrice on adding every time 800 µl of the lysing buffer. The bound fluorescently labeled proteins were eluted from the sorbent by heating the immunoprecipitates with the buffer of a sample for electrophoresis for 5 min at 80°C. The eluted proteins were separated by SDS-PAGE in 12% polyacrylamide gel under reducing conditions in the Laemmli buffer system. Prestained Low Range proteins (Bio-Rad) were used as markers of molecular weight. After electrophoresis, the proteins were visualized in the gel using a Molecular Imager FX Pro fluorescence scanner (Bio-Rad) without an additional fixation, staining, or drying of the gel [17].
Western blotting. The cellular lysate was separated by SDS-PAGE in 12% polyacrylamide gel under non-reducing conditions in the Laemmli buffer system. The separated proteins were transferred onto a PVDF membrane using a Mini Trans-Blot device (Bio-Rad) during 1 h at voltage 100-120 V in the following buffer: 25 mM Tris, 192 mM glycine (pH 8.3), 20% ethanol (v/v), 0.1% SDS. The membranes were blocked overnight at 4°C with 5% dry defatted milk in PBS with 0.1% Tween-20 and incubated for 1 h at room temperature with antibodies CL3, CL4, and CL7 against LPAP or with antibody M2 against the FLAG peptide. The membrane was washed and incubated with goat antibodies against mouse Ig labeled with horseradish peroxidase (Sorbent, Russia). The blot was visualized by chemiluminescence with ECL reagent (Millipore, USA) using a ChemiDoc device (Bio-Rad).
Plasmids and cell transfection. A DNA sequence encoding the full-size human LPAP protein was prepared by us earlier [16]. On the C-terminus of LPAP before the stop codon, the sequence DYKDDDDK (FLAG epitope) was inserted to provide additional recognition of the protein with the anti-FLAG antibody M2. Mutagenesis of LPAP was performed using a standard PCR approach with overlapping primers or by the method of Quick Change PCR Mutagenesis (Stratagene, USA). Shortened forms of LPAP were prepared by inserting stop codons into definite positions. As external primers, the following oligonucleotides were used:
5′-EcoRI-XbaI-LPAP:
CCGAATTCTCTAGACCATGGCTCTGCCCTGCACC,
3′-LPAP-Flag-XmaI:
GGCCCGGGCTACTTGTCGTCATCGTCTTTGTAGTCACCCAGTGCGGTGACATGGAG.
To create mutations, the internal overlapping primers were used as follows:
SSY(58,61,64)AAA:
5′-LPAP-58-61-64:
GCCCGTGACGCAGGGGGCGCCTACCACCCGGCCCGCCTAG,
3′-LPAP-58-61-64:
GCGCCCCCTGCGTCACGGGCGAGGCGGCGCCAGGCCAG;
TY(113,115)AA:
5′-LPAP-113-115:
CAGGACGCAGACGCTGACCACGTCGCGGATGGTGG,
3′-LPAP-113-115:
GTGGTCAGCGTCTGCGTCCTGCTCATCCTCCTGTCG;
S(139)A:
5′-LPAP-139: GAGGCGTCCGCCCCAGAGCAGGTCCCCGTGC,
3′-LPAP-139: GCTCTGGGGCGGACGCCTCTCCACATTGCTGC;
S(153)A:
5′-LPAP-153: GCCAGAGACGCTGACACGGAGGGCGACCTGG,
3′-LPAP-153: CTCCGTGTCAGCGTCTCTGGCTTCCTCAGCCCG;
SSS(163,168,172)AAA:
5′-LPAP-163-168-172:
GCCCCAGGACCAGCGGCCGCAGGGGGCGCTGCTGAGGCCCTGCTGAGTG,
3′-LPAP-163-168-172:
GCGCCCCCTGCGGCCGCTGGTCCTGGGGCGCCGAGGACCAGGTCGCC;
S(99)A:
5′-LPAP S99A: GCTGGGGGCCACAGACAATGACCTTGAGCG,
3′-LPAP S99A: CATTGTCTGTGGCCCCCAGCTCAGCTCGGGC;
S(163)A:
5′-LPAP-163: CCTCGGCGCCCCAGGACCAGCGAGCG,
3′-LPAP-163: GTCCTGGGGCGCCGAGGACCAGGTCGC;
S(168)A:
5′-LPAP-168: GACCAGCGGCCGCAGGGGGCAGTGCTG,
3′-LPAP-168: CCTGCGGCCGCTGGTCCTGGGGAGC;
S(172)A:
5′-LPAP-172: CAGGGGGCGCTGCTGAGGCCCTGCTGAGTG,
3′-LPAP-172: CCTCAGCAGCGCCCCCTGCGCTCGCTG;
Δ122.…..HDVADG:
5′-LPAP-stop122: CGCGGATGGTTAGCTGCAGGCTGACCCTGGGG,
3′-LPAP-stop122: GCCTGCAGCTAACCATCCGCGACGTGGTCATAG;
Δ137.......QCGEA:
5′-LPAP-stop137: GGAGAGGCGTAGAGCCCAGAGCAGGTCCCCG,
3′-LPAP-stop137: CTCTGGGCTCTACGCCTCTCCACATTGCTGCTC;
Δ152.....AEEARD:
5′-LPAP-stop152: GCCAGAGACTAGGACACGGAGGGCGACCTGG,
3′-LPAP-stop152: CCTCCGTGTCCTAGTCTCTGGCTTCCTCAGCCC;
Δ160…..DTEGDL:
5′-LPAP-stop160: GGCGACCTGTAGCTCGGCTCCCCAGGACCAG,
3′-LPAP-stop160: GAGCCGAGCTACAGGTCGCCCTCCGTGTCAC.
PCR products prepared using Pfu-polymerase (SibEnzyme, Russia) were cloned into plasmid pJet 1.2 (Fermentas, Lithuania), sequenced, and then re-cloned into expression plasmid pCMVpA by restriction sites XbaI and XmaI. The plasmids were acquired and isolated using a Qiagen Plasmid Maxi Kit (Qiagen, USA). All sequences of the resulting mutant plasmids were verified by DNA sequencing. Human HEK293T cells were transfected in 12-well plates (Costar, USA). The cells (4·105) in DMEM medium supplemented with 10% fetal calf serum were transfected with 1.5 µg plasmid DNA using 3 µl of LF2000 reagent (Invitrogen, USA) according to the producer’s protocol.
Treatment with proteinase K. The CEM-CCRF cells (4·107) were washed in PBS, suspended in 1 ml of PBS supplemented with 12.5 µM proteinase K (Sigma, USA), and incubated for 15 min at room temperature. To stop the digestion, 1 mM inhibitor PMSF (Sigma) was added, and the cells were washed in PBS.
RESULTS
Mutual inhibition of antibodies reveals at least three epitopes on the LPAP molecule. Earlier, we prepared monoclonal antibodies against LPAP protein that had similar reactivity spectra when tested by immunofluorescence, immunoprecipitation, and Western-blotting [16]. Pronounced differences between the antibodies were revealed by the mutual inhibition test (table). The permeabilized CEM cells were pretreated with inhibiting antibodies at saturating concentration (300 µg/ml) and then stained with FITC-labeled inhibited antibodies.
Cross-inhibition of binding of anti-LPAP antibodies
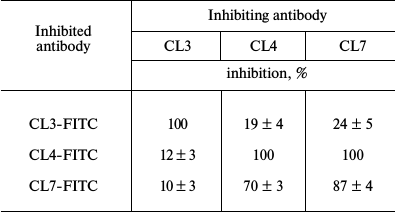
Note: Inhibition was calculated as decrease in mean fluorescence
intensity expressed in % of luminescence intensity on staining the
cells with FITC-labeled antibody without addition of the inhibiting
monoclone. Mean values of inhibition and standard errors are
presented.
The inhibition degree was evaluated by the decrease in cell fluorescence intensity in the presence of inhibiting antibodies in comparison with that of cells not pretreated with the inhibiting unlabeled antibodies. The degree of antibody inhibition indicated the overlapping degree of the epitopes recognized by the inhibiting and inhibited antibodies. Concentrations of the unlabeled antibodies were chosen to cause at least 70% decrease in the inhibition of binding of the same name FITC-labeled antibody. The table shows that antibody CL3 does not inhibit and is not inhibited by any other antibody. Antibodies CL4 and CL7 asymmetrically inhibited each other. Whereas antibody CL7 completely inhibited the binding of CL4, antibody CL4 inhibited the binding of CL7 only partially. Thus, the data on inhibition showed that the three studied antibodies discriminated different or partially overlapping epitopes. Note that the antibodies represent different isotypes: IgG1 (CL4 and CL3) and IgG2a (CL7). Thus, at least three antigenic epitopes exist on the LPAP molecule.
Analysis of antigenic epitopes using LPAP mutants. To compare the revealed antigenic epitopes with the amino acid sequence of LPAP, experiments were performed using mutant variants of this protein. First, the reaction of antibodies with the protein mutants shortened from the C-terminus was studied. Stop codons were inserted in different positions into the plasmid encoding the LPAP protein, and then the modified plasmids were transiently transfected into HEK293T cells. The antibody reaction with LPAP mutants was tested by Western blotting (Fig. 1a) or by immunofluorescence (Fig. 1b). The CL7 antibody reacted with all shortened forms, including the Δ122 mutant with the cytoplasmic domain of LPAP nearly half shortened as compared to the wild type. It seems that the epitope recognized by the CL7 antibody is close to position 122, because the reaction with the mutant Δ122 was slightly weaker than that with mutant Δ160 and the wild-type protein. The CL4 antibody did not react with mutant Δ122, but it bound with the other shortened forms beginning from Δ137. Finally, antibody CL3 did not react with any shortened form.
Fig. 1. Antibody interactions with shortened forms of protein LPAP. HEK293T cells were transiently transfected with plasmids encoding the wild-type protein LPAP (wt) or its shortened forms. a) The cell lysate was analyzed by Western blotting with antibodies CL3, CL4, and CL7 against LPAP; b) cells were stained with the antibodies CL3, CL4, CL7, and anti-FLAG and analyzed by flow cytometry. For comparison, results are presented that were obtained with two alanine point mutants.
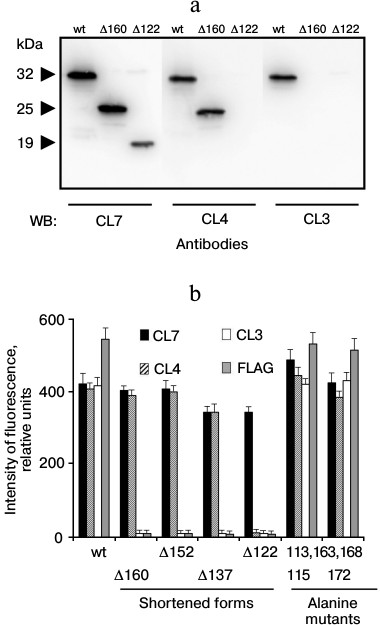
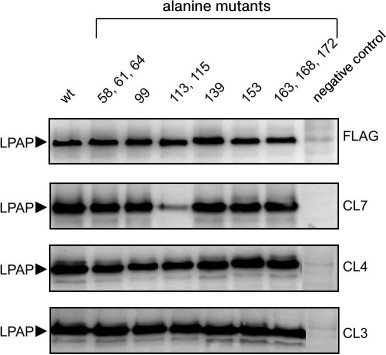
The antigenic epitopes could be located more accurately with point mutants. Six variants were used with alanine mutations in one, two, or three nearby positions (Fig. 2). Staining with antibodies against the FLAG peptide on the C-terminus of the protein was used to control the expression and load on the electrophoretic lane. Antibodies CL3 and CL4 equally recognized all point mutants with which they reacted similarly to the binding with the wild-type protein. As differentiated from this, antibody CL7 in Western blotting weakly recognized mutant T113A,Y115A, but it reacted well with all other point mutants. The low binding of antibody CL7 with mutant T113A,Y115A could not be caused by low expression of this mutant because in parallel samples antibodies CL3 and CL4 and the antibody against the FLAG epitope indicated the normal expression of this mutant. The Western blotting data are somewhat contradicted by the immunofluorescence data, where antibody CL7 bound with mutant T113A,Y115A similarly to binding with the positive control (Fig. 1b). It is very likely that the affinity of antibody CL7 for denatured LPAP is much lower than for the native protein. This could lead to a sharp decrease in binding with the denatured protein in the case of even an insignificant point mutation (Western blotting), but such mutation could have no influence on the reaction with the native protein (immunofluorescence).
Fig. 2. Interactions of antibodies with LPAP carrying point alanine mutations. HEK293T cells were transiently transfected with plasmids encoding the wild-type LPAP (wt) or mutant forms with alanine point mutations. The cell lysate was separated by SDS-PAGE in 12% polyacrylamide gel under reducing conditions in the Laemmli buffer system, transferred onto a membrane, and stained with antibodies against LPAP or FLAG epitopes. Cells transfected with an empty plasmid were used as a negative control.
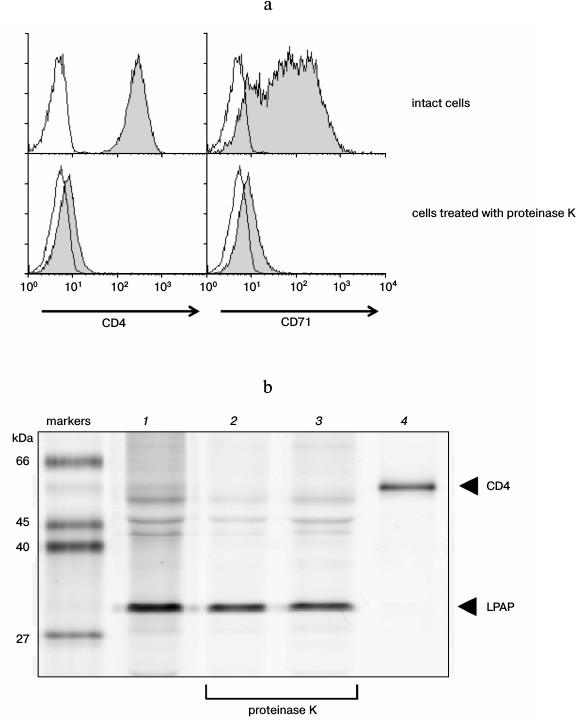
Evaluation of the size of the extracellular part of LPAP. To evaluate the size of the LPAP extracellular part, CEM cells were treated with proteinase K. This enzyme does not penetrate into living cells and is characterized by low specificity to the amino acid sequence, which allows it to remove extracellular domains of transmembrane proteins [18]. The efficiency of proteinase K was demonstrated with respect to well-known transmembrane proteins with pronounced extracellular domains. After the CEM cells were treated with proteinase K, proteins CD4 and CD71 could not be stained with the corresponding antibodies (Fig. 3a). This indicated that the treatment removed extracellular regions of transmembrane proteins that were markedly expressed into the external medium.
The LPAP protein was precipitated from CEM cells using antibody CL7. In the fluorescence immunoprecipitation test, the cell proteins were labeled with fluorescent dye R6G-SE, which reacted with amino groups of lysines. The LPAP protein contains no lysine residues; however, it was stained well with the R6G-SE dye (Fig. 3b, lane 1). The only reacting group capable of LPAP binding with R6G-SE was the N-terminal amine. The treatment with proteinase K after labeling of the cells did not influence the staining intensity of the LPAP band (Fig. 3b, lane 2); consequently, the N-terminal fragment of LPAP on the plasma membrane is unavailable for the enzyme. If the cells were labeled with R6G-SE after treatment with proteinase K, no decrease was observed in the molecular weight of LPAP (Fig. 3b, lane 3), which also confirmed the unavailability of the N-terminal fragment of LPAP for proteinase K.
Fig. 3. Extracellular part of protein LPAP is unavailable for proteolytic action of proteinase K. a) Proteinase K removed proteins CD4 and CD71 from the cell surface. CEM cells were stained with antibodies against antigens CD4 and CD71 and analyzed with flow cytometry (filled histograms). The negative control (the proteins were stained only with secondary antibodies) is represented by the open histograms; b) the CEM cells were labeled with rhodamine 6G and lysed. LPAP was isolated by immunoprecipitation with antibody CL7 (lanes 1-3) or with control antibody anti-CD4 (lane 4). The resulting immunoprecipitates were separated by SDS-PAGE in 12% polyacrylamide gel under reducing conditions in the Laemmli buffer system. Images of the protein bands were obtained upon fluorescence scanning of the gels. The cells were additionally treated with proteinase K (lanes 2 and 3) before labeling with rhodamine 6G (lane 2) or after the labeling (lane 3).
DISCUSSION
The LPAP is a unique molecule. It is not a member of any known family of proteins and does not have analogs in the human genome. The only approach for getting information about its structure is to separate by approaches of bioinformatics domains localized in the extracellular, transmembrane, and cytoplasmic parts of the protein.
The antibodies prepared by us reacted with the native protein within cells (the immunofluorescence test), with the solubilized protein upon lysis of the cells in 1% solution of Triton X-100 (the immunoprecipitation test), and with the denatured protein upon treatment with 2% solution of SDS (Western blotting). These results show that the antibodies used by us recognize linear epitopes of the LPAP protein. In the polypeptide chain of LPAP, at least three regions could be determined that form antigenic epitopes. Antibody CL3 discriminated the most distal epitope located between L159 and the C-terminus. Experiments with shortened forms of LPAP clearly indicated that the antibody CL4 epitope was limited by a region consisting of only 15 a.a. between G122 and A137. And finally, the results obtained with the point and shortened mutants indicate that antibody CL7 binds with the most proximal epitope located close to G121 and including T113 and Y115 (Fig. 4). The proximity of the epitopes of the CL4 and CL7 antibodies explains why these antibodies markedly inhibit each other on binding with LPAP.
Fig. 4. Epitopic map of LPAP protein.
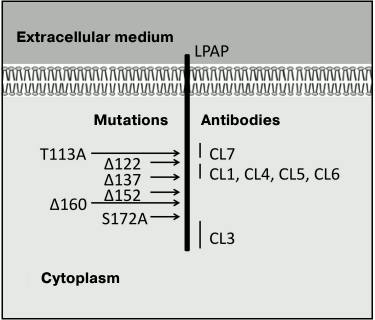
The human LPAP and mouse CD45-AP are highly homologous proteins. Their amino acid sequences are more than 60% coinciding; nevertheless, neither of the prepared antibodies reacted with the mouse CD45-AP (data not presented). It should be noted that the polypeptide sequence of the CD45-AP lacked both T113 and Y115, which participated in formation of epitope CL7. Moreover, the epitope region of antibody CL4 between G122 and A137 was very variable, which completely explains the absence of reaction of the CL4 and CL7 antibodies with mouse CD45-AP. We still cannot localize the CL3 antibody epitope accurately. On one hand, alanine mutations simultaneously in three positions – 163, 168, and 172 – did not limit the binding of antibody CL3, which indicates that the antibody CL3 epitope is located nearer to the C-terminus of the protein and is before S172. On the other hand, the C-terminus of the protein is the most conservative, and the absence of the CL3 antibody reaction with mouse CD45-AP has to be due to minimal differences between the human and mouse proteins in this region.
The N-terminal sequence of proteins is usually subjected to posttranslational modification and determines such features of a protein as localization, processing, and lifetime [19-21]. Theoretical analysis with the Signal P 4.1 program [22] predicted that the N-terminal fragment of LPAP consisting of the first 20 a.a. is a signaling peptide that must be removed from the mature LPAP molecule. On the other hand, different bioinformatic resources [23, 24] predict that the transmembrane domain of LPAP should be located on the region that begins after a.a. 31 or 35 and is continued up to a.a. 54 or 55. Thus, the extracellular part of the mature LPAP molecule can consist of 11-15 a.a. A possibility of the translation beginning from methionine located in the 11th position increases the uncertainty of the size of the extracellular part of LPAP [25, 26]. It seems that the size of the extracellular domain can be assessed based on the molecular weight of the protein measured by SDS-PAGE. However, experiments with recombinant LPAP revealed that this protein has abnormal electrophoretic mobility [16]. The native LPAP also displayed the same feature [1]. At the theoretical weight of 21 kDa, the native LPAP migrated as a 32-kDa protein. And because of such abnormal behavior, it was difficult to determine the size of the extracellular domain of LPAP based on measurement of the electrophoretic mobility of the protein.
To determine the extracellular part of transmembrane proteins, in some cases the cells have been treated with such membrane impermeable proteases as trypsin [11], proteinase K [18], etc. We used this approach combined with fluorescence immunoprecipitation. It was reasonable to expect that the treatment of cells with proteinase K after labeling the proteins with the R6G-SE dye would detach the N-terminal fragment together with the fluorescent label. However, this was not observed. Moreover, the inverse order of treatment with proteinase K and fluorescent labeling caused no shortening of the polypeptide chain. Earlier we prepared monoclonal antibodies against the N-terminal peptide of the mature LPAP molecule (a.a. 21-30). These antibodies reacted with the peptide from the conjugate with proteins BSA and KLH but did not bind with the native LPAP. It seems that the N-terminus of the LPAP protein is insufficiently exposed into the extracellular medium to be recognized by the antibodies. Thus, our findings are in agreement with data indicating that the N-terminal fragment of LPAP has minimal exposure to the extracellular medium. Therefore, the possibility of influencing LPAP with some extracellular ligands, including specific antibodies, casts some doubt. Our findings give more accurate ideas about the structure of the LPAP molecule (Fig. 4). New experimental data on the structure of the protein will be useful for establishing its function, which is still unknown.
This work was supported by the Russian Foundation for Basic Research (project 12-04-00402) and the Russian Scientific Foundation (project 14-15-00702).
REFERENCES
1.Schraven, B., Schoenhaut, D., Bruyns, E., Koretzky,
G., Eckerskorn, C., Wallich, R., Kirchgessner, H., Sakorafas, P.,
Labkovsky, B., Ratnofsky, S., and Meuer, S. (1994) LPAP, a novel 32-kDa
phosphoprotein that interacts with CD45 in human lymphocytes, J.
Biol. Chem., 269, 29102-29111.
2.Takeda, A., Maizel, A. L., Kitamura, K., Ohta, T.,
and Kimura, S. (1994) Molecular cloning of the CD45-associated 30-kDa
protein, J. Biol. Chem., 269, 2357-2360.
3.Takeda, A., Wu, J. J., and Maizel, A. L. (1992)
Evidence for monomeric and dimeric forms of CD45 associated with a
30-kDa phosphorylated protein, J. Biol. Chem., 267,
16651-16659.
4.Mustelin, T., and Tasken, K. (2003) Positive and
negative regulation of T-cell activation through kinases and
phosphatases, Biochem. J., 371, 15-27.
5.Takeda, A., Matsuda, A., Paul, R. M., and Yaseen,
N. R. (2004) CD45-associated protein inhibits CD45 dimerization and
up-regulates its protein tyrosine phosphatase activity, Blood,
103, 3440-3447.
6.Kung, C., Okumura, M., Seavitt, J. R., Noll, M. E.,
White, L. S., Pingel, J. T., and Thomas, M. L. (1999) CD45-associated
protein is not essential for the regulation of antigen
receptor-mediated signal transduction, Eur. J. Immunol.,
29, 3951-3955.
7.Ding, I., Bruyns, E., Li, P., Magada, D., Paskind,
M., Rodman, L., Seshadri, T., Alexander, D., Giese, T., and Schraven,
B. (1999) Biochemical and functional analysis of mice deficient in
expression of the CD45-associated phosphoprotein LPAP, Eur. J.
Immunol., 29, 3956-3961.
8.Matsuda, A., Motoya, S., Kimura, S., McInnis, R.,
Maizel, A. L., and Takeda, A. (1998) Disruption of lymphocyte function
and signaling in CD45-associated protein-null mice, J. Exp.
Med., 187, 1863-1870.
9.Leitenberg, D., Falahati, R., Lu, D. D., and
Takeda, A. (2007) CD45-associated protein promotes the response of
primary CD4 T cells to low-potency T-cell receptor (TCR) stimulation
and facilitates CD45 association with CD3/TCR and Lck,
Immunology, 121, 545-554.
10.Altin, J. G., and Sloan, E. K. (1997) The role of
CD45 and CD45-associated molecules in T-cell activation, Immunol.
Cell Biol., 75, 430-445.
11.Kitamura, K., Maiti, A., Ng, D. H., Johnson, P.,
Maizel, A. L., and Takeda, A. (1995) Characterization of the
interaction between CD45 and CD45-AP, J. Biol. Chem.,
270, 21151-21157.
12.Cahir McFarland, E. D., and Thomas, M. L. (1995)
CD45 protein-tyrosine phosphatase associates with the WW
domain-containing protein, CD45AP, through the transmembrane region,
J. Biol. Chem., 270, 28103-28107.
13.Bruyns, E., Hendricks-Taylor, L. R., Meuer, S.,
Koretzky, G. A., and Schraven, B. (1995) Identification of the sites of
interaction between lymphocyte phosphatase-associated phosphoprotein
(LPAP) and CD45, J. Biol. Chem., 270, 31372-31376.
14.Veillette, A., Soussou, D., Latour, S., Davidson,
D., and Gervais, F. G. (1999) Interactions of CD45-associated protein
with the antigen receptor signaling machinery in T-lymphocytes, J.
Biol. Chem., 274, 14392-14399.
15.Shimizu, Y., Sugiyama, H., Fujii, Y., Sasaki, K.,
Inoue, K., Ogawa, H., Tamaki, H., Miyake, S., Oji, Y., Soma, T.,
Yamagami, T., Hirata, M., Ikeda, K., Monden, T., and Kishimoto, T.
(1997) Lineage- and differentiation stage-specific expression of LSM-1
(LPAP), a possible substrate for CD45, in human hematopoietic cells,
Am. J. Hematol., 54, 1-11.
16.Filatov, A. V., Lavrova, O. I., Mazurov, D. V.,
and Sheval’e, A. F. (2011) Expression of lymphocyte
phosphatase-associated protein on human lymphoid lines,
Immunologiya, 32, 30-34.
17.Filatov, A. V., Krotov, G. I., Zgoda, V. G., and
Volkov, Y. (2007) Fluorescent immunoprecipitation analysis of cell
surface proteins: a methodology compatible with mass-spectrometry,
J. Immunol. Methods, 319, 21-33.
18.Polyak, M. J., Tailor, S. H., and Deans, J. P.
(1998) Identification of a cytoplasmic region of CD20 required for its
redistribution to a detergent-insoluble membrane compartment, J.
Immunol., 161, 3242-3248.
19.Martoglio, B., and Dobberstein, B. (1998) Signal
sequences: more than just greasy peptides, Trends Cell Biol.,
8, 410-415.
20.Meinnel, T., Peynot, P., and Giglione, C. (2005)
Processed N-termini of mature proteins in higher eukaryotes and
their major contribution to dynamic proteomics, Biochimie,
87, 701-712.
21.Polevoda, B., and Sherman, F. (2003)
N-Terminal acetyltransferases and sequence requirements for
N-terminal acetylation of eukaryotic proteins, J. Mol.
Biol., 325, 595-622.
22.Petersen, T. N., Brunak, S., von Heijne, G., and
Nielsen, H. (2011) SignalP 4.0: discriminating signal peptides from
transmembrane regions, Nat. Methods, 8, 785-786.
23.Hirokawa, T., Boon-Chieng, S., and Mitaku, S.
(1998) SOSUI: classification and secondary structure prediction system
for membrane proteins, Bioinformatics, 14, 378-379.
24.Hofmann, K., and Stoffel, W. (1993) TMBASE
– a database of membrane spanning protein segments, Biol.
Chem. Hoppe-Seyler, 374, 166.
25.Bruyns, E., Mincheva, A., Bruyns, R. M.,
Kirchgessner, H., Weitz, S., Lichter, P., Meuer, S., and Schraven, B.
(1996) Sequence, genomic organization, and chromosomal localization of
the human LPAP (PTPRCAP) and mouse CD45-AP/LSM-1 genes,
Genomics, 38, 79-83.
26.Kitamura, K., Matsuda, A., Motoya, S., and
Takeda, A. (1997) CD45-associated protein is a lymphocyte-specific
membrane protein expressed in two distinct forms, Eur. J.
Immunol., 27, 383-388.